Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hidekatsu Yanai | -- | 2389 | 2024-03-18 08:55:15 | | | |
| 2 | Mona Zou | Meta information modification | 2389 | 2024-03-19 10:14:37 | | | | |
| 3 | Mona Zou | -15 word(s) | 2374 | 2024-03-27 09:37:13 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Yanai, H.; Adachi, H.; Hakoshima, M.; Iida, S.; Katsuyama, H. URAT1 and Other UA Transporters with Metabolic Syndrome. Encyclopedia. Available online: https://encyclopedia.pub/entry/56370 (accessed on 25 June 2026).
Yanai H, Adachi H, Hakoshima M, Iida S, Katsuyama H. URAT1 and Other UA Transporters with Metabolic Syndrome. Encyclopedia. Available at: https://encyclopedia.pub/entry/56370. Accessed June 25, 2026.
Yanai, Hidekatsu, Hiroki Adachi, Mariko Hakoshima, Sakura Iida, Hisayuki Katsuyama. "URAT1 and Other UA Transporters with Metabolic Syndrome" Encyclopedia, https://encyclopedia.pub/entry/56370 (accessed June 25, 2026).
Yanai, H., Adachi, H., Hakoshima, M., Iida, S., & Katsuyama, H. (2024, March 18). URAT1 and Other UA Transporters with Metabolic Syndrome. In Encyclopedia. https://encyclopedia.pub/entry/56370
Yanai, Hidekatsu, et al. "URAT1 and Other UA Transporters with Metabolic Syndrome." Encyclopedia. Web. 18 March, 2024.
Copy Citation
Urate transporter 1 (URAT1), which is a urate anion exchanger that regulates serum uric acid (UA) levels in the human kidney, was identified in 2002, and it has been targeted by uricosuric agents. In humans, renal reabsorption of UA into the blood plays an important role in controlling serum UA levels. The UA exchange is mediated by various molecules expressed in the renal proximal tubule. UA enters the proximal tubule epithelial cells in exchange for monocarboxylate via apical URAT1 and for dicarboxylate via the apical organic anion transporter (OAT) 4. OAT1 and OAT3 on the basolateral membrane of epithelial cells transport UA from the renal interstitial into the renal proximal tubule epithelial cells. Renal UA reabsorption is mainly mediated by URAT1 and glucose transporter 9 (GLUT9). Apical GLUT9b plays a significant role in UA reabsorption; the reabsorbed UA exits the proximal tubule epithelial cells into the blood through basolateral GLUT9a. The ATP-binding cassette transporter G2 (ABCG2) has been identified as a high-capacity UA exporter that mediates renal and/or extra-renal (intestinal) UA excretion.
ATP-binding cassette transporter G2
chronic kidney disease
dotinurad
hyperuricemia
organic anion transporter1/3
urate transporter 1
1. Introduction
Urate transporter 1 (URAT1), which is a urate anion exchanger that regulates serum uric acid (UA) levels in the human kidney, was identified in 2002 [1], and it has been targeted by uricosuric agents. In humans, renal reabsorption of UA into the blood plays an important role in controlling serum UA levels. The UA exchange is mediated by various molecules expressed in the renal proximal tubule [2][3] (Figure 1). UA enters the proximal tubule epithelial cells in exchange for monocarboxylate via apical URAT1 and for dicarboxylate via the apical organic anion transporter (OAT) 4 [4]. OAT1 and OAT3 on the basolateral membrane of epithelial cells transport UA from the renal interstitial into the renal proximal tubule epithelial cells [4][5]. Renal UA reabsorption is mainly mediated by URAT1 and glucose transporter 9 (GLUT9) [1][6][7][8]. Apical GLUT9b plays a significant role in UA reabsorption; the reabsorbed UA exits the proximal tubule epithelial cells into the blood through basolateral GLUT9a [4]. The ATP-binding cassette transporter G2 (ABCG2) has been identified as a high-capacity UA exporter that mediates renal and/or extra-renal (intestinal) UA excretion [9][10].
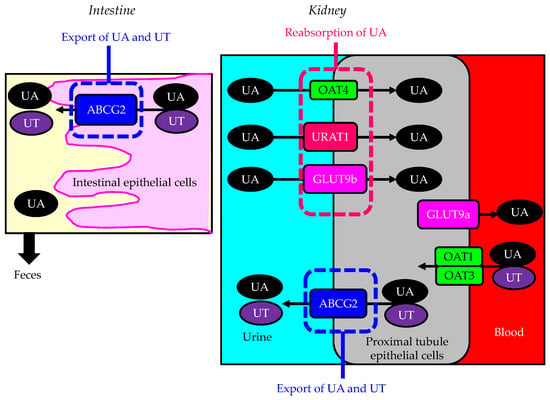
Figure 1. Urate transporters in the kidneys and intestine. Black arrows indicate the flow of uric acid and uremic toxins. ABCG2—ATP-binding cassette transporter G2; GLUT9—glucose transporter 9; OAT—organic anion transporter; UA—uric acid; URAT1—urate transporter 1; UT—uremic toxin.
Uricosuric agents have been developed to target such UA transporters and have been used as therapeutic agents for hyperuricemia. Probenecid inhibits URAT1 and GLUT9 [11]. Benzbromarone also inhibits URAT 1 and GLUT 9 [12]. Lesinurad and arhalofenate inhibit URAT1 and OAT4 [11]. It has been difficult to accurately evaluate the function of URAT1 because the previous uricosuric agents inhibited not only URAT1 but also GLUT9 and OAT4.
2. The Association of URAT1 and Other UA Transporters with Metabolic Syndrome
2.1. Metabolic Syndrome and Hyperuricemia
Hyperuricemia is significantly associated with the development and severity of metabolic syndrome. A meta-analysis showed that higher serum UA levels led to an increased risk of metabolic syndrome, with a linear dose–response relationship [13]. The serum UA concentrations increased with the number of components of metabolic syndrome adjusted for age, sex, creatinine clearance, and alcohol, and diuretic use [14]. Multivariate analyses showed that the visceral fat area (VFA) was the most important determinant of elevation in serum UA and a decrease in UA clearance [15]. The magnitude of the insulin resistance and the serum UA levels were significantly related; insulin resistance was also significantly and inversely related to urinary UA clearance, and urinary UA clearance was significantly and inversely associated with serum UA levels [16]. Insulin resistance due to visceral fat accumulation may increase serum UA by decreasing renal UA clearance in patients with metabolic syndrome.
2.2. The Effect of Insulin Resistance on URAT1 Expression
To elucidate the mechanism of obesity and metabolic syndrome-related hyperuricemia, the expression of URAT1 was investigated [17]. The protein level of URAT1 increased in the kidneys of leptin-deficient mice (ob/ob mice) [17]. Furthermore, the quick fat diet (crude fat content: 13.6%) enhanced the protein level of URAT1 in the kidneys of C57BL/6 mice [17]. Insulin-resistant Otsuka Long–Evans Tokushima Fatty (OLETF) rats and the control, Long–Evans Tokushima Ohtsuka (LETO) rats, were used as a model for acute hyperuricemia [18]. The OLETF rats showed a significantly higher incidence of hyperuricemia compared to the control LETO rats, indicating that insulin resistance induces hyperuricemia following a high-purine load [18]. Following a high-purine load, insulin resistance enhanced UA reabsorption through upregulation of the URAT1 expression [18].
A high-fructose diet (HFD) upregulated the expression of GLUT9 and URAT1 in the kidneys and increased the serum UA concentration in rats [19]. Another study also revealed that long-term HFD significantly upregulated the protein expression of GLUT9 and URAT1 in the kidneys of mice [20]. Resveratrol is a polyphenol that is abundant in plants; it has been reported to exert anti-inflammatory and antioxidative effects, inhibit lipid peroxidation, and extend life in mice [21]. Furthermore, the effects of resveratrol on the amelioration of insulin resistance and liver and kidney pathologies have been shown in several animal models [22][23]. Compared with those in the HFD group, the protein expression levels of GLUT9 and URAT1 were significantly lower in the HFD group treated with resveratrol. Insulin resistance enhanced the expression of URAT1 and GLUT9.
2.3. The Effect of Insulin on UA Transport by Other Urate Transporters
Insulin and hyperinsulinemia reduce the renal fractional excretion of UA and play a key role in the genesis of hyperuricemia and gout. Physiological euglycemic hyperinsulinemia induced by insulin infusion in healthy volunteers acutely reduced urinary UA, suggesting a significant contribution of insulin to the pathogenesis of hyperuricemia [24][25][26]. In rats, insulin decreased urinary UA excretion, with a concurrent increased expression of URAT1 and a decreased expression of ABCG2 [27]. There was an increased expression of GLUT9 in the kidneys of streptozotocin-induced diabetic mice [28]. The heterologous expression of individual UA transporters in Xenopus oocytes revealed that insulin increased UA transport by GLUT9, OAT1, and OAT3 and decreased UA transport by ABCG2 [29].
The effects of insulin resistance and hyperinsulinemia on UA transport by each of the UA transporters are shown in Figure 2. Insulin resistance and hyperinsulinemia increase UA transport by URAT1 and GLUT9 and decrease UA transport by ABCG2, which may induce a decrease in renal UA clearance. Therefore, URAT1, GLUT9, and ABCG2 can be therapeutic targets for uricosuric drugs in patients with insulin resistance and hyperinsulinemia.
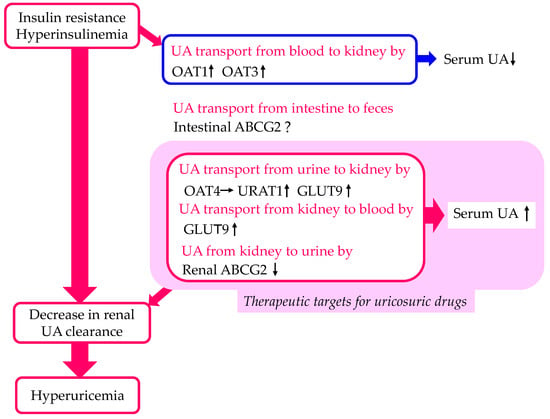
Figure 2. Changes in UA transport by UA transporters in the kidneys and intestine by insulin resistance and hyperinsulinemia. Upward- and downward-facing arrows indicate increase or decrease in substances or expression of molecules, respectively. Right arrow and ? indicate no change and no available data about change of substances or expression of molecules, respectively. ABCG2—ATP-binding cassette transporter G2; GLUT9—glucose transporter 9; OAT—organic anion transporter; UA—uric acid; URAT1—urate transporter 1.
2.4. The Effect of Inhibition of URAT1 on Metabolic Parameters in Humans
The researchers found that dotinurad reduced body weight, blood pressure, HbA1c, serum low-density lipoprotein-cholesterol (LDL-C), triglyceride (TG), and non-high-density lipoprotein-cholesterol (non-HDL-C), as well as serum UA, in patients with CKD and DKD [30]. The researchers speculated that dotinurad selectively inhibits URAT1 and increases the urinary concentration of UA in the proximal tubules; this un-reabsorbed UA may compete with urinary glucose for apical GLUT9b, reducing glucose reabsorption, which may induce improvements in HbA1c, serum lipids, blood pressure, and body weight.
2.5. The Effect of Inhibition of URAT1 on Metabolic Parameters in Mice
Tanaka, Y. et al. found that URAT1 was also expressed in the liver, white adipose tissue (WAT), and brown adipose tissue (BAT) in addition to the kidneys [31]. Dotinurad administration significantly ameliorated high-fat diet-induced obesity and insulin resistance [31]. Serum TG in high-fat diet-fed mice was elevated in comparison with that in normal-fat diet-fed mice, and dotinurad significantly reduced serum TG in both types of mice [31]. Remarkably, a high-fat diet induced nonalcoholic fatty liver disease (NAFLD), which was attenuated by dotinurad [31]. Various factors, such as pro-inflammatory cytokines released from adipose tissues, hypercholesterolemia, and hyperuricemia, contribute to the development of NAFLD in high-fat diet-induced obese mice [32]. Hyperuricemia directly induces fat accumulation and inflammation in hepatocytes through URAT1 [33]. Dotinurad may improve NAFLD by inhibiting extracellular UA uptake in hepatocytes via URAT1, resulting in a reduction in lipid deposition and inflammation. The re-browning of brown adipose tissue (BAT) and the browning of epididymal white adipose tissue (WAT) may be also associated with an improvement in NAFLD via adipokines [34].
WAT could be converted to beige adipose tissue (browning), which increases energy expenditure by activating the uncoupling protein 1 (UCP1), which improves systemic insulin resistance [34][35]. The uptake of UA in WAT by URAT1 leads to WAT dysfunction and the deterioration of systemic insulin resistance [36]. In epididymal WAT, dotinurad significantly increased the UCP1 expression under high-fat diet conditions, indicating that the selective inhibition of URAT1 led to the browning of WAT under high-fat diet conditions [31]. A previous study showed that the enhanced UA uptake into WAT via URAT1 and the elevation in the intracellular UA led to the inhibition of the leptin–AMP-activated protein kinase (AMPK) pathway, which resulted in a reduction in the UCP1 expression in WAT [35].
The upregulation of the expression and activity of UCP1 in BAT plays an important role in the improvement of glucose metabolism and insulin sensitivity [37]. The UCP1 levels in BAT were significantly increased by dotinurad [31]. The uptake of UA can increase the oxidative stress in adipocytes, which induces insulin resistance [38]. The reactive oxygen species (ROS) levels in BAT were significantly reduced by treatment with dotinurad [31].
2.6. The Effects of Other UA-Lowering Drugs on Metabolic Parameters
Allopurinol and febuxostat are xanthine oxidase (XO) inhibitors that reduce the hepatic production of UA. In comparison with no treatment, the allopurinol and febuxostat treatments induced a significant reduction in body weight, systolic blood pressure, blood glucose, insulin, and lipids in rat models of insulin resistance and metabolic syndrome [39].
Allopurinol significantly reduced hepatic steatosis, epididymal fat, serum UA, the homeostatic model assessment for insulin resistance (HOMA-IR), hepatic enzyme levels, and cholesterol in the HFD-fed OLETF rats [40]. The hepatic expression of lipogenic genes, such as sterol regulatory element-binding protein 1c (SREBP-1c) and stearoyl-CoA desaturase 1 (SCD-1), was significantly upregulated in the OLETF and the HFD-fed OLETF rats compared with the LETO rats. However, allopurinol significantly downregulated SREBP-1c and SCD-1 gene expressions in the HFD-fed OLETF rats. Peroxisome proliferator-activated receptor alpha (PPARα) and carnitine palmitoyl-transferase 1 (CPT-1) were significantly downregulated in the OLETF and the HFD-fed OLETF rats compared with the LETO rats [40]. However, allopurinol improved the downregulation of lipid oxidation genes observed in the HFD-fed OLETF rats. The hepatic mRNA expression of tumor necrosis factor-alpha (TNF-α) was significantly increased in the OLETF and the HFD-fed OLETF rats, and this increase was abolished by allopurinol. In addition, allopurinol significantly decreased endoplasmic reticulum (ER) stress-induced protein expression, in comparison with the no-treatment group.
Insulin resistance increases the expression of SREBP-1c, which increases fatty acid (FA) synthesis [41]. Hepatic FA metabolism is controlled by the combination of FA uptake, FA export by very-low-density lipoprotein (VLDL) secretion, FA synthesis by SREBP-1c, and FA oxidation by β-oxidation. The entry of FA into mitochondria depends on CPT-1. One of the major regulators of CPT-1 is PPARα [42][43][44][45]. The activation of PPARα induces the transcription of genes associated with FA oxidation [42][46][47]. SCD1 plays a crucial role in FA oxidation, FA synthesis, and storage [48]. It was proposed that SCD1 plays a crucial role in the development of obesity in Mediterranean countries [49]. In experimental animals, SCD1 was significantly associated with obesity and insulin resistance [50][51]. Therefore, the allopurinol-mediated downregulation of SREBP-1c and SCD-1 genes and the upregulation of PPARα and CPT-1 in the HFD-fed OLETF rats indicate that allopurinol has a beneficial effect on hepatic steatosis in insulin resistance.
The relationship between the decrease in serum UA and VFA reduction in patients with gout was investigated [52]. The UA-lowering therapy (ULT) (febuxostat 20–80 mg/day or benzbromarone 25–50 mg/day) resulted in a decrease in the serum UA level, accompanied by a decrease in VFA. Using the multiple regression model, change in serum UA was a significant determinant of the decrease in VFA (beta, 0.302; p = 0.001). The reduction in serum UA is positively associated with reduced VFA, providing a rationale for clinical trials to affirm whether ULT promotes the loss of visceral fat in patients with gout. The ULT significantly reduced body weight, blood pressure, serum TG and total cholesterol levels, aspartate aminotransferase (ALT), and aspartate aminotransferase (AST).
Treatment with the XO inhibitor, topiroxostat, suppressed weight gain compared to control without any impact on food intake in diabetic obese mice [53]. However, the weight of the fat pads and the hepatic and muscle TG content did not change. Prehypertensive, obese adolescents, aged 11 to 17 years, were randomized to the XO inhibitor, allopurinol, uricosuric, probenecid, or placebo in a randomized, double-blinded, placebo-controlled trial (RCT) [54]. The subjects treated with ULT showed a significantly high reduction in blood pressure.
2.7. The Possible Mechanisms of an Improvement in Metabolic Parameters by Dotinurad
The possible mechanisms of an improvement in metabolic parameters by dotinurad are shown in Figure 3. In the kidney, dotinurad selectively inhibits URAT1 and increases the urinary concentration of UA in the proximal tubules; this un-reabsorbed UA may compete with urinary glucose for apical GLUT9b, reducing glucose reabsorption, which may induce an improvement in HbA1c, serum lipids, blood pressure, and body weight. In the liver, the inhibition by dotinurad of UA entry into the liver via URAT1 may upregulate the genes associated with FA oxidation and may downregulate the genes associated with FA synthesis and inflammation, which improve hepatic steatosis, systemic insulin resistance, and serum lipids. The inhibition of URAT1 in WAT by dotinurad induces the browning of WAT, and the inhibition of URAT1 in BAT increases the expression of UCP-1 and decreases the production of ROS, which may reduce body weight and visceral fat and may improve insulin resistance as well as glucose and lipid metabolism.
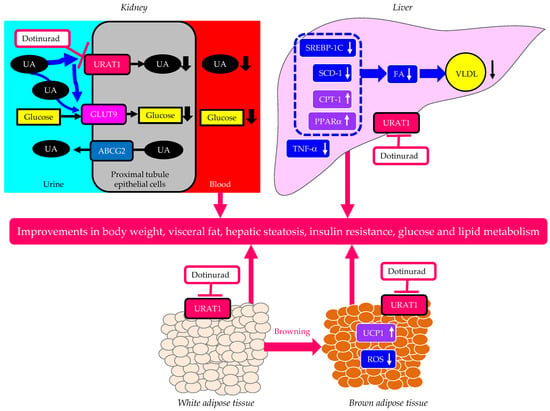
Figure 3. The possible mechanisms of an improvement in metabolic parameters by dotinurad. Upward- and downward-facing arrows indicate increase or decrease in substances or expression of molecules, respectively. ABCG2—ATP-binding cassette transporter G2; CPT-1—carnitine palmitoyl-transferase 1; FA—fatty acid; GLUT9—glucose transporter 9; PPARα—proliferator-activated receptor alpha; ROS—reactive oxygen species; SCD-1—stearoyl-CoA desaturase 1; SREBP-1c—sterol regulatory element-binding protein 1c; TNF-α—tumor necrosis factor-alpha; UA—uric acid; UCP1—uncoupling protein 1; URAT1—urate transporter 1; VLDL—very-low-density lipoprotein.
References
- Enomoto, A.; Kimura, H.; Chairoungdua, A.; Shigeta, Y.; Jutabha, P.; Cha, S.H.; Hosoyamada, M.; Takeda, M.; Sekine, T.; Igarashi, T. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 2002, 417, 447–452.
- Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Molecular Biological and Clinical Understanding of the Pathophysiology and Treatments of Hyperuricemia and Its Association with Metabolic Syndrome, Cardiovascular Diseases and Chronic Kidney Disease. Int. J. Mol. Sci. 2021, 22, 9221.
- Dalbeth, N.; Merriman, T. Crystal ball gazing: New therapeutic targets for hyperuricaemia and gout. Rheumatology 2009, 48, 222–226.
- Merriman, T.R.; Dalbeth, N. The genetic basis of hyperuricaemia and gout. Jt. Bone Spine 2011, 78, 35–40.
- Xu, L.; Shi, Y.; Zhuang, S.; Liu, N. Recent advances on uric acid transporters. Oncotarget 2017, 8, 100852–100862.
- Caulfield, M.J.; Munroe, P.B.; O’Neill, D.; Witkowska, K.; Charchar, F.J.; Doblado, M.; Evans, S.; Eyheramendy, S.; Onipinla, A.; Howard, P.; et al. SLC2A9 is a high-capacity urate transporter in humans. PLoS Med. 2008, 5, e197.
- Li, S.; Sanna, S.; Maschio, A.; Busonero, F.; Usala, G.; Mulas, A.; Lai, S.; Dei, M.; Orrù, M.; Albai, G. The GLUT9 gene is associated with serum uric acid levels in Sardinia and Chianti cohorts. PLoS Genet. 2007, 3, e194.
- Vitart, V.; Rudan, I.; Hayward, C.; Gray, N.K.; Floyd, J.; Palmer, C.N.; Knott, S.A.; Kolcic, I.; Polasek, O.; Graessler, J.; et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat. Genet. 2008, 40, 437–442.
- Woodward, O.M.; Köttgen, A.; Coresh, J.; Boerwinkle, E.; Guggino, W.B.; Köttgen, M. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc. Natl. Acad. Sci. USA 2009, 106, 10338–10342.
- Matsuo, H.; Takada, T.; Ichida, K.; Nakamura, T.; Nakayama, A.; Ikebuchi, Y.; Ito, K.; Kusanagi, Y.; Chiba, T.; Tadokoro, S. Common defects of ABCG2, a high-capacity urate exporter, cause gout: A function-based genetic analysis in a Japanese population. Sci. Transl. Med. 2009, 1, 5ra11.
- Sattui, S.E.; Gaffo, A.L. Treatment of hyperuricemia in gout: Current therapeutic options, latest developments and clinical implications. Ther. Adv. Musculoskelet. Dis. 2016, 8, 145–159.
- Reinders, M.K.; van Roon, E.N.; Houtman, P.M.; Brouwers, J.R.; Jansen, T.L. Biochemical effectiveness of allopurinol and allopurinol-probenecid in previously benzbromarone-treated gout patients. Clin. Rheumatol. 2007, 26, 1459–1465.
- Yuan, H.; Yu, C.; Li, X.; Sun, L.; Zhu, X.; Zhao, C.; Zhang, Z.; Yang, Z. Serum Uric Acid Levels and Risk of Metabolic Syndrome: A Dose-Response Meta-Analysis of Prospective Studies. J. Clin. Endocrinol. Metab. 2015, 100, 4198–4207.
- Hjortnaes, J.; Algra, A.; Olijhoek, J.; Huisman, M.; Jacobs, J.; van der Graaf, Y.; Visseren, F. Serum uric acid levels and risk for vascular diseases in patients with metabolic syndrome. J. Rheumatol. 2007, 34, 1882–1887.
- Takahashi, S.; Yamamoto, T.; Tsutsumi, Z.; Moriwaki, Y.; Yamakita, J.; Higashino, K. Close correlation between visceral fat accumulation and uric acid metabolism in healthy men. Metabolism 1997, 46, 1162–1165.
- Facchini, F.; Chen, Y.D.; Hollenbeck, C.B.; Reaven, G.M. Relationship between resistance to insulin-mediated glucose uptake, urinary uric acid clearance, and plasma uric acid concentration. JAMA 1991, 266, 3008–3011.
- Doshi, M.; Takiue, Y.; Saito, H.; Hosoyamada, M. The increased protein level of URAT1 was observed in obesity/metabolic syndrome model mice. Nucleosides Nucleotides Nucleic Acids 2011, 30, 1290–1294.
- Miao, Z.; Yan, S.; Wang, J.; Wang, B.; Li, Y.; Xing, X.; Yuan, Y.; Meng, D.; Wang, L.; Gu, J. Insulin resistance acts as an independent risk factor exacerbating high-purine diet induced renal injury and knee joint gouty lesions. Inflamm. Res. 2009, 58, 659–668.
- Yang, Y.; Zhang, D.M.; Liu, J.H.; Hu, L.S.; Xue, Q.C.; Ding, X.Q.; Kong, L.D. Wuling San protects kidney dysfunction by inhibiting renal TLR4/MyD88 signaling and NLRP3 inflammasome activation in high fructose-induced hyperuricemic mice. J. Ethnopharmacol. 2015, 169, 49–59.
- Zhang, X.; Nie, Q.; Zhang, Z.; Zhao, J.; Zhang, F.; Wang, C.; Wang, X.; Song, G. Resveratrol affects the expression of uric acid transporter by improving inflammation. Mol. Med. Rep. 2021, 24, 564.
- Thiel, G.; Rössler, O.G. Resveratrol regulates gene transcription via activation of stimulus-responsive transcription factors. Pharmacol. Res. 2017, 117, 166–176.
- Cheng, K.; Song, Z.; Chen, Y.; Li, S.; Zhang, Y.; Zhang, H.; Zhang, L.; Wang, C.; Wang, T. Resveratrol protects against renal damage via attenuation of inflammation and oxidative stress in high-fat-diet-induced obese mice. Inflammation 2019, 42, 937–945.
- Saldanha, J.F.; Leal, V.O.; Stenvinkel, P.; Carraro-Eduardo, J.C.; Mafra, D. Resveratrol: Why is it a promising therapy for chronic kidney disease patients? Oxid. Med. Cell. Longev. 2013, 2013, 963217.
- Quiñones Galvan, A.; Natali, A.; Baldi, S.; Frascerra, S.; Sanna, G.; Ciociaro, D.; Ferrannini, E. Effect of insulin on uric acid excretion in humans. Am. J. Physiol. 1995, 268, E1–E5.
- Muscelli, E.; Natali, A.; Bianchi, S.; Bigazzi, R.; Galvan, A.Q.; Sironi, A.M.; Frascerra, S.; Ciociaro, D.; Ferrannini, E. Effect of insulin on renal sodium and uric acid handling in essential hypertension. Am. J. Hypertens. 1996, 9, 746–752.
- Ter Maaten, J.C.; Voorburg, A.; Heine, R.J.; Ter Wee, P.M.; Donker, A.J.; Gans, R.O. Renal handling of urate and sodium during acute physiological hyperinsulinaemia in healthy subjects. Clin. Sci. 1997, 92, 51–58.
- Toyoki, D.; Shibata, S.; Kuribayashi-Okuma, E.; Xu, N.; Ishizawa, K.; Hosoyamada, M.; Uchida, S. Insulin stimulates uric acid reabsorption via regulating urate transporter 1 and ATP-binding cassette subfamily G member 2. Am. J. Physiol. Renal. Physiol. 2017, 313, F826–F834.
- Keembiyehetty, C.; Augustin, R.; Carayannopoulos, M.O.; Steer, S.; Manolescu, A.; Cheeseman, C.I.; Moley, K.H. Mouse glucose transporter 9 splice variants are expressed in adult liver and kidney and are up-regulated in diabetes. Mol. Endocrinol. 2006, 20, 686–697.
- Mandal, A.K.; Leask, M.P.; Estiverne, C.; Choi, H.K.; Merriman, T.R.; Mount, D.B. Genetic and Physiological Effects of Insulin on Human Urate Homeostasis. Front. Physiol. 2021, 12, 713710.
- Yanai, H.; Katsuyama, H.; Hakoshima, M.; Adachi, H. Urate Transporter 1 Can Be a Therapeutic Target Molecule for Chronic Kidney Disease and Diabetic Kidney Disease: A Retrospective Longitudinal Study. Biomedicines 2023, 11, 567.
- Tanaka, Y.; Nagoshi, T.; Takahashi, H.; Oi, Y.; Yoshii, A.; Kimura, H.; Ito, K.; Kashiwagi, Y.; Tanaka, T.D.; Yoshimura, M. URAT1-selective inhibition ameliorates insulin resistance by attenuating diet-induced hepatic steatosis and brown adipose tissue whitening in mice. Mol. Metab. 2022, 55, 101411.
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922.
- Spiga, R.; Marini, M.A.; Mancuso, E.; Di Fatta, C.; Fuoco, A.; Perticone, F.; Andreozzi, F.; Mannino, G.C.; Sesti, G. Uric Acid Is Associated With Inflammatory Biomarkers and Induces Inflammation Via Activating the NF-κB Signaling Pathway in HepG2 Cells. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1241–1249.
- Czech, M.P. Mechanisms of insulin resistance related to white, beige, and brown adipocytes. Mol. Metab. 2020, 34, 27–42.
- Su, M.; Sun, L.; Li, W.; Liu, H.; Liu, Y.; Wei, Y.; Yuan, Y.; Zheng, L.; Yin, S.; Dai, C.; et al. Metformin alleviates hyperuricaemia-induced serum FFA elevation and insulin resistance by inhibiting adipocyte hypertrophy and reversing suppressed white adipose tissue beiging. Clin. Sci. 2020, 134, 1537–1553.
- Baldwin, W.; McRae, S.; Marek, G.; Wymer, D.; Pannu, V.; Baylis, C.; Johnson, R.J.; Sautin, Y.Y. Hyperuricemia as a mediator of the proinflammatory endocrine imbalance in the adipose tissue in a murine model of the metabolic syndrome. Diabetes 2011, 60, 1258–1269.
- Kwon, M.M.; O’Dwyer, S.M.; Baker, R.K.; Covey, S.D.; Kieffer, T.J. FGF21-mediated improvements in glucose clearance require uncoupling protein 1. Cell Rep. 2015, 13, 1521–1527.
- Sautin, Y.Y.; Nakagawa, T.; Zharikov, S.; Johnson, R.J. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am. J. Physiol. Cell Physiol. 2007, 293, C584–C596.
- Nadwa, E.H.; Morcos, G.N.B.; Salama, N.M.; Shafik, A.N. Comparing the Effects of Febuxostat and Allopurinol in an Animal Model of Metabolic Syndrome. Pharmacology 2021, 106, 564–572.
- Cho, I.J.; Oh, D.H.; Yoo, J.; Hwang, Y.C.; Ahn, K.J.; Chung, H.Y.; Jeong, S.W.; Moon, J.Y.; Lee, S.H.; Lim, S.J.; et al. Allopurinol ameliorates high fructose diet induced hepatic steatosis in diabetic rats through modulation of lipid metabolism, inflammation, and ER stress pathway. Sci. Rep. 2021, 11, 9894.
- Avramoglu, R.K.; Basciano, H.; Adeli, K. Lipid and lipoprotein dysregulation in insulin resistant states. Clin. Chim. Acta 2006, 368, 1–19.
- Hinds, T.D., Jr.; Hosick, P.A.; Chen, S.; Tukey, R.H.; Hankins, M.W.; Nestor-Kalinoski, A.; Stec, D.E. Mice with hyperbilirubinemia due to Gilbert’s syndrome polymorphism are resistant to hepatic steatosis by decreased serine 73 phosphorylation of PPARα. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E244–E252.
- Stec, D.E.; John, K.; Trabbic, C.J.; Luniwal, A.; Hankins, M.W.; Baum, J.; Hinds, T.D., Jr. Bilirubin Binding to PPARα Inhibits Lipid Accumulation. PLoS ONE 2016, 11, e0153427.
- Hinds, T.D., Jr.; Adeosun, S.O.; Alamodi, A.A.; Stec, D.E. Does bilirubin prevent hepatic steatosis through activation of the PPARα nuclear receptor? Med. Hypotheses 2016, 95, 54–57.
- Hinds, T.D., Jr.; Burns, K.A.; Hosick, P.A.; McBeth, L.; Nestor-Kalinoski, A.; Drummond, H.A.; AlAmodi, A.A.; Hankins, M.W.; Heuvel, J.P.V.; Stec, D.E. Biliverdin Reductase A Attenuates Hepatic Steatosis by Inhibition of Glycogen Synthase Kinase (GSK) 3β Phosphorylation of Serine 73 of Peroxisome Proliferator-activated Receptor (PPAR) α. J. Biol. Chem. 2016, 291, 25179–25191.
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.R.; Van Hul, W.; Mertens, I.; et al. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 2015, 63, 164–173.
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int. J. Mol. Sci. 2020, 21, 2061.
- Cohen, P.; Ntambi, J.M.; Friedman, J.M. Stearoyl-CoA desaturase-1 and the metabolic syndrome. Curr. Drug Targets-Immune Endocr. Metab. Disord. 2003, 3, 271–280.
- Soriguer, F.; Rojo-Martínez, G.; de Fonseca, F.R.; García-Escobar, E.; Fuentes, E.G.; Olveira, G. Obesity and the metabolic syndrome in Mediterranean countries: A hypothesis related to olive oil. Mol. Nutr. Food. Res. 2007, 51, 1260–1267.
- Dobrzyn, A.; Ntambi, J.M. The role of stearoyl-CoA desaturase in body weight regulation. Trends Cardiovasc. Med. 2004, 14, 77–81.
- Rahman, S.M.; Dobrzyn, A.; Lee, S.H.; Dobrzyn, P.; Miyazaki, M.; Ntambi, J.M. Stearoyl-CoA desaturase 1 deficiency increases insulin signalling and glycogen accumulation in brown adipose tissue. Am. J. Physiol. Endocrinol. Metab. 2005, 288, 381–387.
- Ran, Z.; Xue, X.; Han, L.; Terkeltaub, R.; Merriman, T.R.; Zhao, T.; He, Y.; Wang, C.; Li, X.; Liu, Z.; et al. Decrease in Serum Urate Level Is Associated With Loss of Visceral Fat in Male Gout Patients. Front. Endocrinol. 2021, 12, 724822.
- Nakamura, T.; Nampei, M.; Murase, T.; Satoh, E.; Akari, S.; Katoh, N.; Mizukami, H. Influence of xanthine oxidoreductase inhibitor, topiroxostat, on body weight of diabetic obese mice. Nutr. Diabetes 2021, 11, 12.
- Soletsky, B.; Feig, D.I. Uric acid reduction rectifies prehypertension in obese adolescents. Hypertension 2012, 60, 1148–1156.
More
Information
Subjects:
Pharmacology & Pharmacy
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
825
Revisions:
3 times
(View History)
Update Date:
27 Mar 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No