Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Luis German Gonzalez-Bonet | -- | 12536 | 2024-03-15 07:34:15 | | | |
| 2 | Peter Tang | Meta information modification | 12536 | 2024-03-15 07:47:39 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Obrador, E.; Moreno-Murciano, P.; Oriol-Caballo, M.; López-Blanch, R.; Pineda, B.; Gutiérrez-Arroyo, J.L.; Loras, A.; Gonzalez-Bonet, L.G.; Martinez-Cadenas, C.; Estrela, J.M.; et al. Glioblastoma Therapy. Encyclopedia. Available online: https://encyclopedia.pub/entry/56318 (accessed on 10 August 2026).
Obrador E, Moreno-Murciano P, Oriol-Caballo M, López-Blanch R, Pineda B, Gutiérrez-Arroyo JL, et al. Glioblastoma Therapy. Encyclopedia. Available at: https://encyclopedia.pub/entry/56318. Accessed August 10, 2026.
Obrador, Elena, Paz Moreno-Murciano, María Oriol-Caballo, Rafael López-Blanch, Begoña Pineda, Julia Lara Gutiérrez-Arroyo, Alba Loras, Luis G. Gonzalez-Bonet, Conrado Martinez-Cadenas, José M. Estrela, et al. "Glioblastoma Therapy" Encyclopedia, https://encyclopedia.pub/entry/56318 (accessed August 10, 2026).
Obrador, E., Moreno-Murciano, P., Oriol-Caballo, M., López-Blanch, R., Pineda, B., Gutiérrez-Arroyo, J.L., Loras, A., Gonzalez-Bonet, L.G., Martinez-Cadenas, C., Estrela, J.M., & Marqués-Torrejón, M.�. (2024, March 15). Glioblastoma Therapy. In Encyclopedia. https://encyclopedia.pub/entry/56318
Obrador, Elena, et al. "Glioblastoma Therapy." Encyclopedia. Web. 15 March, 2024.
Copy Citation
Glioblastoma (GB) stands out as the most prevalent and lethal form of brain cancer. Despite multimodality treatments, recurrence is almost universal with survival rates under 2 years after diagnosis.
glioblastoma
targeted therapy
immunotherapy
nanotherapy
non-ionizing radiation
1. Introduction
1.1. The Nature and Prognosis of Glioblastoma
Glioblastoma (GB), categorized as a grade IV astrocytoma, is the most prevalent, aggressive, and lethal primary brain tumor in adults. In 2021, the World Health Organization (WHO) introduced significant changes in the criteria for the diagnosis of gliomas, focusing on the importance of genetic and molecular alterations. According to these new criteria (Figure 1), GB should be diagnosed in adults as an isocitrate dehydrogenase wild-type (IDHwt) diffuse astrocytic glioma if there is microvascular proliferation or necrosis (the conventional criteria), and/or at least one of the following three criteria: concurrent gain of whole chromosome 7 and loss of whole chromosome 10 (+7/−10), telomerase reverse transcriptase (TERT) promoter mutations, and epidermal growth factor receptor (EGFR) amplification [1][2]. The primary driver behind the change in diagnosis criteria is the IDH mutation status, which results in the following modifications: restricting the diagnosis of GB to tumors that do not have IDH mutations (IDHwt); reclassifying tumors previously identified as IDH-mutated GBs as astrocytomas with IDH mutations (grade IV); and establishing the presence of IDH mutations as a requirement for classifying tumors as astrocytomas or oligodendrogliomas Consequently, due to its more favorable prognosis, the previously designated IDH-mutant GB is now categorized within the astrocytomas group, which covers grades II–IV, thus eliminating the term IDH-mutant GB [3]. Moreover, in IDHwt diffuse astrocytomas occurring in younger people, diagnostic consideration should be given to the different types of diffuse pediatric-type gliomas [1]. Gliosarcoma, epithelioid cell GB and giant cell GB are still registered subtypes of GBs, and the term “glioblastoma multiforme” should not be used [4][5]. New clinical trials will need to be designed with these new distinctions in mind [6].
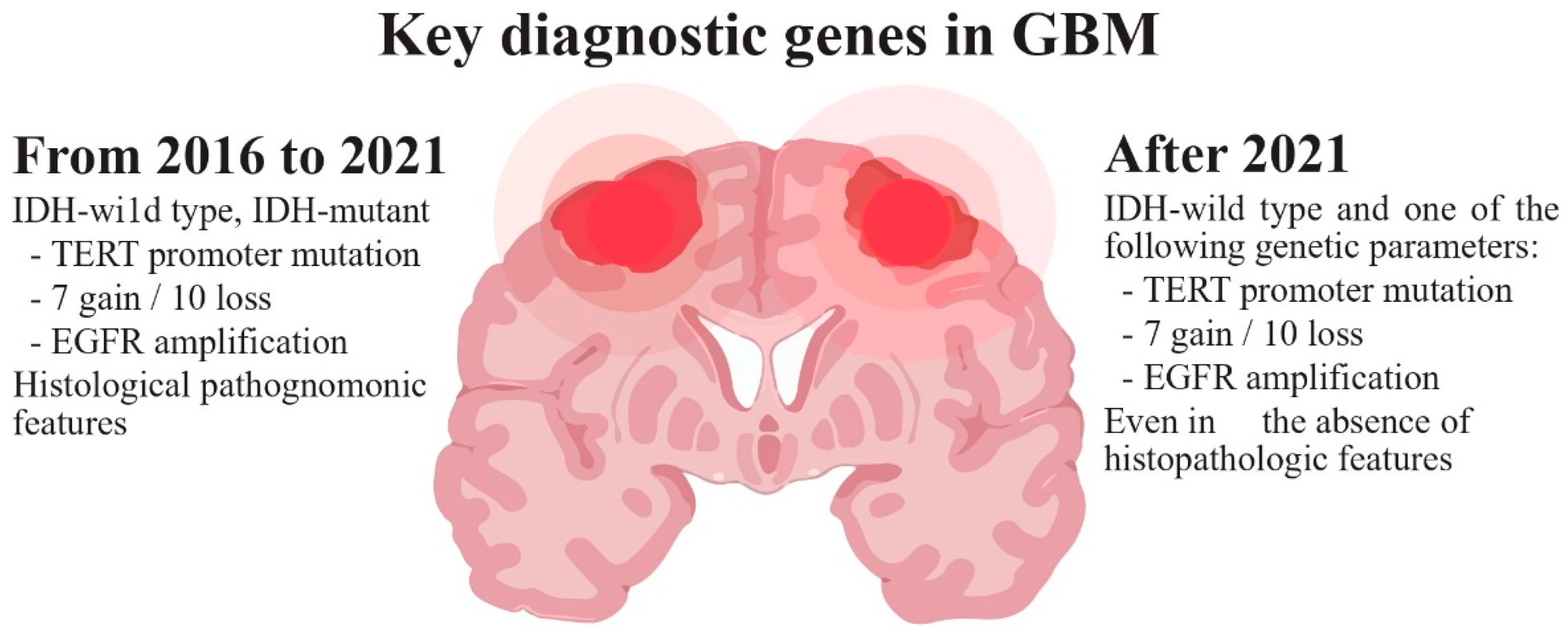
Figure 1. WHO diagnostic criteria for glioblastoma.
GB typically appears in the cerebral hemispheres, with 95% of these tumors arising in the supratentorial region, especially in the frontal and temporal globes. It infiltrates inside the brain parenchyma and usually does not spread to other parts of the body [7][8][9]. Histologically, GBs are characterized by prominent cellular and nuclear atypia, increased mitotic activity, areas of necrosis, and microvascular proliferation. At least one of these two features must be present for a histologic diagnosis of GB [5]. GB causes death in less than 6 months if untreated [10]. Despite advances in neurosurgery, chemotherapy and radiotherapy, GB remains one of the most treatment-resistant malignancies and its relapse is, in practice, inevitable [7][11][12]. Recurrence often implies a more aggressive form and a median survival of less than 18 months in treated patients [13][14]. Survival beyond 5 years is observed in less than 5.8% of patients [7]. Patients with recurrent GB (rGB) show an approx. 6-month progression-free survival (PFS6) in only 15% of the cases, and overall survival (OS) ranging between 24 and 40 weeks. Survival rate decreases with age [11][15].
As suggested by the moniker “multiforme”, GB has a widespread tumoral heterogeneity and plasticity at the cytopathological, transcriptional, and genomic levels [16][17][18][19][20][21]. Moreover, its highly infiltrative nature and the protection by the blood–brain barrier (BBB) have posed significant treatment challenges [9][22][23]. Glioma stem cells (GSCs) are a small subpopulation of cells within the GB, with genomic instability, self-renewal and tumor-initiating capacity, and the ability to differentiate into different GB subpopulations, being responsible for tumor heterogeneity [24][25][26][27]. Moreover, GSCs are resistant to apoptosis [28][29][30][31], can modulate the components of the tumor microenvironment (TME), are involved in angiogenesis activation and immunosuppression and drive radio/chemoresistance [22][32][33]. The inability of current therapies to eliminate specific GSC subpopulations has been considered a major factor contributing to the inevitable recurrence after treatment [33][34].
Verhaak et al. proposed a four-subtype classification of GB (classical, mesenchymal, proneural, and neural) based on the analysis of mutational changes in 601 genes in the context of The Cancer Genome Atlas (TCGA) [35]. Verhaak’s latest update removed the neural subtype attributing its origin to a peripheral contamination of the tumor samples [20]. The proneural subtype is associated with a more favorable outcome with respect to the mesenchymal, but this difference is relative to the more favorable outcome of IDH-mutant GBs which were consistently classified as proneural GBs [20][36]. Moreover, mesenchymal GSCs are enriched with genes associated with angiogenesis, inflammation, and cell migration/invasion. They tend to develop immunosuppression and exhibit increased radio/chemoresistance, all of which are features linked to a worse prognosis [36][37][38][39]. In any of the cases, the survival difference is minimal because both subtypes can coexist in the same tumor, and dynamic transitions from a proneural to a mesenchymal phenotype can be induced by TNF-α, temozolomide (TMZ), or radiation through an NF-κB-dependent mechanism [20][36][40][41]. Meanwhile, multiplatform analyses of the genetic, epigenetic, and transcriptional profiles have proven useful in refining the classification of gliomas and predicting patient outcomes [42][43][44][45].
1.2. Incidence and Risk Factors
GB is the most common (50.1%) among all malignant brain tumors [15]. The annual incidence is low (≈3.19 per 100,000 people in developed countries) but seems to be increasing in some countries owing to aging populations and improvements in diagnosis, among other factors [46]. The median age of diagnosis is approx. 64 and the incidence increases with age reaching its maximum value (15 per 100,000 people) between 75 and 84 years [11][15][47]. It is extremely rare in a pediatric population (0.15 per 100,000), which usually shows longer survivals. The occurrence of GB is 1.6 times more common in males than females and in Caucasians relative to other ethnicities [15][48].
Beyond rare cases of genetic susceptibility and high-dose radiation exposure, there are no known GB risk factors. An increased risk is seen in some specific genetic diseases, such as hereditary retinoblastoma or Cowden, Turcot, Lynch, Li-Fraumeni and Maffucci syndromes. However, less than 1% of GB patients have a known hereditary disease. Radiation-induced GB can be diagnosed several years after radiation therapy for another tumor or condition in children [49], but no increased risk was observed in adults exposed to IR [50]. Patients diagnosed with previous non-neurological cancers may have an overall elevated incidence of GB compared to the general population [51].
1.3. Criteria to Evaluate Treatment Response and Progression
The MacDonald criteria [52] have traditionally been used to determine treatment response and progression by assessing contrast-enhancing tumor size [by computed tomography (CT) or magnetic resonance imaging (MRI)] along with clinical evaluation and corticosteroid dosage. These criteria categorized the response into four groups: complete response (CR), partial response (PR), stable disease (SD), and progressive disease (PD). Nevertheless, these criteria have several limitations. One is the temporary increase in tumor enhancement (known as pseudo-progression), which occurs in 20–30% of the patients treated with chemo/radiotherapy and challenges differentiation with a real tumor progression. Another limitation is the high radiographic response rates seen with anti-angiogenic agents and other treatments [53]. To address these issues in 2010, the Response Assessment in Neuro-Oncology (RANO) criteria were developed to address these limitations. Although the RANO criteria improved therapy evaluation in high-grade glioma, the assessment of treatment-related side effects can hinder accurate response evaluation. The appearance of new lesions is considered a criterion for disease progression according to both RANO and MacDonald criteria. However, neuro-oncology patients receiving immunotherapies may experience the transient appearance of new enhancing lesions, either locally or in distant sites. In such cases, it is advisable to evaluate imaging findings within 6 months of starting immunotherapy, including the development of new lesions or radiographic progression, as long as there is no significant clinical deterioration [54].
To tackle the challenges in assessing immunotherapy response for neuro-oncology, the immunotherapy RANO (iRANO) criteria were introduced. The iRANO criteria combine the response assessment framework of RANO with guidelines for confirming disease progression, as originally proposed by the Immunotherapy Response Criteria in Solid Tumors to assist in clinical decision-making. The aim is to minimize premature discontinuation of potentially beneficial therapies while ensuring patient safety [54]. However, in most recurrence cases, there is a mixture of tumor cells and tissue affected by radiation injury. Radiologists strive to identify the predominant component of the lesion to determine prognostic factors and categorize the findings according to the RANO criteria, thus providing the most appropriate treatment for the patient. To overcome the aforementioned limitations in the follow-up, incorporating changes measured by advanced MRI and positron emission tomography (PET) imaging, which may precede anatomical changes in tumor volume, shows promise [55][56]. PET may also help to differentiate actual progression from pseudo progression [57]. Additionally, 18F-FMISO-PET can localize regions of hypoxia that are thought to drive radio/chemoresistance in GBs and promote immune suppression [58].
2. Lessons Learned in the Pathophysiology of Glioblastoma
2.1. Glioma Stem Cells and Tumor Microenvironment
The two prevailing hypotheses for the origin of GB are the GSC and the astrocyte de-differentiation theories [25][26][37]. Neural stem cells (NSCs), as unique stem cell type in the brain, have the ability to self-renew and can differentiate into neurons, astrocytes, and oligodendrocytes (Figure 2) [25][59]. NSCs are most active during development, but small populations remain functional in specific stem-cell niches in the adult brain. Compelling evidence suggests that GSCs may arise from NSCs located in the adult subventricular zone (SVZ) [60][61][62][63][64][65], and a recent article provided molecular genetic confirmation of this hypothesis in a preclinical model [60]. GSCs express the mutated genes TERT, PTEN, EGFR, TP53, and PDGF present in NSCs. In addition, there is an evident functional overlap and similarity between both types of stem cells, reflected in numerous shared gene expression patterns such as CD133, Sox10, nestin, vimentin, musashi, GFAP, and Olig1/2 [65][66][67]. Due to the migration ability of GSCs and the unique environment of SVZ (the vascular system of SVZ is richer than that of other brain regions), treatment-resistant GSCs easily migrate to and colonize the SVZ [68]. Consequently, numerous retrospective studies have confirmed that GBs in close contact with the SVZ possess more aggressive patterns of recurrence and worse clinical outcomes [67][69][70]. Therefore, new therapy strategies are being assayed with the aim of targeting SVZ to eradicate NSCs or GSCs [71].
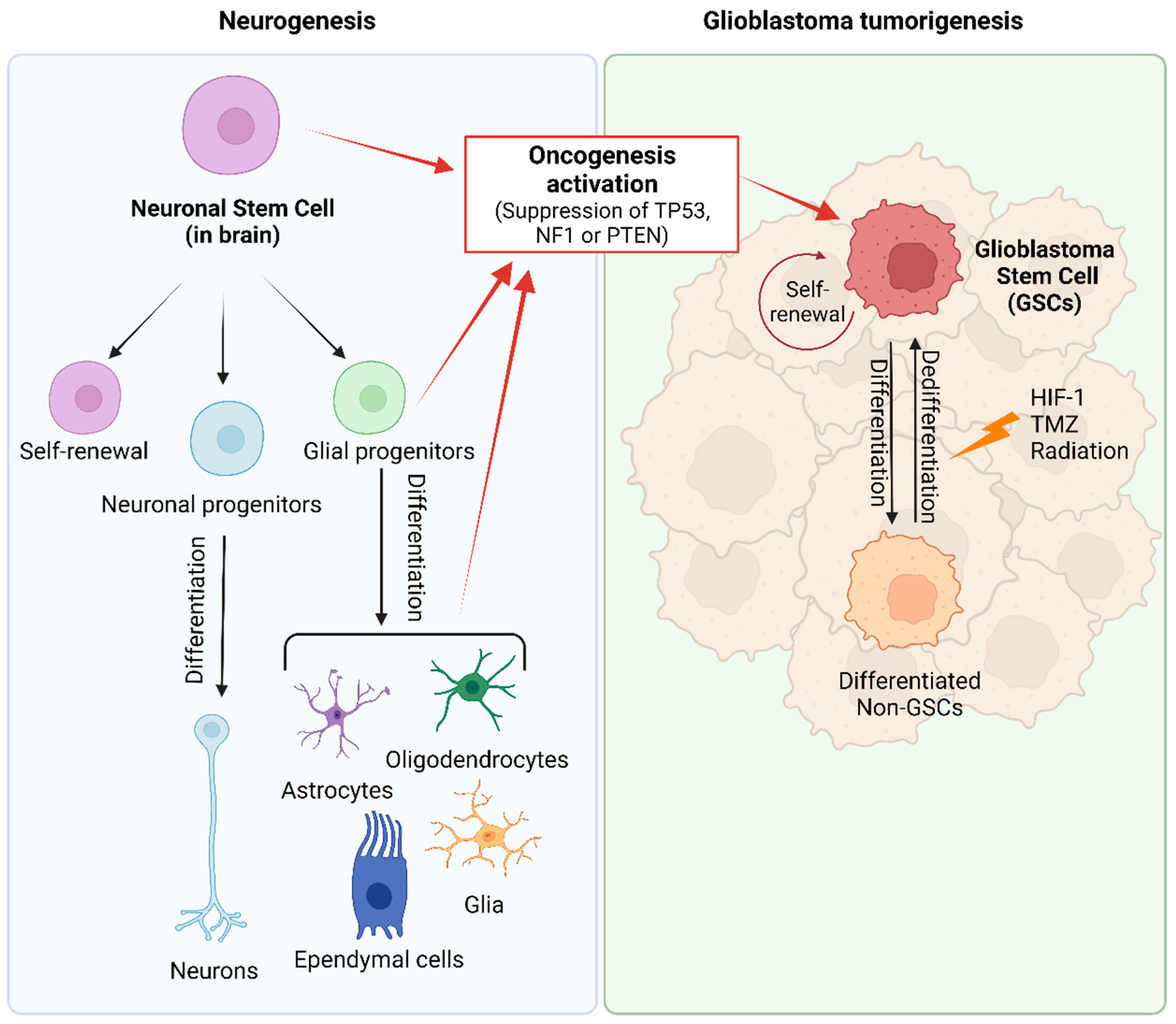
Figure 2. Origin of glioblastoma. During normal embryonic development and in the adult brain, neural stem cells (NSCs) generate glial and neuronal cells. Glioma stem cells (GSCs) arise from NSCs, astrocytes, oligodendrocytes, or glial precursor cells through the activation of oncogenic pathways (inactivation of TP53, NF1 or PTEN). GSCs are described as slow-dividing or quiescent cells, with multilineage differentiation capacity that allows them to differentiate into GB cells and cells with astrocytic, neuronal, and endothelial features and even trans-differentiation abilities. In GB tumors, there exists a dynamic equilibrium between quiescent and proliferative GSCs, and between GSC populations and their lineage-committed counterparts (differentiated non-GSC) that can also dedifferentiate into stem-lineage GSCs. Created with BioRender.com, accessed on 24 January 2024.
The origin of GB, based on the stem cell theory, explains the versatility and plasticity of heterogeneous GB tumor populations. However, several studies provide evidence suggesting that partially differentiated glial cells, including oligodendrocyte and astrocyte precursors, may play a role in or be responsible for tumorigenesis [60][72][73]. The astrocyte de-differentiation theory is supported by experiments demonstrating the formation of tumors that are histologically similar to GB after activation of oncogenes and/or suppression of tumor suppressor genes in astrocytes [24][72][74][75]. Nevertheless, this manipulation in astrocytes results in their acquisition of stem-cell-like characteristics. Consequently, both hypotheses are not mutually exclusive and explain the presence of cancer stem cells within the tumor [64][76][77]. Moreover, the dedifferentiation of non-GSCs to GSCs further complicates the GSC-targeted therapy [25][78].
GSCs represent a very low percentage of cells within GBs, and are functionally defined and distinguished from their differentiated tumor progeny at central transcriptional, epigenetic, and metabolic regulatory levels [79][80]. Recognized markers of GSCs include CD133 (PROM1) [81], CD44 [82], SOX2 and nestin [76][83], but none of them are specific markers of GSC. Other putative biomarkers are CD15 (FUT4), A2B5 antigen, CD90 (THY1), integrin ITGA6, CD171 (L1CAM), S100A4, ATP-binding cassette transporters and the combination of CD44 and ID1 (reviewed in [84]). GSCs develop genetic variability and possess self-renewal capacity and specific characteristics that support tumor development, heterogeneity, recurrence, immunosuppression and radio and chemotherapeutic resistance [85]. Therefore, the heterogeneity of GB tumor cells can be attributed to the clonal evolution and differentiation/dedifferentiation capacity of GSC [25][66][72][77][86][87]. The GSCs’ ability to adapt to different niches implies that they can dynamically restructure their transcriptional program, inducing the transient expression of genes with specific functions for each cell state [18][36]. Furthermore, microglia and endothelial cells of the perivascular niche produce numerous growth factors that contribute to the support of proliferation, migration, and differentiation of NSCs and GSCs [27][88][89]. In turn, GSCs release transforming growth factor β (TGFβ) that enhances the tumor vasculature and can even transdifferentiate and generate endothelial cells or pericytes to form new tumor vascular niches [90][91]. GSCs exhibit elevated migratory and invasive potential, eliciting infiltration into healthy tissue, thus limiting the effect of total surgical resection and radiotherapy [92]. Residual cells have the ability to regenerate GB in brain regions distant from the initial tumor by acquiring new and different driver mutations that make them resistant to treatments [41]. Consequently, GSCs are more radioresistant than GB cells [93], can be resilient to TMZ-mediated cell death [94], and have mutations that facilitate recurrence after therapy [95]. DNA damage repair mechanisms, such as ATM, ATR, CHK1, and PARP1, are upregulated in GSCs, and CHK1 is preferentially activated following irradiation [96][97].Consequently, GSCs exhibit rapid G2-M cell cycle checkpoint activation and enhanced DNA repair [98]. The preferential activation of DNA damage checkpoint responses [34] and the increased expression of drug efflux pumps and antiapoptotic proteins [99] contribute to GSC recruitment after treatment. Interestingly, the inhibition of DNA repair protein RAD51 homolog 1 has been found to delay G2 cell cycle arrest, thereby sensitizing GSCs to radiation [100].
Ionizing radiation also enhances the motility, invasiveness and aggressiveness of GSCs. The increased motility and invasiveness result from the activation of the HIF(hypoxia-inducible factor)-1α, whereas aggressiveness is attributable to a pro-neural-to-mesenchymal transition associated with the activation of the STAT3 transcriptional factor [101]. STAT3 is overexpressed in GSCs [87] and plays a crucial role in sustaining stem-like characteristics [102]. Moreover, it enhances the expression of pro-tumorigenic genes related to cell cycle progression, extracellular matrix remodeling, as well as the secretion of cytokines and growth factors [103]. Consequently, STAT3 deletion or inhibition in GB cell lines markedly decreases tumorigeneses in vitro and in vivo [103][104] and has a radiosensitizing effect [93]. WP1066, one of the most promising STAT3 inhibitors, will be investigated in a phase II clinical trial for patients with recurrent malignant glioma [105].
GB cells have the ability to manipulate the TME to favor immunosuppression and to develop a niche sustaining tumor growth, invasion, migration, and survival [28][106]. GB cells can evade immune surveillance through the release of various soluble mediators such as TGFβ, IL-10, and PGE-2. In the presence of TGFβ, CD4+ T cells upregulate FoxP3 and differentiate into Treg cells with potent immunosuppressive potential. This cytokine inhibits the expression of cytolytic gene products (perforin, granzyme A, granzyme B, Fas ligand, and IFN-γ) which are co-responsible for CD8+ T-cell-mediated tumor cytotoxicity. Increased secretion of IL-10 is associated with enhanced expression of anti-inflammatory cytokines, such as IL-4, CCL2, and TGFβ. In the presence of IL-10, TAMs downregulate the expression of antigen-presenting molecules, thereby impairing CD4+ T cell activation. In turn, PGE-2 has been shown as a key mediator of immunosuppressive activity through the expansion of myeloid-derived suppressor cells (MDSCs) [107]. In fact, GSCs and GB cells play the role in recruiting and activating MDSCs [108] and M2 macrophages to drive immune suppression [109][110][111]. Simultaneously, GSCs protect themselves from T-cell-mediated killing by secreting extracellular vesicles containing programmed death ligand 1 (PD-L1) [112][113].
Consequently, new therapies that effectively target this important population may help to prevent recurrence and improve patient survival, and for sure, no single therapeutic modality will be effective against such a heterogeneous population of cells.
2.2. Metabolic Features Favoring Growth and Resistance
Metabolic reprogramming plays a crucial role in enabling GB invasive cells to generate the energy required for colonizing the surrounding brain tissue and adapting to hypoxic microenvironments [114][115]. The metabolism of GB is characterized by the upregulation of the PI3K/Akt/mTOR signaling pathway, a high rate of glycolysis, and increased lipid storage [116][117]. Aerobic glycolysis along with glucose consumption and lactate production supports rapid GB growth and correlates with a lower survival rate [118]. Nevertheless, GB cells adapt their metabolism according to glucose availability, which gives them extra resistance to hypoxia or altered redox situations. Selective pressure on GB cells makes them overexpress glucose transporters (GLUT1 and, particularly, GLUT3). GLUT3 has a five-fold higher affinity for glucose compared to GLUT1, thus facilitating glucose uptake in environments with lower glucose concentrations. Additionally, the acquisition of a stem cell state is associated with a significant increase in GLUT3 expression in induced pluripotent cells, and this overexpression correlates with poor glioma patient survival [119][120]. When glucose levels are low, HIF-1α guarantees the upregulation of GLUT3 and hexokinase-2, increasing the glycolytic pathway [121][122].
The activation of sterol regulatory element-binding protein-1, a crucial transcription factor controlling fatty acid and cholesterol synthesis, as well as cholesterol uptake, enables GB to obtain significant quantities of lipids essential for its rapid growth [123]. GSCs exhibit high expression of mediators of lipid metabolism, such as brain-fatty-acid-binding protein (FABP7), which leads to an increase in lipid contents that are specifically metabolized under glucose-deprived conditions [116]. GB cells direct significant amounts of lipids into specialized storage organelles known as lipid droplets, thus avoiding lipotoxicity. This process involves the overexpression of diacylglycerol acyltransferase-1 and sterol-O-acyltransferase-1, which convert surplus fatty acids and cholesterol into triacylglycerol and cholesteryl esters, respectively, increasing the storage as neutral lipids within lipid droplets [123].
Amino acids play a crucial role as important fuels for GB growth. Gene expression profiling has shown an upregulation of the L-Gln importer ASCT2 in GB compared to low-grade gliomas, and L-Gln deprivation has slowed tumor growth in some in vitro studies [124]. The L-Gln-derived glutamate and glucose-derived pyruvate are substrates for the glutamate-pyruvate transaminase 2 (GPT2), which synthetizes α-ketoglutarate. Through GPT2 upregulation, the anaplerotic replenishment of the TCA cycle is possible; otherwise, it is impaired by augmented pyruvate conversion to lactate. In other words, the Warburg effect, manifested as increased lactate release, drives L-Gln addiction in order to maintain the TCA cycle function [124]. Moreover, L-Gln has been shown to promote the mTOR-dependent signaling pathway, a potent driver of GB growth and progression [125][126]. Other amino acids are also utilized to fuel bioenergetic reactions and the synthesis of macromolecules in GBs [114]. L-Asp has been shown to be a limiting metabolite for GB cellular proliferation in hypoxic conditions [127]. L-Arg is involved in GB cell adhesion, and thereby in tumor cell migration and invasion [128]. L-Trp and L-Arg metabolism have also been linked to decreased detection by neighboring immune cells, creating a favorable environment [129].
In gliomas, autocrine glutamatergic signaling has been identified as a promoter of invasion [130]. GB cells release high levels of glutamate, which not only enhances tumor invasiveness but also promotes the turnover of GSCs [131]. In other words, GB cells create a positive feedback system whereby an excess of glutamate promotes their own growth and secondarily causes excitotoxicity-induced cell death in surrounding brain tissue [132]. It is probable that such tissue damage contributes to cerebral edema and the neurotoxicity associated with a growing GB. Consequently, the inhibition of glutamatergic signaling has been proposed as a strategy to mitigate GB-induced neurotoxicity [133].
Moreover, the invasive nature of GB is modulated by cell-to-cell crosstalk within the TME and altered expression of specific genes, such as ANXA2 (encoding the protein annexin A2, a Ca2+-dependent phospholipid-binding protein that helps to organize exocytosis of intracellular proteins to the extracellular domain) [134], GBP2 (encoding the guanylate-binding protein 2, which binds to guanine nucleotides and works in intracellular signaling) [135], FN1 (encoding fibronectin, which binds to integrins and facilitates adhesion, growth, migration, and differentiation) [136], PHIP (encoding the pleckstrin homology domain interacting protein, which regulates growth and survival of GB cells) [137], and SLC2A3 (encoding the glucose transporter 3) [114][138].
2.3. Ion Channels
Different studies have demonstrated the upregulation of Ca2+ selective ion channels in GB, contributing to invasion, proliferation and resistance to apoptosis [139]. By blocking L-type voltage-gated Ca2+ channels, cell invasion is inhibited as filopodia (also known as tumor microtubes, TMs) formation is blocked [140]. Indeed, GB cells possess the ability to extract specific signals from healthy neurons using TMs [141]. Furthermore, inhibition of T-type Ca2+ channels has been shown to induce apoptosis in GB cells [142]. Therefore, blocking Ca2+ could prevent tumorigenesis through several mechanisms, i.e., cell cycle progression, induction of apoptosis and inhibition of cell migration.
K+ ion channels play a crucial role in the proliferation and the resistance to apoptosis in GB. Specifically, certain voltage-gated K+ channels are overexpressed in GBs participating in signaling pathways that promote proliferation and inhibit apoptosis [143]. Some of these effects are due to the role of K+ channels in establishing the resting membrane potential, and therefore affecting the cell cycle. Different studies have shown that inhibition of K+ channels improves survival in GB patients, which emphasizes their role in GB development and progression [144]. Consequently, blocking of ion channels could represent an interesting therapeutic approach against GB progression.
2.4. Epigenetics of Glioblastoma
GB progression is associated with different types of epigenetic alterations, including histone modifications, DNA methylation, chromatin remodeling, and aberrant microRNA (miRNA) [145][146], a group of small non-coding RNA (19–22 nucleotide long) molecules that regulate the post-transcriptional degradation of mRNA [147]. Ciafré et al. performed the first experiment related to miRNAs in GB, investigating the expression of 245 miRNAs using microarrays [148]. The most interesting results came from miR-221 upregulation, and a set of brain-enriched miRNAs (miR-128, miR-181a, miR-181b, and miR-181c) that are down-regulated in GB. At the same time, miRNAs have been shown to be important regulators of gene expression and may also regulate cellular processes, including apoptosis, proliferation, invasion, angiogenesis, and chemoresistance [149][150]. Therefore, microRNAs can be classified according to their role in tumorigenesis (i.e., tumor suppressor or oncogenic).
CircRNAs exert their biological effects through four different mechanisms: serving as sponges of RNA binding proteins, modulating parental gene transcription, encoding functional proteins and, most importantly, serving as sponges of miRNAs [151]. As is thoroughly reviewed in [151][152], circRNAs regulate GB proliferation and invasion and are also involved in angiogenesis activation. Good stability, broad distribution and high specificity make circRNAs promising biomarkers for GB prognosis and/or diagnosis, although their clinical implementation still has a long way to go.
Long-noncoding RNAs (lncRNAs) are a class of regulatory noncoding RNAs (>200 nt) that interact with DNA, RNA, and proteins to regulate various biological processes. As reviewed in [153][154][155][156], numerous studies have shown that lncRNA regulates the expression of genes involved in GB tumorigenesis (CHRM3-AS2, DLGAP1-AS1, DGCR10, LINC01057, LINC-PINT, MIR31HG, MIR210HG, NEAT1, NONHSAT079852.2, PVT1, SEMA3B, RBPMS-AS1), progression (ASLNC22381, ASLNC20819, CRNDE, DGCR10, HNF1A-AS1, HOXD-AS2, HRA1B, HOTAIRM1, LINC-PINT, PRADX, NEAT1, OXCT1-AS, TCONS-00004099) and therapeutic resistance (H19, MALAT1, MUF, DANCR, HOTAIR, HOTAIRM1, LINC00511, UCA1, OIP5-AS1, DANCR, FOXO3, HERC2P2) of GB cells. Moreover, lncRNAs exhibit stable secondary structure; thus, some of them (HOTAIR, GAS5, HOXA11-AS, HOTAIRM1, AGAP2-AS1, and AC002456.1) have been proposed as prognostic and diagnostic GB biomarkers [157][158][159][160]. More specifically, SBF2-AS1, MALAT1, CRNDE, TP73-AS1 and LINC00511 have been suggested as biomarkers of TMZ resistance in GB [160]. Lately, some evidence has indicated that lncRNAs also take part in GB cell metabolism. For instance, the lncRNA TP53TG1, under glucose deprivation, may promote cell proliferation and migration by influencing the expression of glucose-metabolism-related genes in glioma cells [161], and the lncRNA LEF1-AS1 facilitates the multiplication of GB cells and impedes apoptosis via the Akt/mTOR pathway [162]. Clinical trials involving the use of lncRNA as biomarkers for GB detection and prognosis are only in the recruitment phase but look promising.
Other epigenetic alterations, such as DNA methylation, histone modifications, and chromatin remodeling, are mechanisms involved in transcriptional activation of critical genes for GB development, lethality and resistance [145][163][164][165]. Thus, several epigenetic agents, including histone methyltransferase inhibitors, DNA methyltransferase inhibitors, histone deacetylase (HDAC) inhibitors, and other agents, are currently being tested for GB treatment in preclinical and clinical trials [146][165]. Protein arginine methyltransferase 5 (PRMT5) is a member of the PRMT family of proteins that plays a key role in the regulation of cellular signaling and gene expression by methylating histones as well as nonhistone proteins [166]. Nuclear expression of PRMT5 negatively correlates with glioma patient survival [167]. Engineered loss of PRMT5 or treatment with CMP5 (a PRMT5 inhibitor) results in apoptosis or loss of self-renewal for differentiated or undifferentiated GB cells, respectively, [168]. CMP5 derails the negative regulation of PTEN by PRMT5, which, in turn, decreases Akt activity in patient-derived GB neurospheres [169].
HDACs have been widely studied in GBM cells due to their relationship with therapeutic resistance, cell proliferation and invasion, angiogenesis and apoptosis [170][171][172][173]. In preclinical studies, HDAC inhibitors (HDACi) have proven to be effective anti-GB agents via multiple mechanisms, such as upregulating the expression of tumor suppressor genes, inhibiting oncogenes, inducing cell cycle arrest, promoting cell apoptosis and differentiation, inhibiting motility/migration, abolishing autophagy and tumor angiogenesis, and upregulating natural killer (NK)-cell-mediated tumor immunity [174][175][176][177]. Additionally, HDACis have demonstrated the capability to reduce cancer stem cell burden in GB tumors by modulating stemness, proliferation, differentiation, cell cycle arrest, apoptosis, autophagy and vasculogenic mimicry of GSCs [171][172][178]. Several HDACis (i.e., valproic acid, voristonat, panobinostat) have been assayed in clinical trials due to their capacity to act as chemo/radio-sensitizers and target GSCs [178]. Voristonat combination regimens with TMZ/radiotherapy and/or bevacizumab (BEV, recombinant humanized monoclonal antibody that blocks VEGFR-A) have proven to be tolerable (NCT01738646), but no statistical improvement in OS and/or PFS was noted [179]. Similar results were obtained with panobinostat in combination with BEV (NCT00859222) [180]. Valproic acid is a potent anticonvulsant that promotes apoptosis and impairs glioma cell proliferation and invasiveness and sensitizes GB cells to several anticancer drugs, such as TMZ, etoposide, gefitinib, nitrosoureas, and radiation therapy [181][182][183]. A meta-analysis [184] and a recent open-label phase II study [185] results seem to confirm that GB patients may experience prolonged survival due to valproic acid administration, providing further justification for a phase III trial of valproic acid/SOC. Levetiracetam, a relatively new antiepileptic drug, modulates HDAC levels ultimately silencing MGMT, thus increasing TMZ effectiveness in GCSs [186]. Retrospective analyses and an open-label phase II study (NCT02815410) seem to evidence that LEV improves GB patients’ PFS and OS [187][188]. So, it is perhaps time to reconsider the results performed in 2016, where a pooled analysis of a large series of cases treated with valproic acid or levetiracetam failed to find an association with patients’ survival [189]. A double-blind randomized clinical trial (ChiCTR2100049941) focusing on the clinical benefits of LEV + TMZ in the treatment of GB is ongoing in China. Nuclear imaging of HDAC expression in GB can be useful to improve the understanding and role of HDAC enzymes in gliomagenesis and identify patients likely to benefit from HDACi-targeted therapy [177][190].
2.5. The Angiogenetic Capacity of Glioblastoma
Aberrant vascular proliferation, necrosis, and infiltration of surrounding brain tissues are considered “hallmarks” of GB. Neo-vessels form from preexisting blood vessels due to VEGF expression by tumor and stromal cells under hypoxic conditions. The combination of VEGF with FGF (fibroblast growth factor)-2 or PDGF (platelet-derived growth factor) is known to synergistically enhance angiogenesis [191]. Vasculogenic mimicry (VM) is a new mechanism of tumor neovascularization in which highly invasive and genetically dysregulated tumor cells acquire vascular cell function, forming de novo vascular-like structures [192]. The involvement of GSCs in VM has been reported by several studies [193][194]. The disruption of GB vasculature through radiation or anti-angiogenic therapies induces a hypoxic microenvironment that promotes VM as an adaptative strategy to assist GB cells in surviving and progressing even when angiogenesis is blocked [195][196]. In keeping with this idea, the inhibition of vasculogenesis, but not sprouting angiogenesis, prevents the recurrence of GB after irradiation in mice [197].
SCs play a crucial role in VM, mainly due to their high plasticity and potential differentiation into endothelial-like cells [198]. The vascular niche is very important for the maintenance of GSCs as it promotes their survival and proliferation [192][199]. Additionally, communication between endothelial and tumor cells allows tumor vasculature formation and tumor cell dissemination [194][200]. Tumor vasculature has been considered a contributor to treatment resistance and relapse [201]. GSCs seem to be attached to the arterioles but not to the capillaries [202]. Arterioles transport, but do not exchange, gasses and nutrients, and promote a peri-hypoxic area. Integrin ligation causes an activation of the integrin-linked kinase leading to increased HIF-1α, as well as increased VEGF production [203]. HIF-1α acts as a potent activator of angiogenesis by stimulating the production of VEGF-A, PDGF and many other factors that initiate endothelial cell proliferation, invasion, and migration [204]. In GB, HIF-1α is not only influenced by oxygen but also by oncogenic signaling pathways, such as MAPK/ERK, p53, and PI3K/Akt/mTOR [205]. Although many approaches have been tried to inhibit HIF-1α, drugs that only target specific components of the hypoxia signaling pathway have generally failed to produce an enduring clinical response in GB. It is thought that the complete inhibition of HIF-1α is necessary to show potent antitumor activity and to promote the activation of the immune system [205]. Inhibition HIF-2α, which can also block the hypoxia pathway, is an alternative attractive strategy for GB treatment. HIF-2α is specifically overexpressed in GB cells and GSCs, but not in normal tissues [206]. Although the HIF-2α inhibitor PT2385 had limited activity in rGB (phase II, NCT03216499) [207], other HIF-2α inhibitors that are currently under research may help in blocking GB progression. It is also important to mention that recent findings suggest that GB hypoxia regulates gene expression in an HIF-independent way. In that sense, Srivastava et al. demonstrated that FAT1 (a FAT atypical cadherin) modulates the epithelial-mesenchymal transition and stemness gene expression in hypoxic GB [208], and hypoxia induces epigenetic regulation of the transmembrane protein odd Oz, altering DNA methylation status and activating the ODZ1-mediated migration of GB cells [164].
Importantly, Aderetti et al. demonstrated the existence of hypoxic peri-arteriolar GSC niches in GB tumor samples [209]. Apparently, GSCs remain attached to peri-arteriolar niches by the same receptor–ligand interactions as hematopoietic stem cells in the bone marrow. GSCs’ infiltration can be promoted via VEGF secreted by endothelial cells, which may induce the trans-differentiation of GSCs into endothelial cells, promoting angiogenesis and invasiveness [86]. Furthermore, a phenomenon of metabolic zonation has been described depending on the relative distance between the tumor cell and the blood vessel [210]. Proximity to the blood vessels promotes the mammalian target of rapamycin mTOR-derived anabolic metabolism and enhances tumor aggressiveness and therapy resistance [210]. Indeed, GB cells located in the perivascular tier exhibit robust anabolic metabolism and deviate from the Warburg principle by extensively engaging in oxidative phosphorylation. These perivascular cancer cells acquire specific functional characteristics, such as heightened tumorigenicity, enhanced migratory and invasive abilities, and surprisingly, remarkable resistance to chemotherapy and radiation; most of these traits are dependent on the mTOR pathway [210].
The BBB is a major obstacle to drug penetration within the brain parenchyma. Only 20% of small molecules/therapeutics agents cross the BBB and reach tumor cells at an effective concentration. GSC or GB cells protected against therapeutic agents by an intact BBB are the source of tumor recurrence [23]. P-glycoprotein, multidrug resistance proteins, organic anion transporters and breast cancer resistance proteins are especially important efflux pumps within the BBB that limit the accumulation of small-molecule-targeted therapies [211]. To make it more difficult, GSCs overexpress ABC transporters, further hindering drug delivery. ABC transporters promote therapy resistance by promoting the efflux of exogenous compounds, such as TMZ, at the cellular and BBB levels [22]. Infiltrating tumor cells are known to compromise the integrity of the BBB, resulting in a vasculature known as the blood–tumor barrier (BTB), which is highly heterogeneous and characterized by numerous distinct features, i.e., non-uniform permeability and active efflux of molecules [211]. Therefore, delivering therapeutic agents across the BBB and BTB, but avoiding their accumulation in the healthy parenchyma, is essential to making significant progress in GB treatment.
3. Present Therapy and Challenges
3.1. Standard of Care in Newly Diagnosed GB Patients
The Stupp protocol became the standard of care (SOC) for newly diagnosed GB (ndGB) patients since a randomized phase III trial evidenced an improved mOS from 12.1 to 14.6 months and an increase in the 2-year survival rate from 10% to 27% [7]. This SOC includes maximal safe resection, radiotherapy with concurrent (75 mg/m2/day × 6 weeks) and adjuvant TMZ (150–200 mg/m2/day × 5 days for six 28-day cycles). Since then, similar results (mOS 15–18 months) have been observed in other clinical studies [212][213][214]. Despite significant advances in the understanding of the molecular biology and pathophysiology of the GB, SOC has remained unchanged, excepting the possibility of adding or not tumor treating fields (TTFields) [13][215][216].
GB mostly recurs within 2–3 cm from the borders of the initial lesion and with multiple lesions, thus, maximal surgical resection improves survival irrespective of the age of the patient or the molecular status of the tumor [214][217]. Preoperative brain mapping techniques such as navigated transcranial magnetic stimulation (nTMS), magnetoencephalography, functional MRI, and diffusion tract imaging (DTI) are used to facilitate safe resections and minimize surgical complications [218]. Compared to non-nTMS techniques, nTMS has been associated in GB patients with smaller craniotomy size, less residual tumor tissue, shorter hospital stays, and improved survival at 3, 6, and 9 months, with no significant difference in surgery-induced neurological deficits [219].
During surgery, various tools are employed to optimize the extent of resection and minimize residual tumor volume. These include functional monitoring, fluorescence-based visualization of the tumor using 5-aminolevulinic acid (5-ALA), ultrasonography, and intraoperative MRI (ioMRI) [217][218][220]. Additionally, techniques like evoked potentials, electromyography, and brain mapping in awake patients, under local anesthesia, are used to monitor and preserve language and cognition during resections in critical brain areas [221]. The use of the amino acid 5-Ala helps to identify tumor volume and areas of neoplastic infiltration through fluorescent visualization, improves PFS, OS, and reduces postoperative neurological damages [218][222][223][224][225]. 5-Ala has also been effectively used in rGB resection, but the risk of false-positive fluorescence for reactive non-tumor tissue is more remarkable in relapse forms, likely due to an altered BBB [226]. Nevertheless, recently, an off-label fluorophore (sodium fluorescein) has become popular due to numerous benefits compared to 5-Ala, including lower cost, non-toxicity, easy administration and a wide indication for other brain tumors [227]. Microsurgical resection of GB using sodium fluorescein has been associated with an increased GTR rate and OS [228], although it is still considered inferior compared to 5-Ala [229].
Intraoperative ultrasound (ioUS) involves the use of sonography to locate tumor tissue during surgery and to delineate it from healthy brain tissue. As opposed to 5-Ala, which can only identify high-grade gliomas, ioUS is able to identify both low- and high-grade gliomas. In practice, 5-Ala and ioUS are considered complementary techniques [218]. Intraoperative magnetic resonance imaging (IoMRI) improves the accuracy and definition of the tumor and provides near real-time information about the dynamic changes occurring during surgery [230]. Analysis of residual GB volumes and neurological outcomes demonstrates that ioMRI is significantly superior to 5-Ala and white-light surgery at comparable peri- and post-operative morbidities [231]. The combination of ioMRI and 5-Ala facilitated achievement of the highest extent of resection (95%), followed by ioMRI alone (94%), 5-Ala alone (74%), and no imaging (73%), and this was associated with fewer post-chirurgic neurological deficits [232][233]. However, the lack of evidence regarding the cost-effectiveness compared to less advanced techniques raises uncertainty [217][234]. Regardless of the technique used, a postoperative contrast-enhanced MRI should be carried out within 48 h to assess the extent of resection and serve as a baseline for further treatments. Additionally, MRIs are performed every 2–3 cycles of TMZ treatment to monitor the tumor’s response [235].
After surgery, the smallest amount of residual tumor correlates with higher survivals [236][237]. However, radical surgical resection is limited by the highly invasive nature of GB cells [238]. Additionally, postoperative complications are a negative prognostic factor, and in this it is essential to prevent permanent neurologic deficits to safeguard the quality of life of the patients [14][47][238]. Carmustine (BCNU) wafers placed in the tumor resection cavity at the time of surgery provide a modest survival advantage (≈2 months) [239]. Wafer implants have been approved by the FDA and the EMA, but are not included in the SOC mainly due to their limited brain penetration, safety and tolerability, and because the treatment may preclude patients from enrolling into clinical trials [240]. Where surgical resection is not possible, stereotactic biopsy or open biopsy are alternative options for histological diagnosis and further molecular testing, which can determine an optional therapy [241][242]. However, this recommendation is not exempt from criticisms since in GB patients with low-performance status and/or advanced age, biopsies imply very little clinical gain [243].
Compared with surgery alone, postoperative radiotherapy is used to control microscopic unresectable disease, delay neurological deterioration and increase survival [7][244]. Radiotherapy is less efficacious in hypoxic TME due to a lower oxidative stress and because cancer cells develop mechanisms to repair DNA [115].
TMZ is a mono-alkylating agent that induces cytotoxic lesions including N7-methylguanine, N3-methyladenine and O6-methylguanine. N7-methylguanine and N3-methyladenine are repaired by base-excision repair (BER) and contribute minimally to the overall cytotoxicity of TMZ, while O6-methylguanine is repaired by O6-methylguanine-DNA-methyl transferase (MGMT) [245]. Methylation of the MGMT gene promoter (40% of GB patients) causes a reduction in MGMT protein expression and activity that results in persistent O6MeG lesions that trigger replicative stress and cytotoxicity via futile cycles of mismatch repair (MMR) [246]. Therefore, MGMT promoter methylation confers a better prognosis and overall survival (OS) associated with a positive response to alkylating agents in GB patients aged <70 years [7][13][247]. Radiotherapy has been shown to upregulate MGMT, whereas prolonged exposure to alkylating agents may suppress MGMT activity making the cells more susceptible to TMZ [248]. Nevertheless, several trials evidence that extending post-radiation TMZ from 6 to 12 months does not improve PFS6 and is associated with greater toxicity, functional deterioration, and poorer quality of life [248][249].
TMZ is a cornerstone of GB treatment, but its effectiveness is limited by the blood–brain and blood–tumor barriers, and the inherently or acquired GB resistance [19][250][251][252]. Upon TMZ treatment, GB and GSC cells induce DNA repair mechanisms, NF-kB signaling mediated antiapoptotic pathways, the expression of anti-apoptotic Bcl-2 family members, EGFR activity, drug efflux by ATP-binding cassette (ABC) transporters, autophagy-mediated resistance, expression of STAT3 and miRNAs, and overexpression of antioxidant proteins [83][250][252][253][254][255][256]. Nitrosoureas, i.e., lomustine (CCNU), carmustine and procarbazine, were widely used before the availability of TMZ, but their use is now limited to the treatment of rGB. Patients in good physical condition with hypermethylated MTMG promoters (NCT01149109) slightly increase their OS survival by the addition of lomustine to SOC (48.1 vs. 31.4 months) [257][258]. Nevertheless, the benefit of this regimen remains unclear since the sample size was small and few patients were able to complete all six cycles of adjuvant treatment due to the greater hematologic toxicity in the lomustine-TMZ arm [259]. The addition of BEV to the SOC improved PFS but not OS in both AVAglio and RTOG 0825 trials (NCT00943826 and NCT00884741) [260][261].
Up to now, TMZ is still commonly used for GBs with unmethylated MGMT promoters, due to the lack of significant benefits of alternative options (BEV plus irinotecan, dose-dense TMZ, BEV+SOC) [247][248][260][261]. Several preclinical studies demonstrated that O6-benzylguanine (O6BG) or O6-bromothenylguanine inactivate MGMT, but the addition of O6BG to radiation and BCNU treatment did not provide further benefit and instead increased toxicity [262].
3.2. Tumor-Treating Fields (TTFields)
TTFields is a non-invasive cancer treatment modality that applies low-intensity (0.7–3 V/cm), intermediate-frequency (100–500 kHz), and alternating electric fields over regions of the body where tumors are localized [263][264]. In growing GB cells, TTFields cause chromosome missegregation, disrupt DNA repair, inhibit mitosis and the cell cycle, and induce apoptosis and autophagy [265][266][267][268][269][270][271][272]. TTFields also interfere with the directionality of cancer migration by inducing changes in the organization and dynamics of microtubules and actin and ablate the primary cilia on GB cells that contribute to tumor growth and chemoresistance to TMZ [273]. TTFields also downregulate the expression levels of VEGF, HIF-1α, and matrix metalloproteinases (MMP2 and MMP9), which are necessary for tumor growth, invasion and metastasis [274]. TTFields also increase the membrane permeability of cancer cells and the BBB [275][276], which can help to increase the uptake and bioefficacy of chemotherapeutic drugs. Although this treatment modality reduces the viability of proliferating T cells [277], it also stimulates maturation and phagocytosis by dendritic cells (DCs) [278], increases CD8 T infiltration in TME [270], promotes the production of type I IFNs in GB cells in a cGAS/STING- and AIM2 inflammasome-dependent mechanism [279] and, thereby, facilitates the immune system response. Interestingly, the combination of hyperthermia and TTFields has shown synergistic effects in GB [280].
Over the past decade, TTFields have emerged as a complementary treatment strategy, which is now part of the SOC in GB treatment [238][281][282][283]. The FDA’s approval of rGB was based on the results from the EF-11 trial (NCT00379470) showing that TTFields monotherapy provided similar efficacy compared to the best physician’s choice chemotherapy in patients with rGB, albeit with better quality of life, less toxicity and a lower incidence of serious adverse events [284]. A randomized clinical trial in ndGB patients (NCT00916409) previously treated with chemoradiotherapy showed that patients treated with the TTFields and TMZ had a median free survival (mPFS) of 6.7 months compared to 4.0 months with TMZ alone. The addition of TTFields to the SOC therapy improved median OS (mOS) from 15.6 to 20.5 months without a negative influence on the health-related quality of life [13][285][286]. In ndGB, TTFields are applied within 6 weeks after the end of the radio-chemotherapy, ideally simultaneously with TMZ monotherapy [13][283]. Patients with compliance > 90% showed extended median and 5-year survival rates [287]. The most common adverse effect is skin irritation, occurring in 43% of patients (2% grade 3 or higher) [13][264][282][288], which is generally managed with array relocation and topical treatments including antibiotics and steroids. The frequency of systemic adverse events was 48% in the TTFields-TMZ group and 44% in the TMZ-alone group. Several limitations should be noted in the NCT00916409 trial: (a) only PF patients after the completion of chemoradiation were enrolled, which excluded those who were more likely to have a poor prognosis; (b) randomization in the EF-14 trial occurred over 2 months after diagnosis, which suggests a selection bias of patients who did not have progression after the initial treatment and would therefore likely have a better survival rate; (c) a “sham” device—to better discern a potential placebo-effect of wearing the device—was not used; (d) second-line therapies (chemotherapies, salvage radiation, radiosurgeries or craniotomies) after tumor progression in both groups were not reported while the TTFields plus TMZ group allowed patients to continue TTFields for up to 24 months or after the second GB progression; (e) molecular markers, such as the IDH1/2 status, were not performed [263][289]. Recently, recognized brain cancer experts concluded that TTFields plus TMZ represents a major advance in the field of GB therapy, though other experts maintain their skepticism regarding the use of the TTFields because of the lack of effect in some patients and because the time lengths required to reach (modest) benefits (at least 18 h per day) limit its utility [263][264][290].
Dexamethasone is administered to GB patients to alleviate cerebral edema and provide symptomatic relief. However, the corticoid-induced immunosuppressive effects may also increase infections and decrease survival [291][292]. In fact, a recent meta-analysis revealed that dexamethasone interferes with the therapeutic effects of TTFields [279]. The threshold dose at which dexamethasone can be used with minimal interactions with the TTFields was 4.1 mg per day or lower [293]. Several ongoing clinical trials are studying the optimal timing for TTFields administration (e.g., NCT04471844, NCT04492163, NCT03705351) and the safety and efficacy of the combination of TTFields with other cancer modalities [263][270][271][294][295]. For instance, PriCoTTFields is a phase I/II clinical trial that evaluates the safety and efficacy of TTFields initiated prior and concomitant to combined radiation and TMZ therapy in ndGB patients [296].
TTFields can reduce the DNA double-strand repair by downregulating the activity of the breast cancer type 1 susceptibility (BRCA1) signaling pathway, thereby increasing the sensitivity to the blockade of DNA repair caused by PARP inhibition [297]. Consequently, an ongoing phase II trial (NCT04221503) will try to determine whether niraparib (a PARP inhibitor) can enhance the effect of TTFields in GB patients (NCT04221503). In addition, the combination of TTFields, TMZ and lomustine has shown benefits in ndGB patients [298] and the triple combination of BEV, irinotecan, and TMZ plus TTFields improved the OS of patients with rGB [299]. Mechanisms involved in the acquisition of TTField resistance include activation of voltage-gated Ca2+ channels linked to cell migration [300]; CDK2NA deletion, mTOR (V2006I) mutations [301], and the upregulation of autophagy which can be reversed by combining TTFields with an autophagy inhibitor [267].
In clinical practice, TTFields are mainly used at a frequency of 200 kHz, but preclinical studies show that different GB cell lines respond to other optimal electric frequencies, as is the case of SF188 (400 kHz) or U87 (100 kHz) [302]. This phenomenon highlights the need for further investigation to individualize “TTFields prescription”. Despite the advances associated with the incorporation of TTFields to GB treatment, its clinical use is still quite restricted. EANO guidelines argue that the clinical benefit of TTFields has not been established yet, which contradicts the ASCO-NSO recommendations [215][303]. Certainly, price, regulation, the increase in the efficacy of combined treatments, and likely the development of novel intracranial electrodes, may assist in increasing the utilization and acceptance of TTFields [271].
3.3. Treatment in Special Patient Populations
Elderly patients (>65 years) or patients with a poor functional status have worse prognosis and are less tolerant to toxicities. Surgical resection is not associated with improved survival [304], but according to a recent retrospective single-center study, BCNU wafer implantation during the surgical resection is safe and improves mOS 39.0 months (≥12 implanted wafers) vs. 16.5 months (<12 implanted wafers) in patients in “extreme” neurosurgical conditions (>80 years and patients with preoperative Karnofsky Performance Status score < 50) [305]. Although just a few patients (6/49) reached that number of implants, these results are impressive, since mOS in the “extreme” conditions subgroup was 10.0 months, and there was a significant improvement in the postoperative KPS score compared to the preoperative KPS score.
The combination of TTFields with maintenance TMZ resulted in improved PFS and OS in ≥ 65-year-old patients with ndGB in the phase III EF-14 trial, without affecting patient quality of life [306]. Nevertheless, clinical trials have shown that standard radiotherapy is associated with poor outcomes, especially in patients older than 70 years [307]. Here, abbreviated courses of radiation therapy must be considered [307][308], although age alone should not represent the sole determining factor for the duration and intensity of the therapy [309]. Hypofractionated radiotherapy schedule (40 Gy delivered in 15) fractions and the addition of concurrent and adjuvant TMZ (NCT00482677) significantly increase survival (9.3 vs. 7.6 months, respectively) without impairing the quality of life [308]. Consequently, partial-brain fractionated radiotherapy with concurrent and adjuvant TMZ is the SOC for elderly patients with good performance status [310][311]. The addition of BEV to radiotherapy had no benefits in elderly patients [312].
A single modality therapy can be considered for patients with poor functional status. RT was more effective than TMZ for unmethylated MGMT-promoter tumors, whereas TMZ was more effective than RT for methylated MGMT-promoter tumors [307][308].
3.4. Options of Treatment in rGB patients
Regardless of the use of multimodality treatments, GB invariably returns after a median interval of less than 10 months, and typically even sooner (≈6 months) in older patients [308]. The genetic and biological changes induced by radiotherapy and/or cytotoxic chemotherapy differentiate rGB from primary tumors. These changes empower GB tumors to navigate the host microenvironment, evade the immune system, and foster intrinsic and acquired resistance to further administration of radiation and/or alkylating agents. Upon recurrence, patients typically exhibit a poor performance status and compromised overall health, with GB tumors often being unresectable, thus requiring substantial use of corticosteroids to manage cerebral edema [313]. This makes rGB prognosis much worse than that of the primary GB.
Actually, although there is no clear SOC salvage therapy for rGB [238], patients who received no salvage treatment had poorer survival than those who received radiation and/or chemotherapy [314]. Therefore, re-resection, re-irradiation and systemic chemotherapy with TMZ rechallenge, nitrosoureas, BEV, and TTFields or clinical trial enrolment to test experimental drugs are considered for all recurrent patients [244][315][316][317][318]. Unfortunately, fewer than 43% of rGB patients were fit enough to be included in clinical trials [319].
Consensus guidelines for selecting candidates for second surgery recommend that patients need to have a good performance status, particularly if more than 6 months have elapsed since the initial surgery [315][320]. According to a retrospective review of the brain tumor database (1997–2016), stereotactic radiosurgery is associated with longer OS and/or PFS in rGB patients with good performance status and small-volume tumor recurrences [321]. In practice, not more than 20–30% of relapsed patients are eligible and only complete resections have any survival benefit (11–17 months) [322][323]. Toxicity to normal brain parenchyma limits re-irradiation in rGB [324]. Radiosurgery or hypofractionated radiotherapy (30–35 Gy in 5–15 fractions) is considered a potentially effective option and is increasingly used for younger patients with good performance status [314][325]. Data from a few prospective studies in rGB suggest that re-irradiation modestly improves PFS compared with systemic treatment alone [314]. OS after re-irradiation (9.7 months) was sufficient to justify this treatment [325][326], but marginal recurrence is significantly more frequent in patients who had prior BEV exposure [327].
Lomustine has become the SOC at relapse in Europe, with thrombocytopenia being the most frequent limiting toxicity [316]. Lomustine is generally preferred to other nitrosoureas given its oral formulation, schedule of administration, and better safety profile. However, lomustine activity is largely restricted to patients with tumors with MGMT promoter methylation and its survival benefit has been found limited: objective response rate was around 10%, mPFS < 2 months, PFS6 was 20%, and OS was 6–9 months [316][328].
One of the most significant features of GB is its hypervascularization, mainly promoted by the hypoxia-facilitated VEGF overexpression in tumor and stromal cells [329]. BEV is an anti-VEGF humanized monoclonal antibody that inhibits tumor-driven angiogenesis and may help in reducing patients’ immune suppression [196][330][331]. rGB with a low apparent diffusion coefficient, large tumor burden, or IDH mutation is more likely to benefit from BEV treatment [332]. BEV gained approval in 2009 for rGB treatment in the US and later in other countries, but BEV is not approved by the EMA as an SOC for rGB [333][334]. BEV has shown promise in extending PFS treating GB, but there is no evidence for its ability to prolong OS [335][336][337][338][339]. The anti-angiogenic effect of BEV decreases contrast uptake during MRI, which can lead to false negatives in recurrences [328]. Despite this, BEV is almost used due to the lack of alternative treatment options, and because it also serves to control brain vasogenic edema [238][333] and to avoid the need for corticoid treatment [340][341]. BEV combined with re-irradiation was found to be safe and tolerable and showed a significant reduction in the incidence of radiation necrosis, patient dependence on corticosteroids and improvement in the Karnofsky score during disease progression-free periods. Survival benefits (10.1 months) have been reported following fractionated stereotactic radiotherapy (35 Gy/10 fractions) and concurrent BEV in a prospective randomized phase II trial [342][343]. A recent retrospective study showed that BEV combined with re-irradiation improved mPFS and mOS to 8 and 13.6 months, respectively [317]. Nevertheless, the validity of these results is constrained by the inclusion of a small number of patients, the heterogeneity of treatment options, and the absence of a control group. Despite these limitations, recent conclusions drawn from a meta-analysis endorse the benefits of this therapeutic option [344]. Lomustine plus BEV for rGB (phase II, NCT01290939) somewhat prolonged PFS but did not confer a survival advantage over treatment with lomustine alone [345]. Although earlier reports suggested that BEV had glucocorticoid-sparing effects, in this trial, the addition of BEV did not reduce the use of glucocorticoids [345].
TTFields did not increase OS (phase III, NCT00379470) but showed efficacy equivalent to chemotherapy commonly used for rGB, with lower toxicity and improved quality of life [284]. According to a recent phase II trial (NCT01894061), the combination of BEV and TTFields is safe and has clinical efficacy in rGB [346].
4. Targeted Therapies
Genetic changes that have been well recognized in GB cells include alterations in the Rb/p16 pathway (> 90%), loss of heterozygosity of 10q (70%), EGFR amplification or mutation (≈50%), TP53 mutations (31%), PDGF receptor gain/amplification (≈25%), mouse double minute homolog 2 (MDM2) gene mutations (10–15%) and the phosphatase and tensin homolog (PTEN) gene mutations (20–34%) [16][347]. Analysis of the large-scale molecular and genomic information present in the Cancer Genome Atlas Program (TCGA) database indicated that p53 pathway (TP53/MDM2/P14arfç), the PI3K/Akt/mTOR pathway, and the RB pathway (CDK4/RB1/P16ink4) are the main signaling pathways involved in GB tumorigenesis, pathophysiology and acquisition of resistance to treatment [16][348][349]. Intrinsically targeting these altered molecules and pathways was seen as a novel avenue in GB treatment. Unfortunately, despite research efforts and clinical trials, except for prolonged PFS afforded by the BEV, no pharmacological intervention has been demonstrated to alter the course of disease [350][351].
5. Immunotherapies
Recent studies show the presence of a variety of immune cell types within the GB TME with a dominance of immunosuppressive cells, i.e., MDSCs, microglia, M2 macrophages, FoxP3+ regulatory T cells (Tregs), and antigen-presenting cells (APCs) (including DCs and bone-marrow-derived macrophages). The presence of M2 macrophages is linked to an increased GB aggressiveness and plays a pivotal role in the acquisition of chemo and radioresistance of GB cells [352][353]. In addition, frequently, CD4+ and CD8+ T cells are functionally deficient, inactivated, or exhausted, often co-expressing immune checkpoint molecules, i.e., programmed cell death receptor 1 (PD-1), lymphocyte activation gene 3 (LAG3) and T cell immunoglobulin mucin 3 (TIM-3) [354]. GB cells secrete immunosuppressive factors such as TGFβ2, PGE-2, IL-1, IL-10 and indoleamine 2,3-dioxygenase (IDO), which work cooperatively to suppress the activity of effector cells and to evading the anti-tumor immune response [355][356]. As described here below, a plethora of novel immunotherapies, i.e., checkpoint inhibitors (ICIs), vaccines, T-cell-based immunotherapies, NK-cell-based therapies, viral therapies, and combined treatments, have been attempted in order to control GB expansion and/or recurrence.
6. Nanotherapies
Invasive (local administration) and noninvasive tools to deliver drugs are continuously evolving to overcome the BBB [23]. As reviewed in previous works [357][358][359][360], several nanostructures, including polymeric nanoparticles (NPs) (e.g., dendrimers, polymer micelles or nanospheres), inorganic NPs (e.g., silica, iron, gold or graphene NPs), lipid-based NPs (e.g., liposomes, emulsions), nanogels, carbon dots and nano-implants, have been developed as drug delivery systems and potential diagnostic agents for GB over the past decades. These elements can contain active anti-GB agents, such as chemotherapeutic/anti-angiogenic drugs, radio or chemosensitizers, or immune cells along with moieties that specifically target GB cellular receptors/angiogenic blood vessels or facilitate opening of the BBB [361][362].
Nanosystems (under 200 nm) may readily cross the BBB and fenestrated arteries, formed during the angiogenesis process, and accumulate within the tumor. This accumulation may be facilitated due to the weak lymphatic drainage system surrounding the tumor [360] or actively through the addition of targeting moieties to the surface of the NP [363]. The surface charge also plays a significant role since the electrostatic interaction between positively charged NPs and the negative surface charge of the BBB endothelial cells facilitates NP internalization through adsorptive-mediated endocytosis [364]. However, positively charged NPs can induce the generation of reactive oxygen species (ROS), which elevates their toxicity and restricts the in vivo efficacy [365]. Moreover, nanocarriers can also undergo dispersal throughout the brain and cause damage [366]. To overcome these limitations, NP surfaces can include targeting ligands that selectively recognize specific or overexpressed receptors on tumoral cells (folate, transferrin, neurokinin-1 or v3 integrin receptors) [360]. Many of these targeting ligands can also interact with receptors present on the BBB, which enhances the ability of these systems to cross the BBB through receptor-mediated transcytosis. Different targeting ligands (i.e., proteins, peptides and aptamers) have been utilized to promote active targeting of nanocarriers specifically to glioma cells [367][368][369].
Other advantages of NPs include the following: (1) nanoencapsulation increases their half-life activity, for instance in the case of TMZ-loaded chitosan NPs from 1.8 to 13.4 h [370]; (2) they can incorporate additional fluorescent/MRI/radioactive compounds that allow the non-invasive monitoring of its biodistribution [371]; (3) they increase hydrophobic drug solubility while favoring a proper biodistribution and evading the mononuclear phagocyte system catabolism; (4) they can combine different additional therapeutic approaches, such as (although not exclusively) radiotherapy sensitization, immune cells stimulation, or induction of heat/ROS [372].
7. Non-Ionizing Energies in GB Therapy
In addition to the use of TTFields, which are part of the SOC for GBs, other types of non-ionizing energies (described here below) are also being developed for therapeutic purposes.
7.1. Laser Interstitial Thermal Therapy
Laser interstitial thermal therapy (LITT) represents a cutting-edge approach for treating brain tumors that are difficult to access through conventional surgery. By inserting a laser catheter into the tumor, LITT may eradicate the tumor by raising its temperature to lethal levels. The catheter implantation process utilizes state-of-the-art computer imaging techniques, ensuring precision and accuracy. Real-time MRI guides the laser through the catheter, enabling neurosurgeons to target the thermal energy solely at the tumor site to minimize damage to surrounding healthy brain tissue. One of the most notable advantages of LITT is its minimally invasive nature and that most patients can return home the day after treatment and quickly resume their normal activities. LITT also offers hope to patients who have not responded to stereotactic radiosurgery or are suffering from radiation necrosis [373][374]. A single-center study [375] and a prospective multicenter registry [376] conclude that LITT can safely reduce intracranial tumor burden in GB patients who have exhausted other adjuvant therapies or are poor candidates for conventional resection techniques. A statistically significant OS advantage was observed in ndGB patients receiving both radiation and chemotherapy within 12 weeks of LITT (16.14 months) versus those who received only one treatment modality or no treatment following LITT (5.36 months) [377]. New neurologic deficits and postprocedural edema (normally resolved with steroid treatment) are the most frequently reported adverse events after LITT [376].
7.2. Focused Ultrasound (LIFU and HIFU)
Focused ultrasound is an early-stage, therapeutic technology that offers possible adjuvant or alternative treatment strategies for GB [378]. This groundbreaking approach involves precisely targeting deep areas of the brain with beams of ultrasonic energy without the need for incisions. This being said, it is important to differentiate between low- and high-intensity (100–10,000 W/cm2) focused ultrasounds (LIFUs and HIFUs). LIFU disrupt the BBB or blood tumor barriers and enhance the uptake of therapeutic agents in the CNS. HIFU can cause thermoablation and mechanical destruction of the tumor. Both can be combined with radiotherapy [379][380].
The advantages of focused ultrasounds over current brain tumor treatments are considerable: (1) eliminating concerns related to surgical wound healing and the risk of infection, making it a safer option for patients; (2) precise targeting; (3) avoiding ionizing radiation exposure; (4) enhancing chemotherapy delivery by temporarily opening the BBB; and (5) their non-invasive nature allows for repeat treatments [379][381].
HIFUs produce frictional heat by causing the vibration of molecules within the tissue. The absorbed energy can quickly elevate the temperature to over 55 °C, which causes protein denaturation, DNA fragmentation and coagulative necrosis when maintained for just a few seconds [382][383]. This thermoablative process further increases tumor sensitivity to radiation by damaging DNA repair enzymes [379]. To date, clinical data are limited to case reports such as those first reported by Coluccia et al. using MRI-guided HIFUs to achieve tumor ablation without inducing neurological deficits or other adverse effects in a patient with rGB [384]. Two phase I clinical trials (NCT01473485 and NCT00147056) have evaluated the safety and efficacy of transcranial MRgFUS (magnetic resonance-guided focused ultrasounds) thermoablation for the treatment of either brain metastasis or recurrent glioma, but the results have not been published. MacDonell et al. have recently proposed an interstitial HIFU device that employs an intraparenchymal catheter to induce hyperthermia directly at the tumor tissue, assisted by an MRI-guided robotic system. The advantages of this interstitial device over external MRI-guided HIFU include avoiding attenuation from the skull, improving treatment margins, and enabling concurrent tissue sampling [385]. Animal studies have demonstrated the feasibility of this technique, but its clinical success has not yet been validated. HIFU technology is approved by the FDA for treatment of several cancers (i.e., prostate, uterine leiomyoma and bone metastasis) and is under investigation for other neoplasms [386]. The primary limitation of its application in GB is the absence of well-circumscribed lesions [383].
In contrast, LIFU uses relatively lower energy pulsed waves (around 500 kHz) relying on mechanical perturbation and acoustic cavitation. Cavitation refers to the oscillation and collapse of gas bubbles in response to the compression and refraction of the ultrasonic pressure wave [386]. LIFU is therefore generally used in conjunction with microbubbles, which can be delivered intravenously and travel to the site targeted by the transducer [387]. These particles oscillate in the presence of the ultrasound wave, expanding and contracting to produce a stable cavitation effect that disrupts the tight junctions of endothelial cells. Thus, LIFU has been explored as a method to transiently increase the permeability of the BBB to enhance therapeutic delivery, limiting the side effects by ensuring that the impermeable state of the BBB is quickly restored [380]. The precision of LIFU can be enhanced using MRI (MRgLIFU), thus minimizing the effects on healthy tissue [388]. Furthermore, the opening of the BBB can be confirmed with contrast-enhanced MRI, allowing real-time monitoring of the biological effects of LIFU [386]. In animal studies, BBB opening is immediate, repeatable, resolves within 6 to 8 h, and does not cause axonal or neuronal injury. The improved delivery of BCNU, TMZ, carboplatin and others, traditionally rendered ineffective by the impermeability of the BBB, has been verified. As a consequence, this treatment not only delayed tumor progression but also enhanced survival in GB animal models [389][390][391]. LIFU has also been used to deliver viruses [392], cells [393] and NPs (loaded with imaging agents, therapeutic agents, or both) [394][395][396]. In addition, the microbubbles can be loaded with tumor antigens, allowing a more focused and effective immune response [397][398]. It is also noteworthy that preclinical studies suggest that LIFU reduces the TME-induced immunosuppression by increasing infiltration of NK and CD8+ T cells, thus facilitating DCs maturation and diminishing the number of Tregs and MDSCs [399][400][401][402]. MRgLIFU-mediated BBB disruption has been utilized in the clinical setting to deliver carboplatin, TMZ, doxorubicin, fluorescein or paclitaxel in GB patients [403][404][405]. The treatment was well tolerated, and disruption resulted in a 15–20% increase in contrast enhancement almost instantaneously, resolving after 20–24 h. A single-center trial (NCT02253212) did not show serious adverse events or carboplatin-related neurotoxicity associated with the implantation of a LIFU device with microbubble injection in rGB patients. Patients with documented BBB disruption relative to patients without or with poor BBB disruption had longer PFS (4.11 vs. 2.73 months) and OS (12.94 vs. 8.64 months) [403]. A small trial (NCT03712293) of six patients with GB treated with multiple cycles of MRgLIFU had an improved penetration of TMZ without immediate or delayed BBB-disruption-related complications. All subjects survived over 1 year, while tumor recurrence was noted in two patients at 11 and 16 months [404]. These recent results evidence that LIFU-mediated BBB opening increases drug delivery in GB, thus improving tumor control and survival, although larger sample sizes are needed to confirm efficacy and the lack of hemorrhagic complications associated with the procedure [406].
At present, a number of clinical trials (NCT03551249, NCT04440358, NCT04417088, NCT05370508, NCT06039709) are ongoing or recruiting patients with GB for focused ultrasound treatment (ClinicalTrials.gov, 4 February 2024).
7.3. Photodynamic and Sonodynamic Therapies
Photodynamic therapy (PDT) and sonodynamic therapy (SDT) are emerging modalities for non-invasive cancer treatment, based on the tumor-selective accumulation of non-toxic molecules [photosensitizers (PS) or sonosensitizers (SS)], which are activated by laser light or ultrasound radiation to produce a localized cytotoxic effect via ROS generation [407][408][409][410]. Apoptotic and necrotic cells elicit the proliferation of effector T cells in the lymph nodes, resulting in further GB eradication. Both techniques can also induce autophagy, endothelial damage, angiogenesis inhibition (associated with ischemia and necrosis), and immune responses [410][411][412][413].
Protoporphyrin IX (PpIX) and fluorescein are most widely used as PS and as SS due to their safety profile and selective accumulation in tumor cells, and most studies in glioma use 5-aminolevulinic acid (5-ALA) as a precursor of PpIX [414][415][416]. After surgery and adjuvant treatment, the residual tumor is predominantly comprised of resistant GSC clones; thus, several strategies have attempted to enhance 5-ALA uptake by this type of cells in order to increase the efficacy of PDT and SDT [415][416]. Moreover, iron chelators, such as deferoxamine and CP94, and ABC transporter inhibitors have been shown to increase PpIX levels in GB cells when utilized as adjuvants [417][418]. GSCs exhibit less accumulation of PpIX than non-GSCs, and deferoxamine-induced iron chelation significantly enhances the 5-ALA-mediated PpIX accumulation in GSCs [417].
Interstitial PDT (iPDT) is a minimally invasive procedure performed in patients whose tumors are present in areas of the brain with readily identifiable neurological function, or in fragile patients who cannot undergo a craniotomy. iPDT is applied via the stereotactic insertion of fiber optic cable(s) into the tumor to deliver photostimulation to the tumor mass after administering a PS to the patient. Intracavitary PDT is applied in the resection cavity at the end of the surgical procedure [408]. The most frequent PDT-associated adverse events are retinal and cutaneous photosensitivity that can last from several days to a few weeks depending on the PS used and time of exposure. Less frequent adverse events include post-operative hemorrhage, neurological deficits (particularly in the case of large tumors), infection, uncontrolled cerebral edema, and even death [408]. The use of iPDT against GB over the last 3 decades demonstrates that, overall, the technique is safe and effective. The mPFS was 14.5 months for ndGB and 14 months for rGB patients, which means there is an improvement compared to historical controls [419]. Different single-center retrospective studies have reported prolonged long-term survivals when using iPDT [420][421][422][423]. However, the lack of a standard control group, restricted sample sizes, and the lack of information about their characteristics, makes it difficult to draw conclusions. Ongoing clinical trials (NCT04469699, NCT03897491, NCT04391062) are actively investigating the potential of PDT in GB treatment [410]. The major disadvantage of PDT is the limited penetration of laser light into deep tissues. This could be mitigated using near-infrared radiation (NIR) that can penetrate 3 cm through skin and bone structures [424]. In fact, NIR photons also diminish both phototoxicity and background autofluorescence, which then leads to improved bio-imaging when compared to traditional fluorescence with visible light [425].
Recent advances in PDT-GB research are as follows: (1) coupled NIR and photo immunotherapy (NIR-PIT), where a photosensitizer is conjugated to a highly specific monoclonal antibody [426][427][428]; (2) NP-based PDT to augment systemic therapies and avoid skin photosensitization [429][430]; (3) NP-based PDT linked to miRNA or other chemotherapeutic agents [431]; (4) strategies to increase PDT efficacy in hypoxic TME conditions [432][433][434][435]. Although some studies have demonstrated the enhanced penetration of light by simultaneous application of ultrasounds [436], combined effects of PDT and SDT did not show any benefit in glioma rat models [437].
SDT requires the interaction of an SS, ultrasounds, and oxygen. The generation of ROS through the stimulated SS and the ultrasound-activated cavitation effects can together induce apoptosis, necrosis, and autophagy, ultimately causing tumor destruction [409]. The major advantage of SDT for GB treatment is the ability of ultrasound energy to penetrate into soft tissues more than 10 cm, and the possibility of delivering a tightly focused ultrasound beam for focal treatment [414]. SDT inhibits tumor growth and increases animal survival in preclinical studies [411][438][439][440]. A significant step forward was made by Raspagliesi et al., who reported, in a porcine animal model, the first intracranial MRI-guided SDT with fluorescein and 5-ALA using the ExAblate system. The porcine model allows a more precise target definition, and better approaches human conditions [441]. The efficacy of STD is limited in larger tumors because ultrasound waves cannot penetrate deep enough into the tumor [442]. Three clinical trials are currently underway to evaluate the safety and feasibility of SDT with 5-ALA in patients with high-grade glioma (NCT04559685), in ndGBs using the ExAblate Model 4000 Type-2 Neuro System (NCT04845919), and the efficacy of SDT using SONALA-001 and Exablate Type-2 devices in subjects with rGB (NCT04988750).
7.4. Microwaves and Pulsed Electric Fields
Microwaves are a form of electromagnetic radiation with wavelengths ranging from about 1 mm to 30 cm, corresponding to frequencies between 1 GHz and 300 GHz, respectively [443]. Microwaves have been introduced in medicine for cancer diagnosis [444] and treatment [445][446]. However, microwaves have well-known adverse effects on the CNS and can affect neurotransmitter release and, thereby, cause a delay in the signaling process [447]. Despite these limitations, the research of Rana et al. is an excellent example of how microwave radiation can therapeutically be used to treat GBs [448]. These authors have shown that a strong electric field (~23 kV/cm) of pulsed high-power microwave (HPM) irradiation causes ROS generation, DNA damage, p53 activation and death in exposed GB U87 cells. Importantly, these authors show that pulse dosage causing damage to GB cells and brain normal cells is different, thus representing a new therapeutic approach that deserves to be tested rapidly in in vivo models. In fact, pulsed microwave-induced thermoacoustic therapy has been recently proposed as a potential alternative modality to precisely and effectively eradicate orthotopic GB. Interestingly, an NP composed of polar amino acids and adenosine-based agonists has been developed having a high microwave absorbance and selective penetration of the BBB. The NP activates the adenosine receptor on the BBB to allow self-passage, and once it is accumulated in the TME, the NP converts absorbed microwaves into ultrasonic shockwaves, which can mechanically destroy tumor cells with minimal damage to adjacent normal brain tissue due to the rapid decay of the ultrasonic shockwave intensity [449].
Pulsed electrical fields [PEF, high voltage/short-duration (nanoseconds-milliseconds) electrical pulses] have emerged as a non-thermal tissue ablation treatment for malignant neoplasms. This technique, where rod/needle-like electrodes are strategically placed directly in or surrounding the tumor, is associated with the terms electroporation and electro-permeabilization [450]. Importantly, reversible permeabilization of the cell membrane can also increase the uptake of chemotherapeutics or facilitate transfection approaches [451]. In the sub-microsecond regime, intracellular effects like nuclear membrane disruption have been observed [452]. The strength and duration of the nanosecond pulse can create nano-sized pores and electroporation effects [453][454][455]. Depending on these parameters, cell electroporation and permeabilization can be reversible, with membrane recovery typically taking minutes, although intracellular repair may require hours [456]. Sub-microsecond pulses have also been found to trigger apoptotic cell death in different cell lines and tumor tissues. Apoptosis induction is associated with caspase activation [457], intracellular Ca2+ release [458], loss of mitochondrial membrane potential [459], and DNA damage [460]. These findings indicate that sub-microsecond PEF protocols, below electroporation thresholds, may offer potential therapeutic benefits for GB treatment and should be explored further [461].
The first demonstration using irreversible electroporation (IRE) against a dog malignant intracranial glioma was published in 2011 by Garcia et al. [462]. Canine malignant gliomas share similarities with GB in various clinical, biological, pathologic, molecular, and genetic aspects, making them a valuable model [463]. In the referenced study, IRE delivery resulted in an approximately 74% reduction in tumor volume. IRE treatment was well tolerated and achieved safe tumor ablation when combined with radiotherapy, anti-edema treatment, and anticonvulsants, with minimal hemorrhage [462]. More recently, further investigations using the Nanoknife procedure in seven dog glioma models revealed that IRE treatment was successful in six out of seven dogs without inducing or exacerbating edema or causing a significant hemorrhage [464]. While most adverse effects were minimal or typical of post-operative surgery, one dog experienced severe cerebral edema (due to the tumor location being close to periventricular regions, a common site of occurrence in human glioma). This highlights the importance of considering tumor location and potential effects during the pre-treatment and planning of IRE in the brain. IRE procedures have shown preservation of critical structures and major blood vessels in humans which is an advantage over microwave or radiofrequency ablation methods [465].
High-frequency irreversible electroporation (H-FIRE, or 2nd generation of IRE) works by delivering short, 1–10 μs pulses in a series of bursts, equivalent to a single monopolar 100 μs pulse used in traditional IRE. However, this approach requires much greater field strength to achieve the same lesion size [466][467]. Latouche et al. conducted an experiment using H-FIRE treatment to selectively ablate intracranial meningioma in three dogs. One of the dogs was alive after 6 months without evidence of the presence of a tumor. Another dog was alive but required increased anticonvulsants to control seizure activity, and there were suspicions of residual or recurrent tumors on an MRI 5 months after treatment. Unfortunately, the third died after 76 days due to a recurrent status epilepticus. This study showed no post-operative adverse effects attributed to H-FIRE [468]. Recently, Campelo et al. reported further evidence that H-FIRE improves survival and immune cell infiltration in rodents with malignant gliomas [469]. Although the application of these techniques is still in its infancy, available results indicate the potential of H-FIRE’s for brain tumor ablation, thus representing an exciting opportunity for clinical applications.
7.5. Targeted Radionuclide Therapy
Targeted radionuclide therapy (TRT) is based on the use of a molecule labeled with a radionuclide to deliver (through systemic or local administration) a toxic level of radiation to cancer cells. This is achieved by employing a biochemical vector, which is linked to a radionuclide and, in most cases, allows both diagnostic and also therapeutic applications. The energy, range of radiation, and type of emission are critical in targeted radionuclide therapy. Unlike molecular imaging, which involves the use of highly penetrating γ- and positron (β+)-emitting radionuclides, TRT employs β−, α, or auger electron emitters with lower penetration capacity but higher ionizing energy [470]. β− particle-emitting radionuclides (e.g., 131I, 90Y, 186/188Re and 177Lu) can irradiate tissue volumes with multicellular dimensions and induce radical formation leading to DNA single-strand breaks. For small tumors, micrometastatic lesions, or residual disease, α-particles (emitted by 213Bi, 225Ac, or 211At) are considered a better option, owing to their short travel distance in tissue (only a few diameters) and high linear energy transfer (LET) (50–230 keV/μm). α-particles induce DNA DSBs that directly trigger cell death, independently of the cell cycle phase, the cell oxygenation level and the MGMT gene promoter methylation status [471].
Radiolabeled small molecules, radioimmunotherapy (RIT), peptide radionuclide therapy (PRT) and radioNPs are four different modalities of TRT [472][473][474]. RIT uses a monoclonal antibody to achieve targeted vectorization of a radionuclide. Clinical trials for certain antigen targets, like EGFR [475][476], tenascin [477][478][479][480], or DNA histone H1 complex [481], have shown positive outcomes. Tenascin targeting appears to be one of the most promising RITs for GB. mOS of GB patients treated by fractionated intracavitary radioimmunotherapy with 131I- or 90Y-labeled anti-tenascin monoclonal antibody reached 25.3 months, thus markedly exceeding that of historical controls, being adverse events well controllable [482]. In the radiopeptide approach, an agonist or antagonist peptide is used to vectorize the radionuclide to a specific receptor overexpressed in cancer cells. In GB clinical trials, radiolabeled somatostatin analogs have been used to target the somatostatin receptor [483], radiolabeled substance P has been used to target the neurokinin receptor type 1 [484][485][486], and TM-601 (a recombinant version of chlorotoxin) has been used to target the matrix metalloproteinase [487]. In most of these assays, partial remissions and an improved OS have been observed. TAM and microglia involved in TME immunosuppression are characterized by the upregulation of somatostatin receptor 2; therefore, targeting this receptor has the additional advantage of increasing the immune response against GB [471]. RadioNP can passively accumulate in the tumor or can have a biologically active peptide or antibody for specific targeting in the same way as the molecular radiopharmaceuticals used in RIT and PRRT.
RadioNPs can be delivered passively or actively using liposomes, metallofullerenes, or lipid nanocapsules. Recently, Georgiou et al. administered 177Lu-AuNPs by CED to treat orthotopic U251-Luc human GB tumors in NRG mice. A high proportion of 177Lu-AuNPs was retained in the U251-Luc tumor for up to 21 days with minimal redistribution to the brain or healthy tissues. The radiation dose in the tumor was 599 Gy, whereas in the surrounding brain, it was 93-fold lower (6.4 Gy), and 2000–3000-fold lower doses were calculated for the contralateral left cerebral hemisphere (0.3 Gy). MRI at 28 days post-treatment showed no visible tumor in mice treated with 177Lu-AuNPs, and 5/8 of them survived up to 150 days, whereas controls had large tumors and required sacrifice within 45 days post-treatment. The results of this study are promising
TRT holds the potential to serve as a potent and supplementary treatment following SOC therapy for primary GBs. It can also be employed as an auxiliary treatment option in cases where the tumor tissue shows resistance to radiation and/or chemotherapy [471][488]. Radiopharmaceuticals can be administered systemically or intratumorally/ intracavitarily to circumvent the BBB [472][473]. The lack of serious adverse effects and promising results of the previously mentioned phase I/II trials makes TRT an attractive treatment modality that should be considered for phase III trials assays integrated into combined-modality regimens [489][490]. As an added advantage, TRT allows interesting theragnostic approaches, thus permitting personalized therapy [472][491].
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro-Oncology 2021, 23, 1231–1251.
- Berger, T.R.; Wen, P.Y.; Lang-Orsini, M.; Chukwueke, U.N. World Health Organization 2021 Classification of Central Nervous System Tumors and Implications for Therapy for Adult-Type Gliomas: A Review. JAMA Oncol. 2022, 8, 1493–1501.
- Kurokawa, R.; Kurokawa, M.; Baba, A.; Ota, Y.; Pinarbasi, E.; Camelo-Piragua, S.; Capizzano, A.A.; Liao, E.; Srinivasan, A.; Moritani, T. Major Changes in 2021 World Health Organization Classification of Central Nervous System Tumors. RadioGraphics 2022, 42, 1474–1493.
- Torp, S.H.; Solheim, O.; Skjulsvik, A.J. The WHO 2021 Classification of Central Nervous System Tumours: A Practical Update on What Neurosurgeons Need to Know—A Minireview. Acta Neurochir 2022, 164, 2453–2464.
- Thomas, D.L. 2021 Updates to the World Health Organization Classification of Adult-Type and Pediatric-Type Diffuse Gliomas: A Clinical Practice Review. Chin. Clin. Oncol. 2023, 12, 7.
- Singer, L.S.; Feldman, A.Z.; Buerki, R.A.; Horbinski, C.M.; Lukas, R.V.; Stupp, R. The Impact of the Molecular Classification of Glioblastoma on the Interpretation of Therapeutic Clinical Trial Results. Chin. Clin. Oncol. 2021, 10, 38.
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996.
- Vehlow, A.; Cordes, N. Invasion as Target for Therapy of Glioblastoma Multiforme. Biochim. Biophys. Acta (BBA) Rev. Cancer 2013, 1836, 236–244.
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2-8.
- Cantrell, J.N.; Waddle, M.R.; Rotman, M.; Peterson, J.L.; Ruiz-Garcia, H.; Heckman, M.G.; Quiñones-Hinojosa, A.; Rosenfeld, S.S.; Brown, P.D.; Trifiletti, D.M. Progress Toward Long-Term Survivors of Glioblastoma. Mayo Clin. Proc. 2019, 94, 1278–1286.
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and Molecular Prognostic Review of Glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996.
- Aldoghachi, A.F.; Aldoghachi, A.F.; Breyne, K.; Ling, K.-H.; Cheah, P.-S. Recent Advances in the Therapeutic Strategies of Glioblastoma Multiforme. Neuroscience 2022, 491, 240–270.
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316.
- Revilla-Pacheco, F.; Rodríguez-Salgado, P.; Barrera-Ramírez, M.; Morales-Ruiz, M.P.; Loyo-Varela, M.; Rubalcava-Ortega, J.; Herrada-Pineda, T. Extent of Resection and Survival in Patients with Glioblastoma Multiforme: Systematic Review and Meta-Analysis. Medicine 2021, 100, e26432.
- Ostrom, Q.T.; Price, M.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2015–2019. Neuro Oncol. 2022, 24, v1–v95.
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477.
- Perrin, S.L.; Samuel, M.S.; Koszyca, B.; Brown, M.P.; Ebert, L.M.; Oksdath, M.; Gomez, G.A. Glioblastoma Heterogeneity and the Tumour Microenvironment: Implications for Preclinical Research and Development of New Treatments. Biochem. Soc. Trans. 2019, 47, 625–638.
- Jovčevska, I. Sequencing the next Generation of Glioblastomas. Crit. Rev. Clin. Lab. Sci. 2018, 55, 264–282.
- Kim, H.; Zheng, S.; Amini, S.S.; Virk, S.M.; Mikkelsen, T.; Brat, D.J.; Grimsby, J.; Sougnez, C.; Muller, F.; Hu, J.; et al. Whole-Genome and Multisector Exome Sequencing of Primary and Post-Treatment Glioblastoma Reveals Patterns of Tumor Evolution. Genome Res. 2015, 25, 316–327.
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6.
- Becker, A.P.; Sells, B.E.; Haque, S.J.; Chakravarti, A. Tumor Heterogeneity in Glioblastomas: From Light Microscopy to Molecular Pathology. Cancers 2021, 13, 761.
- Wu, W.; Klockow, J.L.; Zhang, M.; Lafortune, F.; Chang, E.; Jin, L.; Wu, Y.; Daldrup-Link, H.E. Glioblastoma Multiforme (GBM): An Overview of Current Therapies and Mechanisms of Resistance. Pharmacol. Res. 2021, 171, 105780.
- Ahmed, M.H.; Canney, M.; Carpentier, A.; Thanou, M.; Idbaih, A. Unveiling the Enigma of the Blood–Brain Barrier in Glioblastoma: Current Advances from Preclinical and Clinical Studies. Curr. Opin. Oncol. 2023, 35, 522–528.
- Stiles, C.D.; Rowitch, D.H. Glioma Stem Cells: A Midterm Exam. Neuron 2008, 58, 832–846.
- Safa, A.R.; Saadatzadeh, M.R.; Cohen-Gadol, A.A.; Pollok, K.E.; Bijangi-Vishehsaraei, K. Glioblastoma Stem Cells (GSCs) Epigenetic Plasticity and Interconversion between Differentiated Non-GSCs and GSCs. Genes Dis. 2015, 2, 152–163.
- Xie, X.P.; Laks, D.R.; Sun, D.; Ganbold, M.; Wang, Z.; Pedraza, A.M.; Bale, T.; Tabar, V.; Brennan, C.; Zhou, X.; et al. Quiescent Human Glioblastoma Cancer Stem Cells Drive Tumor Initiation, Expansion, and Recurrence Following Chemotherapy. Dev. Cell 2022, 57, 32–46.e8.
- Hu, Y.; Li, Z.; Zhang, Y.; Wu, Y.; Liu, Z.; Zeng, J.; Hao, Z.; Li, J.; Ren, J.; Yao, M. The Evolution of Tumor Microenvironment in Gliomas and Its Implication for Target Therapy. Int. J. Biol. Sci. 2023, 19, 4311–4326.
- Di Nunno, V.; Franceschi, E.; Tosoni, A.; Gatto, L.; Bartolini, S.; Brandes, A.A. Glioblastoma Microenvironment: From an Inviolable Defense to a Therapeutic Chance. Front. Oncol. 2022, 12, 852950.
- Schaff, L.R.; Mellinghoff, I.K. Glioblastoma and Other Primary Brain Malignancies in Adults: A Review. JAMA 2023, 329, 574–587.
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant Astrocytic Glioma: Genetics, Biology, and Paths to Treatment. Genes Dev. 2007, 21, 2683–2710.
- Atkins, R.J.; Ng, W.; Stylli, S.S.; Hovens, C.M.; Kaye, A.H. Repair Mechanisms Help Glioblastoma Resist Treatment. J. Clin. Neurosci. 2015, 22, 14–20.
- Yalamarty, S.S.K.; Filipczak, N.; Li, X.; Subhan, M.A.; Parveen, F.; Ataide, J.A.; Rajmalani, B.A.; Torchilin, V.P. Mechanisms of Resistance and Current Treatment Options for Glioblastoma Multiforme (GBM). Cancers 2023, 15, 2116.
- Eckerdt, F.; Platanias, L.C. Emerging Role of Glioma Stem Cells in Mechanisms of Therapy Resistance. Cancers 2023, 15, 3458.
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma Stem Cells Promote Radioresistance by Preferential Activation of the DNA Damage Response. Nature 2006, 444, 756–760.
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110.
- Steponaitis, G.; Tamasauskas, A. Mesenchymal and Proneural Subtypes of Glioblastoma Disclose Branching Based on GSC Associated Signature. Int. J. Mol. Sci. 2021, 22, 4964.
- Fayzullin, A.; Sandberg, C.J.; Spreadbury, M.; Saberniak, B.M.; Grieg, Z.; Skaga, E.; Langmoen, I.A.; Vik-Mo, E.O. Phenotypic and Expressional Heterogeneity in the Invasive Glioma Cells. Transl. Oncol. 2019, 12, 122–133.
- Seliger, C.; Meyer, A.-L.; Leidgens, V.; Rauer, L.; Moeckel, S.; Jachnik, B.; Proske, J.; Dettmer, K.; Rothhammer-Hampl, T.; Kaulen, L.D.; et al. Metabolic Heterogeneity of Brain Tumor Cells of Proneural and Mesenchymal Origin. Int. J. Mol. Sci. 2022, 23, 11629.
- Ah-Pine, F.; Khettab, M.; Bedoui, Y.; Slama, Y.; Daniel, M.; Doray, B.; Gasque, P. On the Origin and Development of Glioblastoma: Multifaceted Role of Perivascular Mesenchymal Stromal Cells. Acta Neuropathol. Commun. 2023, 11, 104.
- Teo, W.-Y.; Sekar, K.; Seshachalam, P.; Shen, J.; Chow, W.-Y.; Lau, C.C.; Yang, H.; Park, J.; Kang, S.-G.; Li, X.; et al. Relevance of a TCGA-Derived Glioblastoma Subtype Gene-Classifier among Patient Populations. Sci. Rep. 2019, 9, 7442.
- Uribe, D.; Niechi, I.; Rackov, G.; Erices, J.I.; San Martín, R.; Quezada, C. Adapt to Persist: Glioblastoma Microenvironment and Epigenetic Regulation on Cell Plasticity. Biology 2022, 11, 313.
- Lee, E.; Yong, R.L.; Paddison, P.; Zhu, J. Comparison of Glioblastoma (GBM) Molecular Classification Methods. Semin. Cancer Biol. 2018, 53, 201–211.
- Wang, Z.; Sun, D.; Chen, Y.-J.; Xie, X.; Shi, Y.; Tabar, V.; Brennan, C.W.; Bale, T.A.; Jayewickreme, C.D.; Laks, D.R.; et al. Cell Lineage-Based Stratification for Glioblastoma. Cancer Cell 2020, 38, 366–379.e8.
- Yanovich-Arad, G.; Ofek, P.; Yeini, E.; Mardamshina, M.; Danilevsky, A.; Shomron, N.; Grossman, R.; Satchi-Fainaro, R.; Geiger, T. Proteogenomics of Glioblastoma Associates Molecular Patterns with Survival. Cell Rep. 2021, 34, 108787.
- Steponaitis, G.; Kucinskas, V.; Golubickaite, I.; Skauminas, K.; Saudargiene, A. Glioblastoma Molecular Classification Tool Based on mRNA Analysis: From Wet-Lab to Subtype. Int. J. Mol. Sci. 2022, 23, 15875.
- Grech, N.; Dalli, T.; Mizzi, S.; Meilak, L.; Calleja, N.; Zrinzo, A. Rising Incidence of Glioblastoma Multiforme in a Well-Defined Population. Cureus 2020, 12, e8195.
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of Glioblastoma: State of the Art and Future Directions. CA A Cancer J. Clin. 2020, 70, 299–312.
- Carrano, A.; Juarez, J.J.; Incontri, D.; Ibarra, A.; Cazares, H.G. Sex-Specific Differences in Glioblastoma. Cells 2021, 10, 1783.
- Ohgaki, H. Epidemiology of Brain Tumors. In Cancer Epidemiology: Modifiable Factors; Verma, M., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2009; pp. 323–342. ISBN 978-1-60327-492-0.
- Lopes, J.; Baudin, C.; Leuraud, K.; Klokov, D.; Bernier, M.-O. Ionizing Radiation Exposure during Adulthood and Risk of Developing Central Nervous System Tumors: Systematic Review and Meta-Analysis. Sci. Rep. 2022, 12, 16209.
- Dadey, D.Y.A.; Medress, Z.A.; Sharma, M.; Ugiliweneza, B.; Wang, D.; Rodrigues, A.; Parker, J.; Burton, E.; Williams, B.; Han, S.S.; et al. Risk of Developing Glioblastoma Following Non-CNS Primary Cancer: A SEER Analysis between 2000 and 2018. J. Neurooncol. 2023, 164, 655–662.
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. Corrigendum to: CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013-2017. Neuro Oncol. 2020, noaa269.
- Wick, W.; Chinot, O.L.; Bendszus, M.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Revil, C.; Kerloeguen, Y.; Cloughesy, T. Evaluation of Pseudoprogression Rates and Tumor Progression Patterns in a Phase III Trial of Bevacizumab plus Radiotherapy/Temozolomide for Newly Diagnosed Glioblastoma. Neuro Oncol. 2016, 18, 1434–1441.
- Okada, H.; Weller, M.; Huang, R.; Finocchiaro, G.; Gilbert, M.R.; Wick, W.; Ellingson, B.M.; Hashimoto, N.; Pollack, I.F.; Brandes, A.A.; et al. Immunotherapy Response Assessment in Neuro-Oncology (iRANO): A Report of the RANO Working Group. Lancet Oncol. 2015, 16, e534–e542.
- Leao, D.J.; Craig, P.G.; Godoy, L.F.; Leite, C.C.; Policeni, B. Response Assessment in Neuro-Oncology Criteria for Gliomas: Practical Approach Using Conventional and Advanced Techniques. AJNR Am. J. Neuroradiol. 2020, 41, 10–20.
- Hooper, G.W.; Ansari, S.; Johnson, J.M.; Ginat, D.T. Advances in the Radiological Evaluation of and Theranostics for Glioblastoma. Cancers 2023, 15, 4162.
- Soni, N.; Ora, M.; Jena, A.; Rana, P.; Mangla, R.; Ellika, S.; Almast, J.; Puri, S.; Meyers, S.P. Amino Acid Tracer PET MRI in Glioma Management: What a Neuroradiologist Needs to Know. Am. J. Neuroradiol. 2023.
- Reeves, K.M.; Song, P.N.; Angermeier, A.; Manna, D.D.; Li, Y.; Wang, J.; Yang, E.S.; Sorace, A.G.; Larimer, B.M. 18F-FMISO PET Imaging Identifies Hypoxia and Immunosuppressive Tumor Microenvironments and Guides Targeted Evofosfamide Therapy in Tumors Refractory to PD-1 and CTLA-4 Inhibition. Clin. Cancer Res. 2022, 28, 327–337.
- Ma, D.K.; Bonaguidi, M.A.; Ming, G.; Song, H. Adult Neural Stem Cells in the Mammalian Central Nervous System. Cell Res. 2009, 19, 672–682.
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.-Y.; Kim, W.K.; Lee, J.-K.; Park, J.; et al. Human Glioblastoma Arises from Subventricular Zone Cells with Low-Level Driver Mutations. Nature 2018, 560, 243–247.
- Altmann, C.; Keller, S.; Schmidt, M.H.H. The Role of SVZ Stem Cells in Glioblastoma. Cancers 2019, 11, 448.
- Kwan, K.; Schneider, J.R.; Patel, N.V.; Boockvar, J.A. Tracing the Origin of Glioblastoma: Subventricular Zone Neural Stem Cells. Neurosurgery 2019, 84, E15–E16.
- Hira, V.V.V.; Molenaar, R.J.; Breznik, B.; Lah, T.; Aronica, E.; Van Noorden, C.J.F. Immunohistochemical Detection of Neural Stem Cells and Glioblastoma Stem Cells in the Subventricular Zone of Glioblastoma Patients. J. Histochem. Cytochem. 2021, 69, 349–364.
- Beiriger, J.; Habib, A.; Jovanovich, N.; Kodavali, C.V.; Edwards, L.; Amankulor, N.; Zinn, P.O. The Subventricular Zone in Glioblastoma: Genesis, Maintenance, and Modeling. Front. Oncol. 2022, 12, 790976.
- Loras, A.; Gonzalez-Bonet, L.G.; Gutierrez-Arroyo, J.L.; Martinez-Cadenas, C.; Marques-Torrejon, M.A. Neural Stem Cells as Potential Glioblastoma Cells of Origin. Life 2023, 13, 905.
- Matarredona, E.R.; Pastor, A.M. Neural Stem Cells of the Subventricular Zone as the Origin of Human Glioblastoma Stem Cells. Therapeutic Implications. Front. Oncol. 2019, 9, 779.
- Yamaki, T.; Shibahra, I.; Matsuda, K.-I.; Kanemura, Y.; Konta, T.; Kanamori, M.; Yamakawa, M.; Tominaga, T.; Sonoda, Y. Relationships between Recurrence Patterns and Subventricular Zone Involvement or CD133 Expression in Glioblastoma. J. Neurooncol. 2020, 146, 489–499.
- Brockman, A.A.; Mobley, B.C.; Ihrie, R.A. Histological Studies of the Ventricular-Subventricular Zone as Neural Stem Cell and Glioma Stem Cell Niche. J. Histochem. Cytochem. 2021, 69, 819–834.
- Mistry, A.M.; Hale, A.T.; Chambless, L.B.; Weaver, K.D.; Thompson, R.C.; Ihrie, R.A. Influence of Glioblastoma Contact with the Lateral Ventricle on Survival: A Meta-Analysis. J. Neurooncol. 2017, 131, 125–133.
- Huang, R.; Wang, T.; Liao, Z.; Wang, Z.; Ye, M.; Zhou, D.; Xie, H.; Bai, Y.; Qiu, Y.; Liu, Y. A Retrospective Analysis of the Risk Factors Affecting Recurrence Time in Patients with Recurrent Glioblastoma. Ann. Palliat. Med. 2021, 10, 5391–5399.
- Li, S.; Dong, L.; Pan, Z.; Yang, G. Targeting the Neural Stem Cells in Subventricular Zone for the Treatment of Glioblastoma: An Update from Preclinical Evidence to Clinical Interventions. Stem Cell Res. Ther. 2023, 14, 125.
- Friedmann-Morvinski, D.; Bushong, E.A.; Ke, E.; Soda, Y.; Marumoto, T.; Singer, O.; Ellisman, M.H.; Verma, I.M. Dedifferentiation of Neurons and Astrocytes by Oncogenes Can Induce Gliomas in Mice. Science 2012, 338, 1080–1084.
- Shahar, T.; Rozovski, U.; Hess, K.R.; Hossain, A.; Gumin, J.; Gao, F.; Fuller, G.N.; Goodman, L.; Sulman, E.P.; Lang, F.F. Percentage of Mesenchymal Stem Cells in High-Grade Glioma Tumor Samples Correlates with Patient Survival. Neuro Oncol. 2017, 19, 660–668.
- Uhrbom, L.; Dai, C.; Celestino, J.C.; Rosenblum, M.K.; Fuller, G.N.; Holland, E.C. Ink4a-Arf Loss Cooperates with KRas Activation in Astrocytes and Neural Progenitors to Generate Glioblastomas of Various Morphologies Depending on Activated Akt1. Cancer Res. 2002, 62, 5551–5558.
- Li, F.; Liu, X.; Sampson, J.H.; Bigner, D.D.; Li, C.-Y. Rapid Reprogramming of Primary Human Astrocytes into Potent Tumor-Initiating Cells with Defined Genetic Factors. Cancer Res. 2016, 76, 5143–5150.
- Bradshaw, A.; Wickremsekera, A.; Tan, S.T.; Peng, L.; Davis, P.F.; Itinteang, T. Cancer Stem Cell Hierarchy in Glioblastoma Multiforme. Front. Surg. 2016, 3, 21.
- Lan, X.; Jörg, D.J.; Cavalli, F.M.G.; Richards, L.M.; Nguyen, L.V.; Vanner, R.J.; Guilhamon, P.; Lee, L.; Kushida, M.M.; Pellacani, D.; et al. Fate Mapping of Human Glioblastoma Reveals an Invariant Stem Cell Hierarchy. Nature 2017, 549, 227–232.
- Schneider, M.; Ströbele, S.; Nonnenmacher, L.; Siegelin, M.D.; Tepper, M.; Stroh, S.; Hasslacher, S.; Enzenmüller, S.; Strauss, G.; Baumann, B.; et al. A Paired Comparison between Glioblastoma “Stem Cells” and Differentiated Cells. Int. J. Cancer 2016, 138, 1709–1718.
- Liau, B.B.; Sievers, C.; Donohue, L.K.; Gillespie, S.M.; Flavahan, W.A.; Miller, T.E.; Venteicher, A.S.; Hebert, C.H.; Carey, C.D.; Rodig, S.J.; et al. Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell 2017, 20, 233–246.e7.
- Gimple, R.C.; Bhargava, S.; Dixit, D.; Rich, J.N. Glioblastoma Stem Cells: Lessons from the Tumor Hierarchy in a Lethal Cancer. Genes Dev. 2019, 33, 591–609.
- Glumac, P.M.; LeBeau, A.M. The Role of CD133 in Cancer: A Concise Review. Clin. Transl. Med. 2018, 7, 18.
- Si, D.; Yin, F.; Peng, J.; Zhang, G. High Expression of CD44 Predicts a Poor Prognosis in Glioblastomas. Cancer Manag. Res. 2020, 12, 769–775.
- Dréan, A.; Rosenberg, S.; Lejeune, F.-X.; Goli, L.; Nadaradjane, A.A.; Guehennec, J.; Schmitt, C.; Verreault, M.; Bielle, F.; Mokhtari, K.; et al. ATP Binding Cassette (ABC) Transporters: Expression and Clinical Value in Glioblastoma. J. Neurooncol. 2018, 138, 479–486.
- Vieira de Castro, J.; Gonçalves, C.S.; Hormigo, A.; Costa, B.M. Exploiting the Complexities of Glioblastoma Stem Cells: Insights for Cancer Initiation and Therapeutic Targeting. Int. J. Mol. Sci. 2020, 21, 5278.
- Zhang, G.-L.; Wang, C.-F.; Qian, C.; Ji, Y.-X.; Wang, Y.-Z. Role and Mechanism of Neural Stem Cells of the Subventricular Zone in Glioblastoma. World J. Stem Cells 2021, 13, 877–893.
- Soda, Y.; Marumoto, T.; Friedmann-Morvinski, D.; Soda, M.; Liu, F.; Michiue, H.; Pastorino, S.; Yang, M.; Hoffman, R.M.; Kesari, S.; et al. Transdifferentiation of Glioblastoma Cells into Vascular Endothelial Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4274–4280.
- Garner, J.M.; Ellison, D.W.; Finkelstein, D.; Ganguly, D.; Du, Z.; Sims, M.; Yang, C.H.; Interiano, R.B.; Davidoff, A.M.; Pfeffer, L.M. Molecular Heterogeneity in a Patient-Derived Glioblastoma Xenoline Is Regulated by Different Cancer Stem Cell Populations. PLoS ONE 2015, 10, e0125838.
- Matarredona, E.R.; Talaverón, R.; Pastor, A.M. Interactions Between Neural Progenitor Cells and Microglia in the Subventricular Zone: Physiological Implications in the Neurogenic Niche and After Implantation in the Injured Brain. Front. Cell Neurosci. 2018, 12, 268.
- David-Bercholz, J.; Kuo, C.T.; Deneen, B. Astrocyte and Oligodendrocyte Responses From the Subventricular Zone After Injury. Front. Cell Neurosci. 2021, 15, 797553.
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour Vascularization via Endothelial Differentiation of Glioblastoma Stem-like Cells. Nature 2010, 468, 824–828.
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma Stem Cells Generate Vascular Pericytes to Support Vessel Function and Tumor Growth. Cell 2013, 153, 139–152.
- Molina, J.R.; Hayashi, Y.; Stephens, C.; Georgescu, M.-M. Invasive Glioblastoma Cells Acquire Stemness and Increased Akt Activation. Neoplasia 2010, 12, 453–463.
- Masliantsev, K.; Pinel, B.; Balbous, A.; Guichet, P.-O.; Tachon, G.; Milin, S.; Godet, J.; Duchesne, M.; Berger, A.; Petropoulos, C.; et al. Impact of STAT3 Phosphorylation in Glioblastoma Stem Cells Radiosensitization and Patient Outcome. Oncotarget 2018, 9, 3968–3979.
- Beier, D.; Schriefer, B.; Brawanski, K.; Hau, P.; Weis, J.; Schulz, J.B.; Beier, C.P. Efficacy of Clinically Relevant Temozolomide Dosing Schemes in Glioblastoma Cancer Stem Cell Lines. J. Neurooncol. 2012, 109, 45–52.
- McCord, M.; Bartom, E.; Burdett, K.; Baran, A.; Eckerdt, F.D.; Balyasnikova, I.V.; McCortney, K.; Sears, T.; Cheng, S.-Y.; Sarkaria, J.N.; et al. Modeling Therapy-Driven Evolution of Glioblastoma with Patient-Derived Xenografts. Cancers 2022, 14, 5494.
- Pavlova, G.; Belyashova, A.; Savchenko, E.; Panteleev, D.; Shamadykova, D.; Nikolaeva, A.; Pavlova, S.; Revishchin, A.; Golbin, D.; Potapov, A.; et al. Reparative Properties of Human Glioblastoma Cells after Single Exposure to a Wide Range of X-Ray Doses. Front. Oncol. 2022, 12, 912741.
- Vilar, J.B.; Christmann, M.; Tomicic, M.T. Alterations in Molecular Profiles Affecting Glioblastoma Resistance to Radiochemotherapy: Where Does the Good Go? Cancers 2022, 14, 2416.
- Ahmed, S.U.; Carruthers, R.; Gilmour, L.; Yildirim, S.; Watts, C.; Chalmers, A.J. Selective Inhibition of Parallel DNA Damage Response Pathways Optimizes Radiosensitization of Glioblastoma Stem-like Cells. Cancer Res. 2015, 75, 4416–4428.
- Dean, M.; Fojo, T.; Bates, S. Tumour Stem Cells and Drug Resistance. Nat. Rev. Cancer 2005, 5, 275–284.
- Tachon, G.; Cortes, U.; Guichet, P.-O.; Rivet, P.; Balbous, A.; Masliantsev, K.; Berger, A.; Boissonnade, O.; Wager, M.; Karayan-Tapon, L. Cell Cycle Changes after Glioblastoma Stem Cell Irradiation: The Major Role of RAD51. Int. J. Mol. Sci. 2018, 19, 3018.
- Lau, J.; Ilkhanizadeh, S.; Wang, S.; Miroshnikova, Y.A.; Salvatierra, N.A.; Wong, R.A.; Schmidt, C.; Weaver, V.M.; Weiss, W.A.; Persson, A.I. STAT3 Blockade Inhibits Radiation-Induced Malignant Progression in Glioma. Cancer Res. 2015, 75, 4302–4311.
- Sherry, M.M.; Reeves, A.; Wu, J.K.; Cochran, B.H. STAT3 Is Required for Proliferation and Maintenance of Multipotency in Glioblastoma Stem Cells. Stem Cells 2009, 27, 2383–2392.
- Ganguly, D.; Fan, M.; Yang, C.H.; Zbytek, B.; Finkelstein, D.; Roussel, M.F.; Pfeffer, L.M. The Critical Role That STAT3 Plays in Glioma-Initiating Cells: STAT3 Addiction in Glioma. Oncotarget 2018, 9, 22095–22112.
- Wang, Y.; Yang, C.; Sims, M.M.; Sacher, J.R.; Raje, M.; Deokar, H.; Yue, P.; Turkson, J.; Buolamwini, J.K.; Pfeffer, L.M. SS-4 Is a Highly Selective Small Molecule Inhibitor of STAT3 Tyrosine Phosphorylation That Potently Inhibits GBM Tumorigenesis in Vitro and in Vivo. Cancer Lett. 2022, 533, 215614.
- Groot, J.d.; Ott, M.; Wei, J.; Kassab, C.; Fang, D.; Najem, H.; O’Brien, B.; Weathers, S.-P.; Matsouka, C.K.; Majd, N.K.; et al. A First-in-Human Phase I Trial of the Oral p-STAT3 Inhibitor WP1066 in Patients with Recurrent Malignant Glioma. CNS Oncol. 2022, 11, CNS87.
- Sharma, P.; Aaroe, A.; Liang, J.; Puduvalli, V.K. Tumor Microenvironment in Glioblastoma: Current and Emerging Concepts. Neurooncol. Adv. 2023, 5, vdad009.
- Rocha Pinheiro, S.L.; Lemos, F.F.B.; Marques, H.S.; Silva Luz, M.; de Oliveira Silva, L.G.; Faria Souza Mendes dos Santos, C.; da Costa Evangelista, K.; Calmon, M.S.; Sande Loureiro, M.; Freire de Melo, F. Immunotherapy in Glioblastoma Treatment: Current State and Future Prospects. World J. Clin. Oncol. 2023, 14, 138–159.
- Otvos, B.; Silver, D.J.; Mulkearns-Hubert, E.E.; Alvarado, A.G.; Turaga, S.M.; Sorensen, M.D.; Rayman, P.; Flavahan, W.A.; Hale, J.S.; Stoltz, K.; et al. Cancer Stem Cell-Secreted Macrophage Migration Inhibitory Factor Stimulates Myeloid Derived Suppressor Cell Function and Facilitates Glioblastoma Immune Evasion. Stem Cells 2016, 34, 2026–2039.
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin Secreted by Glioblastoma Stem Cells Recruits M2 Tumour-Associated Macrophages and Promotes Malignant Growth. Nat. Cell Biol. 2015, 17, 170–182.
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional Communication in the Microenvirons of Glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495.
- Wei, J.; Marisetty, A.; Schrand, B.; Gabrusiewicz, K.; Hashimoto, Y.; Ott, M.; Grami, Z.; Kong, L.-Y.; Ling, X.; Caruso, H.; et al. Osteopontin Mediates Glioblastoma-Associated Macrophage Infiltration and Is a Potential Therapeutic Target. J. Clin. Investig. 2019, 129, 137–149.
- Ricklefs, F.L.; Alayo, Q.; Krenzlin, H.; Mahmoud, A.B.; Speranza, M.C.; Nakashima, H.; Hayes, J.L.; Lee, K.; Balaj, L.; Passaro, C.; et al. Immune Evasion Mediated by PD-L1 on Glioblastoma-Derived Extracellular Vesicles. Sci. Adv. 2018, 4, eaar2766.
- Himes, B.T.; Peterson, T.E.; de Mooij, T.; Garcia, L.M.C.; Jung, M.-Y.; Uhm, S.; Yan, D.; Tyson, J.; Jin-Lee, H.J.; Parney, D.; et al. The Role of Extracellular Vesicles and PD-L1 in Glioblastoma-Mediated Immunosuppressive Monocyte Induction. Neuro Oncol. 2020, 22, 967–978.
- Garcia, J.H.; Jain, S.; Aghi, M.K. Metabolic Drivers of Invasion in Glioblastoma. Front. Cell Dev. Biol. 2021, 9.
- Erices, J.I.; Bizama, C.; Niechi, I.; Uribe, D.; Rosales, A.; Fabres, K.; Navarro-Martínez, G.; Torres, Á.; San Martín, R.; Roa, J.C.; et al. Glioblastoma Microenvironment and Invasiveness: New Insights and Therapeutic Targets. Int. J. Mol. Sci. 2023, 24, 7047.
- Hoang-Minh, L.B.; Siebzehnrubl, F.A.; Yang, C.; Suzuki-Hatano, S.; Dajac, K.; Loche, T.; Andrews, N.; Schmoll Massari, M.; Patel, J.; Amin, K.; et al. Infiltrative and Drug-Resistant Slow-Cycling Cells Support Metabolic Heterogeneity in Glioblastoma. EMBO J. 2018, 37, e98772.
- Verdugo, E.; Puerto, I.; Medina, M.Á. An Update on the Molecular Biology of Glioblastoma, with Clinical Implications and Progress in Its Treatment. Cancer Commun. 2022, 42, 1083–1111.
- Stanke, K.M.; Wilson, C.; Kidambi, S. High Expression of Glycolytic Genes in Clinical Glioblastoma Patients Correlates With Lower Survival. Front. Mol. Biosci. 2021, 8.
- Flavahan, W.A.; Wu, Q.; Hitomi, M.; Rahim, N.; Kim, Y.; Sloan, A.E.; Weil, R.J.; Nakano, I.; Sarkaria, J.N.; Stringer, B.W.; et al. Brain Tumor Initiating Cells Adapt to Restricted Nutrition through Preferential Glucose Uptake. Nat. Neurosci. 2013, 16, 1373–1382.
- Libby, C.J.; Gc, S.; Benavides, G.A.; Fisher, J.L.; Williford, S.E.; Zhang, S.; Tran, A.N.; Gordon, E.R.; Jones, A.B.; Tuy, K.; et al. A Role for GLUT3 in Glioblastoma Cell Invasion That Is Not Recapitulated by GLUT1. Cell Adh Migr. 2021, 15, 101–115.
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 Is a Key Mediator of Aerobic Glycolysis and Promotes Tumor Growth in Human Glioblastoma Multiforme. J. Exp. Med. 2011, 208, 313–326.
- Labak, C.M.; Wang, P.Y.; Arora, R.; Guda, M.R.; Asuthkar, S.; Tsung, A.J.; Velpula, K.K. Glucose Transport: Meeting the Metabolic Demands of Cancer, and Applications in Glioblastoma Treatment. Am. J. Cancer Res. 2016, 6, 1599–1608.
- Kou, Y.; Geng, F.; Guo, D. Lipid Metabolism in Glioblastoma: From De Novo Synthesis to Storage. Biomedicines 2022, 10, 1943.
- Obara-Michlewska, M.; Szeliga, M. Targeting Glutamine Addiction in Gliomas. Cancers 2020, 12, 310.
- Natarajan, S.K.; Venneti, S. Glutamine Metabolism in Brain Tumors. Cancers 2019, 11, 1628.
- Barzegar Behrooz, A.; Talaie, Z.; Jusheghani, F.; Łos, M.J.; Klonisch, T.; Ghavami, S. Wnt and PI3K/Akt/mTOR Survival Pathways as Therapeutic Targets in Glioblastoma. Int. J. Mol. Sci. 2022, 23, 1353.
- Garcia-Bermudez, J.; Baudrier, L.; La, K.; Zhu, X.G.; Fidelin, J.; Sviderskiy, V.O.; Papagiannakopoulos, T.; Molina, H.; Snuderl, M.; Lewis, C.A.; et al. Aspartate Is a Limiting Metabolite for Cancer Cell Proliferation under Hypoxia and in Tumours. Nat. Cell Biol. 2018, 20, 775–781.
- Karatsai, O.; Stasyk, O.; Redowicz, M.J. Effects of Arginine and Its Deprivation on Human Glioblastoma Physiology and Signaling. Adv. Exp. Med. Biol. 2020, 1202, 243–258.
- Kesarwani, P.; Prabhu, A.; Kant, S.; Kumar, P.; Graham, S.F.; Buelow, K.L.; Wilson, G.D.; Miller, C.R.; Chinnaiyan, P. Tryptophan Metabolism Contributes to Radiation-Induced Immune Checkpoint Reactivation in Glioblastoma. Clin. Cancer Res. 2018, 24, 3632–3643.
- Lyons, S.A.; Chung, W.J.; Weaver, A.K.; Ogunrinu, T.; Sontheimer, H. Autocrine Glutamate Signaling Promotes Glioma Cell Invasion. Cancer Res. 2007, 67, 9463–9471.
- Corsi, L.; Mescola, A.; Alessandrini, A. Glutamate Receptors and Glioblastoma Multiforme: An Old “Route” for New Perspectives. Int. J. Mol. Sci. 2019, 20, 1796.
- Savaskan, N.E.; Fan, Z.; Broggini, T.; Buchfelder, M.; Eyüpoglu, I.Y. Neurodegeneration in the Brain Tumor Microenvironment: Glutamate in the Limelight. Curr. Neuropharmacol. 2015, 13, 258–265.
- Kumaria, A.; Ashkan, K. Novel Therapeutic Strategies in Glioma Targeting Glutamatergic Neurotransmission. Brain Res. 2023, 1818, 148515.
- Maule, F.; Bresolin, S.; Rampazzo, E.; Boso, D.; Puppa, A.D.; Esposito, G.; Porcù, E.; Mitola, S.; Lombardi, G.; Accordi, B.; et al. Annexin 2A Sustains Glioblastoma Cell Dissemination and Proliferation. Oncotarget 2016, 7, 54632–54649.
- Yu, S.; Yu, X.; Sun, L.; Zheng, Y.; Chen, L.; Xu, H.; Jin, J.; Lan, Q.; Chen, C.C.; Li, M. GBP2 Enhances Glioblastoma Invasion through Stat3/Fibronectin Pathway. Oncogene 2020, 39, 5042–5055.
- Wu, S.; Liu, C.; Wei, X.; Nong, W.-X.; Lin, L.-N.; Li, F.; Xie, X.-X.; Liao, X.-S.; Luo, B.; Zhang, Q.-M.; et al. High Expression of Fibronectin 1 Predicts a Poor Prognosis in Glioblastoma. Curr. Med. Sci. 2022, 42, 1055–1065.
- de Semir, D.; Bezrookove, V.; Nosrati, M.; Scanlon, K.R.; Singer, E.; Judkins, J.; Rieken, C.; Wu, C.; Shen, J.; Schmudermayer, C.; et al. PHIP Drives Glioblastoma Motility and Invasion by Regulating the Focal Adhesion Complex. Proc. Natl. Acad. Sci. USA 2020, 117, 9064–9073.
- Cosset, É.; Ilmjärv, S.; Dutoit, V.; Elliott, K.; von Schalscha, T.; Camargo, M.F.; Reiss, A.; Moroishi, T.; Seguin, L.; Gomez, G.; et al. Glut3 Addiction Is a Druggable Vulnerability for a Molecularly Defined Subpopulation of Glioblastoma. Cancer Cell 2017, 32, 856–868.e5.
- Santoni, G.; Santoni, M.; Nabissi, M. Functional Role of T-Type Calcium Channels in Tumour Growth and Progression: Prospective in Cancer Therapy. Br. J. Pharmacol. 2012, 166, 1244–1246.
- Jacquemet, G.; Baghirov, H.; Georgiadou, M.; Sihto, H.; Peuhu, E.; Cettour-Janet, P.; He, T.; Perälä, M.; Kronqvist, P.; Joensuu, H.; et al. L-Type Calcium Channels Regulate Filopodia Stability and Cancer Cell Invasion Downstream of Integrin Signalling. Nat. Commun. 2016, 7, 13297.
- Portela, M.; Casas-Tintó, S. New Cellular Dimensions on Glioblastoma Progression. Neurosci. Insights 2020, 15, 2633105520923076.
- Kuga, T.; Sadoshima, J.; Tomoike, H.; Kanaide, H.; Akaike, N.; Nakamura, M. Actions of Ca2+ Antagonists on Two Types of Ca2+ Channels in Rat Aorta Smooth Muscle Cells in Primary Culture. Circ. Res. 1990, 67, 469–480.
- Pointer, K.B.; Clark, P.A.; Eliceiri, K.W.; Salamat, M.S.; Robertson, G.A.; Kuo, J.S. Administration of Non-Torsadogenic Human Ether-à-Go-Go-Related Gene Inhibitors Is Associated with Better Survival for High hERG-Expressing Glioblastoma Patients. Clin. Cancer Res. 2017, 23, 73–80.
- Chen, D.; Song, M.; Mohamad, O.; Yu, S.P. Inhibition of Na+/K+-ATPase Induces Hybrid Cell Death and Enhanced Sensitivity to Chemotherapy in Human Glioblastoma Cells. BMC Cancer 2014, 14, 716.
- Dong, Z.; Cui, H. Epigenetic Modulation of Metabolism in Glioblastoma. Semin. Cancer Biol. 2019, 57, 45–51.
- Uddin, M.S.; Mamun, A.A.; Alghamdi, B.S.; Tewari, D.; Jeandet, P.; Sarwar, M.S.; Ashraf, G.M. Epigenetics of Glioblastoma Multiforme: From Molecular Mechanisms to Therapeutic Approaches. Semin. Cancer Biol. 2022, 83, 100–120.
- Zhao, D. Single Nucleotide Alterations in MicroRNAs and Human Cancer-A Not Fully Explored Field. Non-Coding RNA Res. 2020, 5, 27–31.
- Ciafrè, S.A.; Galardi, S.; Mangiola, A.; Ferracin, M.; Liu, C.-G.; Sabatino, G.; Negrini, M.; Maira, G.; Croce, C.M.; Farace, M.G. Extensive Modulation of a Set of microRNAs in Primary Glioblastoma. Biochem. Biophys. Res. Commun. 2005, 334, 1351–1358.
- Shea, A.; Harish, V.; Afzal, Z.; Chijioke, J.; Kedir, H.; Dusmatova, S.; Roy, A.; Ramalinga, M.; Harris, B.; Blancato, J.; et al. MicroRNAs in Glioblastoma Multiforme Pathogenesis and Therapeutics. Cancer Med. 2016, 5, 1917–1946.
- Chen, M.; Medarova, Z.; Moore, A. Role of microRNAs in Glioblastoma. Oncotarget 2021, 12, 1707–1723.
- Zhang, L.; Zhang, Y.; Gao, H.; Li, X.; Li, P. Underlying Mechanisms and Clinical Potential of circRNAs in Glioblastoma. Oncol. Res. 2023, 31, 449–462.
- Guo, X.; Piao, H. Research Progress of circRNAs in Glioblastoma. Front. Cell Dev. Biol. 2021, 9, 791892.
- Peng, Z.; Liu, C.; Wu, M. New Insights into Long Noncoding RNAs and Their Roles in Glioma. Mol. Cancer 2018, 17, 61.
- Zottel, A.; Šamec, N.; Videtič Paska, A.; Jovčevska, I. Coding of Glioblastoma Progression and Therapy Resistance through Long Noncoding RNAs. Cancers 2020, 12, 1842.
- Dai, L.; Liang, W.; Shi, Z.; Li, X.; Zhou, S.; Hu, W.; Yang, Z.; Wang, X. Systematic Characterization and Biological Functions of Non-coding RNAs in Glioblastoma. Cell Prolif. 2022, 56, e13375.
- Yadav, G.; Kulshreshtha, R. Metastasis Associated Long Noncoding RNAs in Glioblastoma: Biomarkers and Therapeutic Targets. J. Cell. Physiol. 2022, 237, 401–420.
- Shen, J.; Hodges, T.R.; Song, R.; Gong, Y.; Calin, G.A.; Heimberger, A.B.; Zhao, H. Serum HOTAIR and GAS5 Levels as Predictors of Survival in Patients with Glioblastoma. Mol. Carcinog. 2018, 57, 137–141.
- Xi, J.; Sun, Q.; Ma, L.; Kang, J. Long Non-Coding RNAs in Glioma Progression. Cancer Lett. 2018, 419, 203–209.
- Yu, W.; Ma, Y.; Hou, W.; Wang, F.; Cheng, W.; Qiu, F.; Wu, P.; Zhang, G. Identification of Immune-Related lncRNA Prognostic Signature and Molecular Subtypes for Glioblastoma. Front. Immunol. 2021, 12, 706936.
- Eraky, A.M.; Keles, A.; Goodman, S.L.; Baskaya, M.K. Serum Long Non-Coding RNAs as Potential Noninvasive Biomarkers for Glioblastoma Diagnosis, Prognosis, and Chemoresistance. JIN 2022, 21, 111.
- Chen, X.; Gao, Y.; Li, D.; Cao, Y.; Hao, B. LncRNA-TP53TG1 Participated in the Stress Response Under Glucose Deprivation in Glioma. J. Cell. Biochem. 2017, 118, 4897–4904.
- Wang, J.; Liu, X.; Yan, C.; Liu, J.; Wang, S.; Hong, Y.; Gu, A.; Zhao, P. LEF1-AS1, a Long-Noncoding RNA, Promotes Malignancy in Glioblastoma. OncoTargets Ther. 2017, 10, 4251–4260.
- Hegi, M.E.; Liu, L.; Herman, J.G.; Stupp, R.; Wick, W.; Weller, M.; Mehta, M.P.; Gilbert, M.R. Correlation of O 6-Methylguanine Methyltransferase (MGMT) Promoter Methylation With Clinical Outcomes in Glioblastoma and Clinical Strategies to Modulate MGMT Activity. JCO 2008, 26, 4189–4199.
- Velásquez, C.; Mansouri, S.; Gutiérrez, O.; Mamatjan, Y.; Mollinedo, P.; Karimi, S.; Singh, O.; Terán, N.; Martino, J.; Zadeh, G.; et al. Hypoxia Can Induce Migration of Glioblastoma Cells Through a Methylation-Dependent Control of ODZ1 Gene Expression. Front. Oncol. 2019, 9, 1036.
- Wu, Q.; Berglund, A.E.; Etame, A.B. The Impact of Epigenetic Modifications on Adaptive Resistance Evolution in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 8324.
- Scoumanne, A.; Zhang, J.; Chen, X. PRMT5 Is Required for Cell-Cycle Progression and P53 Tumor Suppressor Function. Nucleic Acids Res. 2009, 37, 4965–4976.
- Yan, F.; Alinari, L.; Lustberg, M.E.; Martin, L.K.; Cordero-Nieves, H.M.; Banasavadi-Siddegowda, Y.; Virk, S.; Barnholtz-Sloan, J.; Bell, E.H.; Wojton, J.; et al. Genetic Validation of the Protein Arginine Methyltransferase PRMT5 as a Candidate Therapeutic Target in Glioblastoma. Cancer Res. 2014, 74, 1752–1765.
- Banasavadi-Siddegowda, Y.K.; Russell, L.; Frair, E.; Karkhanis, V.A.; Relation, T.; Yoo, J.Y.; Zhang, J.; Sif, S.; Imitola, J.; Baiocchi, R.; et al. PRMT5-PTEN Molecular Pathway Regulates Senescence and Self-Renewal of Primary Glioblastoma Neurosphere Cells. Oncogene 2017, 36, 263–274.
- Banasavadi-Siddegowda, Y.K.; Welker, A.M.; An, M.; Yang, X.; Zhou, W.; Shi, G.; Imitola, J.; Li, C.; Hsu, S.; Wang, J.; et al. PRMT5 as a Druggable Target for Glioblastoma Therapy. Neuro-Oncology 2018, 20, 753–763.
- Yang, W.-B.; Wu, A.-C.; Hsu, T.-I.; Liou, J.-P.; Lo, W.-L.; Chang, K.-Y.; Chen, P.-Y.; Kikkawa, U.; Yang, S.-T.; Kao, T.-J.; et al. Histone Deacetylase 6 Acts Upstream of DNA Damage Response Activation to Support the Survival of Glioblastoma Cells. Cell Death Dis. 2021, 12, 884.
- Pastorino, O.; Gentile, M.T.; Mancini, A.; Del Gaudio, N.; Di Costanzo, A.; Bajetto, A.; Franco, P.; Altucci, L.; Florio, T.; Stoppelli, M.P.; et al. Histone Deacetylase Inhibitors Impair Vasculogenic Mimicry from Glioblastoma Cells. Cancers 2019, 11, 747.
- Yang, W.; Liu, Y.; Gao, R.; Yu, H.; Sun, T. HDAC6 Inhibition Induces Glioma Stem Cells Differentiation and Enhances Cellular Radiation Sensitivity through the SHH/Gli1 Signaling Pathway. Cancer Lett. 2018, 415, 164–176.
- Zhang, I.; Beus, M.; Stochaj, U.; Le, P.U.; Zorc, B.; Rajić, Z.; Petrecca, K.; Maysinger, D. Inhibition of Glioblastoma Cell Proliferation, Invasion, and Mechanism of Action of a Novel Hydroxamic Acid Hybrid Molecule. Cell Death Discov. 2018, 4, 41.
- LEE, P.; MURPHY, B.; MILLER, R.; MENON, V.; BANIK, N.L.; GIGLIO, P.; LINDHORST, S.M.; VARMA, A.K.; VANDERGRIFT, W.A.; PATEL, S.J.; et al. Mechanisms and Clinical Significance of Histone Deacetylase Inhibitors: Epigenetic Glioblastoma Therapy. Anticancer. Res. 2015, 35, 615–625.
- Rahman, M.A.; Engelsen, A.S.T.; Sarowar, S.; Bindesbøll, C.; Birkeland, E.; Goplen, D.; Lotsberg, M.L.; Knappskog, S.; Simonsen, A.; Chekenya, M. Bortezomib Abrogates Temozolomide-Induced Autophagic Flux through an ATG5 Dependent Pathway. Front. Cell Dev. Biol. 2022, 10, 1022191.
- Rampazzo, E.; Manfreda, L.; Bresolin, S.; Cani, A.; Mariotto, E.; Bortolozzi, R.; Della Puppa, A.; Viola, G.; Persano, L. Histone Deacetylase Inhibitors Impair Glioblastoma Cell Motility and Proliferation. Cancers 2022, 14, 1897.
- Everix, L.; Seane, E.N.; Ebenhan, T.; Goethals, I.; Bolcaen, J. Introducing HDAC-Targeting Radiopharmaceuticals for Glioblastoma Imaging and Therapy. Pharmaceuticals 2023, 16, 227.
- Reddy, R.G.; Bhat, U.A.; Chakravarty, S.; Kumar, A. Advances in Histone Deacetylase Inhibitors in Targeting Glioblastoma Stem Cells. Cancer Chemother. Pharmacol. 2020, 86, 165–179.
- Ghiaseddin, A.; Reardon, D.; Massey, W.; Mannerino, A.; Lipp, E.S.; Herndon, J.E.; McSherry, F.; Desjardins, A.; Randazzo, D.; Friedman, H.S.; et al. Phase II Study of Bevacizumab and Vorinostat for Patients with Recurrent World Health Organization Grade 4 Malignant Glioma. Oncologist 2018, 23, 157-e21.
- Lee, E.Q.; Reardon, D.A.; Schiff, D.; Drappatz, J.; Muzikansky, A.; Grimm, S.A.; Norden, A.D.; Nayak, L.; Beroukhim, R.; Rinne, M.L.; et al. Phase II Study of Panobinostat in Combination with Bevacizumab for Recurrent Glioblastoma and Anaplastic Glioma. Neuro Oncol. 2015, 17, 862–867.
- Tsai, H.-C.; Wei, K.-C.; Chen, P.-Y.; Huang, C.-Y.; Chen, K.-T.; Lin, Y.-J.; Cheng, H.-W.; Chen, Y.-R.; Wang, H.-T. Valproic Acid Enhanced Temozolomide-Induced Anticancer Activity in Human Glioma Through the P53-PUMA Apoptosis Pathway. Front. Oncol. 2021, 11, 722754.
- Stella, M.; Baiardi, G.; Pasquariello, S.; Sacco, F.; Dellacasagrande, I.; Corsaro, A.; Mattioli, F.; Barbieri, F. Antitumor Potential of Antiepileptic Drugs in Human Glioblastoma: Pharmacological Targets and Clinical Benefits. Biomedicines 2023, 11, 582.
- Sullivan, J.K.; Fahey, P.P.; Agho, K.E.; Hurley, S.P.; Feng, Z.; Day, R.O.; Lim, D. Valproic Acid as a Radio-Sensitizer in Glioma: A Systematic Review and Meta-Analysis. Neurooncol. Pract. 2023, 10, 13–23.
- Yuan, Y.; Xiang, W.; Qing, M.; Yanhui, L.; Jiewen, L.; Yunhe, M. Survival Analysis for Valproic Acid Use in Adult Glioblastoma Multiforme: A Meta-Analysis of Individual Patient Data and a Systematic Review. Seizure 2014, 23, 830–835.
- Krauze, A.V.; Megan, M.; Theresa, C.-Z.; Peter, M.; Shih, J.H.; Tofilon, P.J.; Rowe, L.; Gilbert, M.; Camphausen, K. The Addition of Valproic Acid to Concurrent Radiation Therapy and Temozolomide Improves Patient Outcome: A Correlative Analysis of RTOG 0525, SEER and a Phase II NCI Trial. Cancer Stud. Ther. 2020, 5.
- Scicchitano, B.M.; Sorrentino, S.; Proietti, G.; Lama, G.; Dobrowolny, G.; Catizone, A.; Binda, E.; Larocca, L.M.; Sica, G. Levetiracetam Enhances the Temozolomide Effect on Glioblastoma Stem Cell Proliferation and Apoptosis. Cancer Cell Int. 2018, 18.
- Hwang, K.; Kim, J.; Kang, S.; Jung, T.; Kim, J.H.; Kim, S.; Kang, S.; Hong, Y.; Kim, T.M.; Kim, Y.J.; et al. Levetiracetam as a Sensitizer of Concurrent Chemoradiotherapy in Newly Diagnosed Glioblastoma: An Open-label Phase 2 Study. Cancer Med. 2021, 11, 371–379.
- Pallud, J.; Huberfeld, G.; Dezamis, E.; Peeters, S.; Moiraghi, A.; Gavaret, M.; Guinard, E.; Dhermain, F.; Varlet, P.; Oppenheim, C.; et al. Effect of Levetiracetam Use Duration on Overall Survival of Isocitrate Dehydrogenase Wild-Type Glioblastoma in Adults: An Observational Study. Neurology 2022, 98, e125–e140.
- Happold, C.; Gorlia, T.; Chinot, O.; Gilbert, M.R.; Nabors, L.B.; Wick, W.; Pugh, S.L.; Hegi, M.; Cloughesy, T.; Roth, P.; et al. Does Valproic Acid or Levetiracetam Improve Survival in Glioblastoma? A Pooled Analysis of Prospective Clinical Trials in Newly Diagnosed Glioblastoma. J. Clin. Oncol. 2016, 34, 731–739.
- Laws, M.T.; Bonomi, R.E.; Kamal, S.; Gelovani, D.J.; Llaniguez, J.; Potukutchi, S.; Lu, X.; Mangner, T.; Gelovani, J.G. Molecular Imaging HDACs Class IIa Expression-Activity and Pharmacologic Inhibition in Intracerebral Glioma Models in Rats Using PET/CT/(MRI) with TFAHA. Sci. Rep. 2019, 9, 3595.
- Cao, R.; Bråkenhielm, E.; Pawliuk, R.; Wariaro, D.; Post, M.J.; Wahlberg, E.; Leboulch, P.; Cao, Y. Angiogenic Synergism, Vascular Stability and Improvement of Hind-Limb Ischemia by a Combination of PDGF-BB and FGF-2. Nat. Med. 2003, 9, 604–613.
- Buccarelli, M.; Castellani, G.; Ricci-Vitiani, L. Glioblastoma-Specific Strategies of Vascularization: Implications in Anti-Angiogenic Therapy Resistance. J. Pers. Med. 2022, 12, 1625.
- Chiao, M.-T.; Yang, Y.-C.; Cheng, W.-Y.; Shen, C.-C.; Ko, J.-L. CD133+ Glioblastoma Stem-like Cells Induce Vascular Mimicry in Vivo. Curr. Neurovascular Res. 2011, 8, 210–219.
- Rosińska, S.; Gavard, J. Tumor Vessels Fuel the Fire in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 6514.
- El Hallani, S.; Boisselier, B.; Peglion, F.; Rousseau, A.; Colin, C.; Idbaih, A.; Marie, Y.; Mokhtari, K.; Thomas, J.-L.; Eichmann, A.; et al. A New Alternative Mechanism in Glioblastoma Vascularization: Tubular Vasculogenic Mimicry. Brain 2010, 133, 973–982.
- Das, S.; Marsden, P.A. Angiogenesis in Glioblastoma. N. Engl. J. Med. 2013, 369, 1561–1563.
- Kioi, M.; Vogel, H.; Schultz, G.; Hoffman, R.M.; Harsh, G.R.; Brown, J.M. Inhibition of Vasculogenesis, but Not Angiogenesis, Prevents the Recurrence of Glioblastoma after Irradiation in Mice. J. Clin. Investig. 2010, 120, 694–705.
- Mei, X.; Chen, Y.-S.; Chen, F.-R.; Xi, S.-Y.; Chen, Z.-P. Glioblastoma Stem Cell Differentiation into Endothelial Cells Evidenced through Live-Cell Imaging. Neuro Oncol. 2017, 19, 1109–1118.
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A Perivascular Niche for Brain Tumor Stem Cells. Cancer Cell 2007, 11, 69–82.
- Jhaveri, N.; Chen, T.C.; Hofman, F.M. Tumor Vasculature and Glioma Stem Cells: Contributions to Glioma Progression. Cancer Lett. 2016, 380, 545–551.
- Charles, N.; Holland, E.C. The Perivascular Niche Microenvironment in Brain Tumor Progression. Cell Cycle 2010, 9, 3012–3021.
- Hira, V.V.V.; Aderetti, D.A.; van Noorden, C.J.F. Glioma Stem Cell Niches in Human Glioblastoma Are Periarteriolar. J. Histochem. Cytochem. 2018, 66, 349–358.
- Tan, C.; Cruet-Hennequart, S.; Troussard, A.; Fazli, L.; Costello, P.; Sutton, K.; Wheeler, J.; Gleave, M.; Sanghera, J.; Dedhar, S. Regulation of Tumor Angiogenesis by Integrin-Linked Kinase (ILK). Cancer Cell 2004, 5, 79–90.
- Zagzag, D.; Lukyanov, Y.; Lan, L.; Ali, M.A.; Esencay, M.; Mendez, O.; Yee, H.; Voura, E.B.; Newcomb, E.W. Hypoxia-Inducible Factor 1 and VEGF Upregulate CXCR4 in Glioblastoma: Implications for Angiogenesis and Glioma Cell Invasion. Lab. Investig. 2006, 86, 1221–1232.
- Domènech, M.; Hernández, A.; Plaja, A.; Martínez-Balibrea, E.; Balañà, C. Hypoxia: The Cornerstone of Glioblastoma. Int. J. Mol. Sci. 2021, 22, 12608.
- Renfrow, J.J.; Soike, M.H.; West, J.L.; Ramkissoon, S.H.; Metheny-Barlow, L.; Mott, R.T.; Kittel, C.A.; D’Agostino, R.B.; Tatter, S.B.; Laxton, A.W.; et al. Attenuating Hypoxia Driven Malignant Behavior in Glioblastoma with a Novel Hypoxia-Inducible Factor 2 Alpha Inhibitor. Sci. Rep. 2020, 10, 15195.
- Strowd, R.; Ellingson, B.; Raymond, C.; Yao, J.; Wen, P.Y.; Ahluwalia, M.; Piotrowski, A.; Desai, A.; Clarke, J.L.; Lieberman, F.S.; et al. Activity of a First-in-Class Oral HIF2-Alpha Inhibitor, PT2385, in Patients with First Recurrence of Glioblastoma. J. Neurooncol. 2023, 165, 101–112.
- Srivastava, C.; Irshad, K.; Dikshit, B.; Chattopadhyay, P.; Sarkar, C.; Gupta, D.K.; Sinha, S.; Chosdol, K. FAT1 Modulates EMT and Stemness Genes Expression in Hypoxic Glioblastoma. Int. J. Cancer 2018, 142, 805–812.
- Aderetti, D.A.; Hira, V.V.V.; Molenaar, R.J.; van Noorden, C.J.F. The Hypoxic Peri-Arteriolar Glioma Stem Cell Niche, an Integrated Concept of Five Types of Niches in Human Glioblastoma. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 346–354.
- Kumar, S.; Sharife, H.; Kreisel, T.; Mogilevsky, M.; Bar-Lev, L.; Grunewald, M.; Aizenshtein, E.; Karni, R.; Paldor, I.; Shlomi, T.; et al. Intra-Tumoral Metabolic Zonation and Resultant Phenotypic Diversification Are Dictated by Blood Vessel Proximity. Cell Metab. 2019, 30, 201–211.e6.
- Da Ros, M.; De Gregorio, V.; Iorio, A.L.; Giunti, L.; Guidi, M.; de Martino, M.; Genitori, L.; Sardi, I. Glioblastoma Chemoresistance: The Double Play by Microenvironment and Blood-Brain Barrier. Int. J. Mol. Sci. 2018, 19, 2879.
- Faustino, A.C.; Viani, G.A.; Hamamura, A.C. Patterns of Recurrence and Outcomes of Glioblastoma Multiforme Treated with Chemoradiation and Adjuvant Temozolomide. Clinics 2020, 75, e1553.
- Lakomy, R.; Kazda, T.; Selingerova, I.; Poprach, A.; Pospisil, P.; Belanova, R.; Fadrus, P.; Vybihal, V.; Smrcka, M.; Jancalek, R.; et al. Real-World Evidence in Glioblastoma: Stupp’s Regimen After a Decade. Front. Oncol. 2020, 10, 840.
- AbdelFatah, M.A.R.; Kotb, A.; Said, M.A.; Abouelmaaty, E.M.H. Impact of Extent of Resection of Newly Diagnosed Glioblastomas on Survival: A Meta-Analysis. Egypt. J. Neurosurg. 2022, 37, 3.
- Weller, M.; van den Bent, M.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. EANO Guidelines on the Diagnosis and Treatment of Diffuse Gliomas of Adulthood. Nat. Rev. Clin. Oncol. 2021, 18, 170–186.
- Mohile, N.A.; Messersmith, H.; Gatson, N.T.; Hottinger, A.F.; Lassman, A.; Morton, J.; Ney, D.; Nghiemphu, P.L.; Olar, A.; Olson, J.; et al. Therapy for Diffuse Astrocytic and Oligodendroglial Tumors in Adults: ASCO-SNO Guideline. J. Clin. Oncol. 2022, 40, 403–426.
- Bonosi, L.; Marrone, S.; Benigno, U.E.; Buscemi, F.; Musso, S.; Porzio, M.; Silven, M.P.; Torregrossa, F.; Grasso, G. Maximal Safe Resection in Glioblastoma Surgery: A Systematic Review of Advanced Intraoperative Image-Guided Techniques. Brain Sci. 2023, 13, 216.
- Gerritsen, J.K.W.; Broekman, M.L.D.; De Vleeschouwer, S.; Schucht, P.; Nahed, B.V.; Berger, M.S.; Vincent, A.J.P.E. Safe Surgery for Glioblastoma: Recent Advances and Modern Challenges. Neurooncol. Pract. 2022, 9, 364–379.
- Ille, S.; Sollmann, N.; Hauck, T.; Maurer, S.; Tanigawa, N.; Obermueller, T.; Negwer, C.; Droese, D.; Zimmer, C.; Meyer, B.; et al. Combined Noninvasive Language Mapping by Navigated Transcranial Magnetic Stimulation and Functional MRI and Its Comparison with Direct Cortical Stimulation. J. Neurosurg. 2015, 123, 212–225.
- Stummer, W.; Tonn, J.-C.; Mehdorn, H.M.; Nestler, U.; Franz, K.; Goetz, C.; Bink, A.; Pichlmeier, U.; ALA-Glioma Study Group. Counterbalancing Risks and Gains from Extended Resections in Malignant Glioma Surgery: A Supplemental Analysis from the Randomized 5-Aminolevulinic Acid Glioma Resection Study. Clinical Article. J. Neurosurg. 2011, 114, 613–623.
- Al-Adli, N.N.; Young, J.S.; Sibih, Y.E.; Berger, M.S. Technical Aspects of Motor and Language Mapping in Glioma Patients. Cancers 2023, 15, 2173.
- Stummer, W.; Pichlmeier, U.; Meinel, T.; Wiestler, O.D.; Zanella, F.; Reulen, H.-J. ALA-Glioma Study Group Fluorescence-Guided Surgery with 5-Aminolevulinic Acid for Resection of Malignant Glioma: A Randomised Controlled Multicentre Phase III Trial. Lancet Oncol. 2006, 7, 392–401.
- Eljamel, S. 5-ALA Fluorescence Image Guided Resection of Glioblastoma Multiforme: A Meta-Analysis of the Literature. Int. J. Mol. Sci. 2015, 16, 10443–10456.
- Kiesel, B.; Wadiura, L.I.; Mischkulnig, M.; Makolli, J.; Sperl, V.; Borkovec, M.; Freund, J.; Lang, A.; Millesi, M.; Berghoff, A.S.; et al. Efficacy, Outcome, and Safety of Elderly Patients with Glioblastoma in the 5-ALA Era: Single Center Experience of More Than 10 Years. Cancers 2021, 13, 6119.
- Baig Mirza, A.; Christodoulides, I.; Lavrador, J.P.; Giamouriadis, A.; Vastani, A.; Boardman, T.; Ahmed, R.; Norman, I.; Murphy, C.; Devi, S.; et al. 5-Aminolevulinic Acid-Guided Resection Improves the Overall Survival of Patients with Glioblastoma—A Comparative Cohort Study of 343 Patients. Neuro-Oncology Adv. 2021, 3, vdab047.
- Chohan, M.O.; Berger, M.S. 5-Aminolevulinic Acid Fluorescence Guided Surgery for Recurrent High-Grade Gliomas. J. Neurooncol. 2019, 141, 517–522.
- Smith, E.J.; Gohil, K.; Thompson, C.M.; Naik, A.; Hassaneen, W. Fluorescein-Guided Resection of High Grade Gliomas: A Meta-Analysis. World Neurosurg. 2021, 155, 181–188.e7.
- Acerbi, F.; Broggi, M.; Schebesch, K.-M.; Höhne, J.; Cavallo, C.; De Laurentis, C.; Eoli, M.; Anghileri, E.; Servida, M.; Boffano, C.; et al. Fluorescein-Guided Surgery for Resection of High-Grade Gliomas: A Multicentric Prospective Phase II Study (FLUOGLIO). Clin. Cancer Res. 2018, 24, 52–61.
- Ahrens, L.C.; Krabbenhøft, M.G.; Hansen, R.W.; Mikic, N.; Pedersen, C.B.; Poulsen, F.R.; Korshoej, A.R. Effect of 5-Aminolevulinic Acid and Sodium Fluorescein on the Extent of Resection in High-Grade Gliomas and Brain Metastasis. Cancers 2022, 14, 617.
- Noh, T.; Mustroph, M.; Golby, A.J. Intraoperative Imaging for High-Grade Glioma Surgery. Neurosurg. Clin. N. Am. 2021, 32, 47–54.
- Roder, C.; Bisdas, S.; Ebner, F.H.; Honegger, J.; Naegele, T.; Ernemann, U.; Tatagiba, M. Maximizing the Extent of Resection and Survival Benefit of Patients in Glioblastoma Surgery: High-Field iMRI versus Conventional and 5-ALA-Assisted Surgery. Eur. J. Surg. Oncol. (EJSO) 2014, 40, 297–304.
- Coburger, J.; Hagel, V.; Wirtz, C.R.; König, R. Surgery for Glioblastoma: Impact of the Combined Use of 5-Aminolevulinic Acid and Intraoperative MRI on Extent of Resection and Survival. PLoS ONE 2015, 10, e0131872.
- Nickel, K.; Renovanz, M.; König, J.; Stöckelmaier, L.; Hickmann, A.-K.; Nadji-Ohl, M.; Engelke, J.; Weimann, E.; Freudenstein, D.; Ganslandt, O.; et al. The Patients’ View: Impact of the Extent of Resection, Intraoperative Imaging, and Awake Surgery on Health-Related Quality of Life in High-Grade Glioma Patients—Results of a Multicenter Cross-Sectional Study. Neurosurg. Rev. 2018, 41, 207–219.
- Golub, D.; Hyde, J.; Dogra, S.; Nicholson, J.; Kirkwood, K.A.; Gohel, P.; Loftus, S.; Schwartz, T.H. Intraoperative MRI versus 5-ALA in High-Grade Glioma Resection: A Network Meta-Analysis. J. Neurosurg. 2020, 1–15.
- Hottinger, A.F.; Stupp, R.; Homicsko, K. Standards of Care and Novel Approaches in the Management of Glioblastoma Multiforme. Chin. J. Cancer 2014, 33, 32–39.
- Brown, T.J.; Brennan, M.C.; Li, M.; Church, E.W.; Brandmeir, N.J.; Rakszawski, K.L.; Patel, A.S.; Rizk, E.B.; Suki, D.; Sawaya, R.; et al. Association of the Extent of Resection With Survival in Glioblastoma: A Systematic Review and Meta-Analysis. JAMA Oncol. 2016, 2, 1460–1469.
- Shah, A.H.; Heiss, J.D. Neurosurgical Clinical Trials for Glioblastoma: Current and Future Directions. Brain Sci. 2022, 12, 787.
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in Adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) Consensus Review on Current Management and Future Directions. Neuro-Oncology 2020, 22, 1073–1113.
- Westphal, M.; Hilt, D.C.; Bortey, E.; Delavault, P.; Olivares, R.; Warnke, P.C.; Whittle, I.R.; Jääskeläinen, J.; Ram, Z. A Phase 3 Trial of Local Chemotherapy with Biodegradable Carmustine (BCNU) Wafers (Gliadel Wafers) in Patients with Primary Malignant Glioma. Neuro Oncol. 2003, 5, 79–88.
- Fisher, J.P.; Adamson, D.C. Current FDA-Approved Therapies for High-Grade Malignant Gliomas. Biomedicines 2021, 9, 324.
- Jung, I.-H.; Chang, K.W.; Park, S.H.; Moon, J.H.; Kim, E.H.; Jung, H.H.; Kang, S.-G.; Chang, J.H.; Chang, J.W.; Chang, W.S. Stereotactic Biopsy for Adult Brainstem Lesions: A Surgical Approach and Its Diagnostic Value According to the 2016 World Health Organization Classification. Cancer Med. 2021, 10, 7514–7524.
- Katzendobler, S.; Do, A.; Weller, J.; Dorostkar, M.M.; Albert, N.L.; Forbrig, R.; Niyazi, M.; Egensperger, R.; Thon, N.; Tonn, J.C.; et al. Diagnostic Yield and Complication Rate of Stereotactic Biopsies in Precision Medicine of Gliomas. Front. Neurol. 2022, 13, 822362.
- Peters, D.R.; Asher, A.L.; Wait, S.D.; Smith, M.D.; Peters, D.R.; Asher, A.L.; Kelly, R.; Boltes, M.; Wait, S.D.; Smith, M.D.; et al. Is There a Role for Stereotactic Biopsy of Unresectable High-Grade Gliomas? A Retrospective Cohort Study of Short-Term Morbidity and Mortality. TNN 2021, 4, 1–7.
- Kazmi, F.; Soon, Y.Y.; Leong, Y.H.; Koh, W.Y.; Vellayappan, B. Re-Irradiation for Recurrent Glioblastoma (GBM): A Systematic Review and Meta-Analysis. J. Neurooncol. 2019, 142, 79–90.
- Zhang, J.; Stevens, M.F.G.; Bradshaw, T.D. Temozolomide: Mechanisms of Action, Repair and Resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114.
- McLendon, R.; Friedman, A.; Bigner, D.; Van Meir, E.G.; Brat, D.J.; Mastrogianakis, G.M.; Olson, J.J.; Mikkelsen, T.; Lehman, N.; Aldape, K.; et al. Comprehensive Genomic Characterization Defines Human Glioblastoma Genes and Core Pathways. Nature 2008, 455, 1061–1068.
- Liu, D.; Yang, T.; Ma, W.; Wang, Y. Clinical Strategies to Manage Adult Glioblastoma Patients without MGMT Hypermethylation. J. Cancer 2022, 13, 354–363.
- Gilbert, M.R.; Wang, M.; Aldape, K.D.; Stupp, R.; Hegi, M.E.; Jaeckle, K.A.; Armstrong, T.S.; Wefel, J.S.; Won, M.; Blumenthal, D.T.; et al. Dose-Dense Temozolomide for Newly Diagnosed Glioblastoma: A Randomized Phase III Clinical Trial. J. Clin. Oncol. 2013, 31, 4085–4091.
- Gately, L.; Mesía, C.; Sepúlveda, J.M.; Del Barco, S.; Pineda, E.; Gironés, R.; Fuster, J.; Hong, W.; Dumas, M.; Gill, S.; et al. A Combined Analysis of Two Prospective Randomised Studies Exploring the Impact of Extended Post-Radiation Temozolomide on Survival Outcomes in Newly Diagnosed Glioblastoma. J. Neurooncol. 2023.
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of Temozolomide Resistance in Glioblastoma—A Comprehensive Review. Cancer Drug Resist. 2021, 4, 17–43.
- Arora, A.; Somasundaram, K. Glioblastoma vs Temozolomide: Can the Red Queen Race Be Won? Cancer Biol. Ther. 2019, 20, 1083–1090.
- Almeida Lima, K.; Osawa, I.Y.A.; Ramalho, M.C.C.; de Souza, I.; Guedes, C.B.; de Souza Filho, C.H.D.; Monteiro, L.K.S.; Latancia, M.T.; Rocha, C.R.R. Temozolomide Resistance in Glioblastoma by NRF2: Protecting the Evil. Biomedicines 2023, 11, 1081.
- Lee, S.Y. Temozolomide Resistance in Glioblastoma Multiforme. Genes Dis. 2016, 3, 198–210.
- Pang, L.Y.; Saunders, L.; Argyle, D.J. Epidermal Growth Factor Receptor Activity Is Elevated in Glioma Cancer Stem Cells and Is Required to Maintain Chemotherapy and Radiation Resistance. Oncotarget 2017, 8, 72494–72512.
- Campos-Sandoval, J.A.; Gómez-García, M.C.; Santos-Jiménez, J.d.L.; Matés, J.M.; Alonso, F.J.; Márquez, J. Antioxidant Responses Related to Temozolomide Resistance in Glioblastoma. Neurochem. Int. 2021, 149, 105136.
- Ortiz, R.; Perazzoli, G.; Cabeza, L.; Jiménez-Luna, C.; Luque, R.; Prados, J.; Melguizo, C. Temozolomide: An Updated Overview of Resistance Mechanisms, Nanotechnology Advances and Clinical Applications. Curr. Neuropharmacol. 2021, 19, 513–537.
- Herrlinger, U.; Tzaridis, T.; Mack, F.; Steinbach, J.P.; Schlegel, U.; Sabel, M.; Hau, P.; Kortmann, R.-D.; Krex, D.; Grauer, O.; et al. Lomustine-Temozolomide Combination Therapy versus Standard Temozolomide Therapy in Patients with Newly Diagnosed Glioblastoma with Methylated MGMT Promoter (CeTeG/NOA-09): A Randomised, Open-Label, Phase 3 Trial. Lancet 2019, 393, 678–688.
- Weller, J.; Tzaridis, T.; Mack, F.; Steinbach, J.P.; Schlegel, U.; Hau, P.; Krex, D.; Grauer, O.; Goldbrunner, R.; Bähr, O.; et al. Health-Related Quality of Life and Neurocognitive Functioning with Lomustine-Temozolomide versus Temozolomide in Patients with Newly Diagnosed, MGMT-Methylated Glioblastoma (CeTeG/NOA-09): A Randomised, Multicentre, Open-Label, Phase 3 Trial. Lancet Oncol. 2019, 20, 1444–1453.
- Stupp, R.; Lukas, R.V.; Hegi, M.E. Improving Survival in Molecularly Selected Glioblastoma. Lancet 2019, 393, 615–617.
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus Radiotherapy-Temozolomide for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 709–722.
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708.
- Blumenthal, D.T.; Rankin, C.; Stelzer, K.J.; Spence, A.M.; Sloan, A.E.; Moore, D.F.; Padula, G.D.A.; Schulman, S.B.; Wade, M.L.; Rushing, E.J. A Phase III Study of Radiation Therapy (RT) and O6-Benzylguanine + BCNU versus RT and BCNU Alone and Methylation Status in Newly Diagnosed Glioblastoma and Gliosarcoma: Southwest Oncology Group (SWOG) Study S0001. Int. J. Clin. Oncol. 2015, 20, 650–658.
- Mehta, M.; Wen, P.; Nishikawa, R.; Reardon, D.; Peters, K. Critical Review of the Addition of Tumor Treating Fields (TTFields) to the Existing Standard of Care for Newly Diagnosed Glioblastoma Patients. Crit. Rev. Oncol. /Hematol. 2017, 111, 60–65.
- Burri, S.H.; Gondi, V.; Brown, P.D.; Mehta, M.P. The Evolving Role of Tumor Treating Fields in Managing Glioblastoma: Guide for Oncologists. Am. J. Clin. Oncol. 2018, 41, 191–196.
- Giladi, M.; Munster, M.; Schneiderman, R.S.; Voloshin, T.; Porat, Y.; Blat, R.; Zielinska-Chomej, K.; Hååg, P.; Bomzon, Z.; Kirson, E.D.; et al. Tumor Treating Fields (TTFields) Delay DNA Damage Repair Following Radiation Treatment of Glioma Cells. Radiat. Oncol. 2017, 12.
- Mun, E.J.; Babiker, H.M.; Weinberg, U.; Kirson, E.D.; Von Hoff, D.D. Tumor-Treating Fields: A Fourth Modality in Cancer Treatment. Clin. Cancer Res. 2018, 24, 266–275.
- Shteingauz, A.; Porat, Y.; Voloshin, T.; Schneiderman, R.S.; Munster, M.; Zeevi, E.; Kaynan, N.; Gotlib, K.; Giladi, M.; Kirson, E.D.; et al. AMPK-Dependent Autophagy Upregulation Serves as a Survival Mechanism in Response to Tumor Treating Fields (TTFields). Cell Death Dis. 2018, 9, 1074.
- Kim, E.H.; Jo, Y.; Sai, S.; Park, M.-J.; Kim, J.-Y.; Kim, J.S.; Lee, Y.-J.; Cho, J.-M.; Kwak, S.-Y.; Baek, J.-H.; et al. Tumor-Treating Fields Induce Autophagy by Blocking the Akt2/miR29b Axis in Glioblastoma Cells. Oncogene 2019, 38, 6630–6646.
- Kissling, C.; Di Santo, S. Tumor Treating Fields—Behind and Beyond Inhibiting the Cancer Cell Cycle. CNS Neurol. Disord. Drug Targets 2020, 19, 599–610.
- Wu, H.; Yang, L.; Liu, H.; Zhou, D.; Chen, D.; Zheng, X.; Yang, H.; Li, C.; Chang, J.; Wu, A.; et al. Exploring the Efficacy of Tumor Electric Field Therapy against Glioblastoma: An in Vivo and in Vitro Study. CNS Neurosci. Ther. 2021, 27, 1587–1604.
- Guo, X.; Yang, X.; Wu, J.; Yang, H.; Li, Y.; Li, J.; Liu, Q.; Wu, C.; Xing, H.; Liu, P.; et al. Tumor-Treating Fields in Glioblastomas: Past, Present, and Future. Cancers 2022, 14, 3669.
- Tanzhu, G.; Chen, L.; Xiao, G.; Shi, W.; Peng, H.; Chen, D.; Zhou, R. The Schemes, Mechanisms and Molecular Pathway Changes of Tumor Treating Fields (TTFields) Alone or in Combination with Radiotherapy and Chemotherapy. Cell Death Discov. 2022, 8, 416.
- Shi, P.; Tian, J.; Ulm, B.S.; Mallinger, J.C.; Khoshbouei, H.; Deleyrolle, L.P.; Sarkisian, M.R. Tumor Treating Fields Suppression of Ciliogenesis Enhances Temozolomide Toxicity. Front. Oncol. 2022, 12, 837589.
- Kim, E.H.; Song, H.S.; Yoo, S.H.; Yoon, M. Tumor Treating Fields Inhibit Glioblastoma Cell Migration, Invasion and Angiogenesis. Oncotarget 2016, 7, 65125.
- Chang, E.; Patel, C.B.; Pohling, C.; Young, C.; Song, J.; Flores, T.A.; Zeng, Y.; Joubert, L.-M.; Arami, H.; Natarajan, A.; et al. Tumor Treating Fields Increases Membrane Permeability in Glioblastoma Cells. Cell Death Discov. 2018, 4, 113.
- Salvador, E.; Kessler, A.F.; Domröse, D.; Hörmann, J.; Schaeffer, C.; Giniunaite, A.; Burek, M.; Tempel-Brami, C.; Voloshin, T.; Volodin, A.; et al. Tumor Treating Fields (TTFields) Reversibly Permeabilize the Blood-Brain Barrier In Vitro and In Vivo. Biomolecules 2022, 12, 1348.
- Diamant, G.; Simchony Goldman, H.; Gasri Plotnitsky, L.; Roitman, M.; Shiloach, T.; Globerson-Levin, A.; Eshhar, Z.; Haim, O.; Pencovich, N.; Grossman, R.; et al. T Cells Retain Pivotal Antitumoral Functions under Tumor-Treating Electric Fields. J. Immunol. 2021, 207, 709–719.
- Voloshin, T.; Davidi, S.; Porat, Y.; Shteingauz, A.; Munster, M.; Kaynan, N.; Giladi, M.; Kirson, E.; Weinberg, U.; Plati, Y. IMMU-52. TUMOR TREATING FIELDS (TTFIELDS) INDUCE IMMUNOGENIC CELL DEATH RESULTING IN ENHANCED ANTITUMOR EFFICACY WHEN COMBINED WITH ANTI-PD-1 THERAPY. Neuro Oncol. 2018, 20, vi133.
- Chen, D.; Le, S.B.; Hutchinson, T.E.; Calinescu, A.-A.; Sebastian, M.; Jin, D.; Liu, T.; Ghiaseddin, A.; Rahman, M.; Tran, D.D. Tumor Treating Fields Dually Activate STING and AIM2 Inflammasomes to Induce Adjuvant Immunity in Glioblastoma. J. Clin. Investig. 2022, 132, e149258.
- Jo, Y.; Han, Y.I.; Lee, E.; Seo, J.; Oh, G.; Sung, H.; Gi, Y.; Kim, H.; Park, S.; Yoon, M. The Combination of Tumor Treating Fields and Hyperthermia Has Synergistic Therapeutic Effects in Glioblastoma Cells by Downregulating STAT3. Am. J. Cancer Res. 2022, 12, 1423–1432.
- Pandey, M.; Xiu, J.; Mittal, S.; Zeng, J.; Saul, M.; Kesari, S.; Azadi, A.; Newton, H.; Deniz, K.; Ladner, K.; et al. Molecular Alterations Associated with Improved Outcome in Patients with Glioblastoma Treated with Tumor-Treating Fields. Neuro-Oncology Adv. 2022, 4, vdac096.
- Bokstein, F.; Blumenthal, D.; Limon, D.; Harosh, C.B.; Ram, Z.; Grossman, R. Concurrent Tumor Treating Fields (TTFields) and Radiation Therapy for Newly Diagnosed Glioblastoma: A Prospective Safety and Feasibility Study. Front. Oncol. 2020, 10, 411.
- Krigers, A.; Pinggera, D.; Demetz, M.; Kornberger, L.-M.; Kerschbaumer, J.; Thomé, C.; Freyschlag, C.F. The Routine Application of Tumor-Treating Fields in the Treatment of Glioblastoma WHO° IV. Front. Neurol. 2022, 13.
- Stupp, R.; Wong, E.T.; Kanner, A.A.; Steinberg, D.; Engelhard, H.; Heidecke, V.; Kirson, E.D.; Taillibert, S.; Liebermann, F.; Dbalý, V.; et al. NovoTTF-100A versus Physician’s Choice Chemotherapy in Recurrent Glioblastoma: A Randomised Phase III Trial of a Novel Treatment Modality. Eur. J. Cancer 2012, 48, 2192–2202.
- Taphoorn, M.J.B.; Dirven, L.; Kanner, A.A.; Lavy-Shahaf, G.; Weinberg, U.; Taillibert, S.; Toms, S.A.; Honnorat, J.; Chen, T.C.; Sroubek, J.; et al. Influence of Treatment With Tumor-Treating Fields on Health-Related Quality of Life of Patients With Newly Diagnosed Glioblastoma: A Secondary Analysis of a Randomized Clinical Trial. JAMA Oncol. 2018, 4, 495–504.
- Vymazal, J.; Kazda, T.; Novak, T.; Slanina, P.; Sroubek, J.; Klener, J.; Hrbac, T.; Syrucek, M.; Rulseh, A.M. Eighteen Years’ Experience with Tumor Treating Fields in the Treatment of Newly Diagnosed Glioblastoma. Front. Oncol. 2023, 12, 1014455.
- Toms, S.A.; Kim, C.Y.; Nicholas, G.; Ram, Z. Increased Compliance with Tumor Treating Fields Therapy Is Prognostic for Improved Survival in the Treatment of Glioblastoma: A Subgroup Analysis of the EF-14 Phase III Trial. J. Neurooncol. 2019, 141, 467–473.
- Stupp, R.; Taillibert, S.; Kanner, A.A.; Kesari, S.; Steinberg, D.M.; Toms, S.A.; Taylor, L.P.; Lieberman, F.; Silvani, A.; Fink, K.L.; et al. Maintenance Therapy With Tumor-Treating Fields Plus Temozolomide vs Temozolomide Alone for Glioblastoma: A Randomized Clinical Trial. JAMA 2015, 314, 2535–2543.
- Zhu, P.; Zhu, J.-J. Tumor Treating Fields: A Novel and Effective Therapy for Glioblastoma: Mechanism, Efficacy, Safety and Future Perspectives. Chin. Clin. Oncol. 2017, 6, 41.
- Rominiyi, O.; Vanderlinden, A.; Clenton, S.J.; Bridgewater, C.; Al-Tamimi, Y.; Collis, S.J. Tumour Treating Fields Therapy for Glioblastoma: Current Advances and Future Directions. Br. J. Cancer 2021, 124, 697–709.
- Wong, E.T.; Lok, E.; Gautam, S.; Swanson, K.D. Dexamethasone Exerts Profound Immunologic Interference on Treatment Efficacy for Recurrent Glioblastoma. Br. J. Cancer 2015, 113, 232–241.
- Swildens, K.X.; Sillevis Smitt, P.A.E.; van den Bent, M.J.; French, P.J.; Geurts, M. The Effect of Dexamethasone on the Microenvironment and Efficacy of Checkpoint Inhibitors in Glioblastoma: A Systematic Review. Neurooncol. Adv. 2022, 4, vdac087.
- Li, S.; Dong, J.; Wang, X.; Meng, X.; Jiang, C.; Cai, J. Dexamethasone and Compliance Affect TTFields Efficacy to Glioblastoma Patients: A Systematic Review and Meta-Analysis. Chin. Neurosurg. J. 2022, 8, 24.
- Wang, M.; Zhang, C.; Wang, X.; Yu, H.; Zhang, H.; Xu, J.; Zhao, J.; Jiang, X. Tumor-Treating Fields (TTFields)-Based Cocktail Therapy: A Novel Blueprint for Glioblastoma Treatment. Am. J. Cancer Res. 2021, 11, 1069–1086.
- Karanam, N.K.; Story, M.D. An Overview of Potential Novel Mechanisms of Action Underlying Tumor Treating Fields-Induced Cancer Cell Death and Their Clinical Implications. Int. J. Radiat. Biol. 2021, 97, 1044–1054.
- Glas, M.; Scheffler, B.; Lazaridis, L.; Herrlinger, U.; Pierscianek, D.; Sure, U.; Proescholdt, M.; Hau, P.; Hense, J.; Kleinschnitz, C.; et al. ACTR-49. PriCoTTF: A PHASE I/II TRIAL OF TUMOR TREATING FIELDS PRIOR AND CONCOMITANT TO RADIOTHERAPY IN NEWLY DIAGNOSED GLIOBLASTOMA. Neuro Oncol. 2018, 20, vi22–vi23.
- Karanam, N.K.; Srinivasan, K.; Ding, L.; Sishc, B.; Saha, D.; Story, M.D. Tumor-Treating Fields Elicit a Conditional Vulnerability to Ionizing Radiation via the Downregulation of BRCA1 Signaling and Reduced DNA Double-Strand Break Repair Capacity in Non-Small Cell Lung Cancer Cell Lines. Cell Death Dis. 2017, 8, e2711.
- Lazaridis, L.; Schäfer, N.; Teuber-Hanselmann, S.; Blau, T.; Schmidt, T.; Oster, C.; Weller, J.; Tzaridis, T.; Pierscianek, D.; Keyvani, K.; et al. Tumour Treating Fields (TTFields) in Combination with Lomustine and Temozolomide in Patients with Newly Diagnosed Glioblastoma. J. Cancer Res. Clin. Oncol. 2020, 146, 787–792.
- Lu, G.; Rao, M.; Zhu, P.; Liang, B.; El-Nazer, R.T.; Fonkem, E.; Bhattacharjee, M.B.; Zhu, J.-J. Triple-Drug Therapy With Bevacizumab, Irinotecan, and Temozolomide Plus Tumor Treating Fields for Recurrent Glioblastoma: A Retrospective Study. Front. Neurol. 2019, 10, 42.
- Catacuzzeno, L.; Franciolini, F. Role of KCa3.1 Channels in Modulating Ca2+ Oscillations during Glioblastoma Cell Migration and Invasion. Int. J. Mol. Sci. 2018, 19, 2970.
- Robins, H.I.; Nguyen, H.N.; Field, A.; Howard, S.; Salamat, S.; Deming, D.A. Molecular Evolution of a Glioblastoma Controlled With Tumor Treating Fields and Concomitant Temozolomide. Front. Oncol. 2018, 8, 451.
- Ahmadi-Zeidabadi, M.; Akbarnejad, Z.; Esmaeeli, M.; Masoumi-Ardakani, Y.; Mohammadipoor-Ghasemabad, L.; Eskandary, H. Impact of Extremely Low-Frequency Electromagnetic Field (100 Hz, 100 G) Exposure on Human Glioblastoma U87 Cells during Temozolomide Administration. Electromagn. Biol. Med. 2019, 38, 198–209.
- Cruz, N.; Herculano-Carvalho, M.; Roque, D.; Faria, C.C.; Cascão, R.; Ferreira, H.A.; Reis, C.P.; Matela, N. Highlighted Advances in Therapies for Difficult-To-Treat Brain Tumours Such as Glioblastoma. Pharmaceutics 2023, 15, 928.
- Baumgarten, P.; Prange, G.; Kamp, M.A.; Monden, D.; Neef, V.; Schwarzer, F.; Dubinski, D.; Dinc, N.; Weber, K.J.; Czabanka, M.; et al. Treatment of Very Elderly Glioblastoma Patients ≥ 75 Years of Age: Whom to Treat. J. Neurooncol. 2023, 165, 509–515.
- Roux, A.; Aboubakr, O.; Elia, A.; Moiraghi, A.; Benevello, C.; Fathallah, H.; Parraga, E.; Oppenheim, C.; Chretien, F.; Dezamis, E.; et al. Carmustine Wafer Implantation for Supratentorial Glioblastomas, IDH-Wildtype in “Extreme” Neurosurgical Conditions. Neurosurg. Rev. 2023, 46, 140.
- Ram, Z.; Kim, C.-Y.; Hottinger, A.F.; Idbaih, A.; Nicholas, G.; Zhu, J.-J. Efficacy and Safety of Tumor Treating Fields (TTFields) in Elderly Patients with Newly Diagnosed Glioblastoma: Subgroup Analysis of the Phase 3 EF-14 Clinical Trial. Front. Oncol. 2021, 11, 671972.
- Malmström, A.; Grønberg, B.H.; Marosi, C.; Stupp, R.; Frappaz, D.; Schultz, H.; Abacioglu, U.; Tavelin, B.; Lhermitte, B.; Hegi, M.E.; et al. Temozolomide versus Standard 6-Week Radiotherapy versus Hypofractionated Radiotherapy in Patients Older than 60 Years with Glioblastoma: The Nordic Randomised, Phase 3 Trial. Lancet Oncol. 2012, 13, 916–926.
- Perry, J.R.; Laperriere, N.; O’Callaghan, C.J.; Brandes, A.A.; Menten, J.; Phillips, C.; Fay, M.; Nishikawa, R.; Cairncross, J.G.; Roa, W.; et al. Short-Course Radiation plus Temozolomide in Elderly Patients with Glioblastoma. N. Engl. J. Med. 2017, 376, 1027–1037.
- Wick, A.; Kessler, T.; Elia, A.E.H.; Winkler, F.; Batchelor, T.T.; Platten, M.; Wick, W. Glioblastoma in Elderly Patients: Solid Conclusions Built on Shifting Sand? Neuro Oncol. 2018, 20, 174–183.
- Sulman, E.P.; Ismaila, N.; Armstrong, T.S.; Tsien, C.; Batchelor, T.T.; Cloughesy, T.; Galanis, E.; Gilbert, M.; Gondi, V.; Lovely, M.; et al. Radiation Therapy for Glioblastoma: American Society of Clinical Oncology Clinical Practice Guideline Endorsement of the American Society for Radiation Oncology Guideline. J. Clin. Oncol. 2017, 35, 361–369.
- Wee, C.W. Radiotherapy for Newly Diagnosed Glioblastoma in the Elderly: What Is the Standard? Brain Tumor Res. Treat. 2022, 10, 12–21.
- Wirsching, H.-G.; Tabatabai, G.; Roelcke, U.; Hottinger, A.F.; Jörger, F.; Schmid, A.; Plasswilm, L.; Schrimpf, D.; Mancao, C.; Capper, D.; et al. Bevacizumab plus Hypofractionated Radiotherapy versus Radiotherapy Alone in Elderly Patients with Glioblastoma: The Randomized, Open-Label, Phase II ARTE Trial. Ann. Oncol. 2018, 29, 1423–1430.
- Theeler, B.J.; Gilbert, M.R. Advances in the Treatment of Newly Diagnosed Glioblastoma. BMC Med. 2015, 13, 293.
- Shi, W.; Scannell Bryan, M.; Gilbert, M.R.; Mehta, M.P.; Blumenthal, D.T.; Brown, P.D.; Valeinis, E.; Hopkins, K.; Souhami, L.; Andrews, D.W.; et al. Investigating the Effect of Reirradiation or Systemic Therapy in Patients With Glioblastoma After Tumor Progression: A Secondary Analysis of NRG Oncology/Radiation Therapy Oncology Group Trial 0525. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 38–44.
- Ringel, F.; Pape, H.; Sabel, M.; Krex, D.; Bock, H.C.; Misch, M.; Weyerbrock, A.; Westermaier, T.; Senft, C.; Schucht, P.; et al. Clinical Benefit from Resection of Recurrent Glioblastomas: Results of a Multicenter Study Including 503 Patients with Recurrent Glioblastomas Undergoing Surgical Resection. Neuro Oncol. 2016, 18, 96–104.
- Weller, M.; Le Rhun, E. How Did Lomustine Become Standard of Care in Recurrent Glioblastoma? Cancer Treat. Rev. 2020, 87, 102029.
- She, L.; Su, L.; Liu, C. Bevacizumab Combined with Re-Irradiation in Recurrent Glioblastoma. Front. Oncol. 2022, 12, 961014.
- Neth, B.J.; Webb, M.J.; Parney, I.F.; Sener, U.T. The Current Status, Challenges, and Future Potential of Therapeutic Vaccination in Glioblastoma. Pharmaceutics 2023, 15, 1134.
- Skaga, E.; Skretteberg, M.A.; Johannesen, T.B.; Brandal, P.; Vik-Mo, E.O.; Helseth, E.; Langmoen, I.A. Real-World Validity of Randomized Controlled Phase III Trials in Newly Diagnosed Glioblastoma: To Whom Do the Results of the Trials Apply? Neurooncol. Adv. 2021, 3, vdab008.
- Brennan, P.M.; Borchert, R.; Coulter, C.; Critchley, G.R.; Hall, B.; Holliman, D.; Phang, I.; Jefferies, S.J.; Keni, S.; Lee, L.; et al. Second Surgery for Progressive Glioblastoma: A Multi-Centre Questionnaire and Cohort-Based Review of Clinical Decision-Making and Patient Outcomes in Current Practice. J. Neurooncol. 2021, 153, 99–107.
- Sharma, M.; Schroeder, J.L.; Elson, P.; Meola, A.; Barnett, G.H.; Vogelbaum, M.A.; Suh, J.H.; Chao, S.T.; Mohammadi, A.M.; Stevens, G.H.J.; et al. Outcomes and Prognostic Stratification of Patients with Recurrent Glioblastoma Treated with Salvage Stereotactic Radiosurgery. J. Neurosurg. 2018, 131, 489–499.
- Suchorska, B.; Weller, M.; Tabatabai, G.; Senft, C.; Hau, P.; Sabel, M.C.; Herrlinger, U.; Ketter, R.; Schlegel, U.; Marosi, C.; et al. Complete Resection of Contrast-Enhancing Tumor Volume Is Associated with Improved Survival in Recurrent Glioblastoma-Results from the DIRECTOR Trial. Neuro Oncol. 2016, 18, 549–556.
- Wann, A.; Tully, P.A.; Barnes, E.H.; Lwin, Z.; Jeffree, R.; Drummond, K.J.; Gan, H.; Khasraw, M. Outcomes after Second Surgery for Recurrent Glioblastoma: A Retrospective Case-Control Study. J. Neurooncol. 2018, 137, 409–415.
- Kamiya-Matsuoka, C.; Gilbert, M.R. Treating Recurrent Glioblastoma: An Update. CNS Oncol. 2015, 4, 91–104.
- Minniti, G.; Niyazi, M.; Alongi, F.; Navarria, P.; Belka, C. Current Status and Recent Advances in Reirradiation of Glioblastoma. Radiat. Oncol. 2021, 16, 36.
- Post, C.C.B.; Kramer, M.C.A.; Smid, E.J.; van der Weide, H.L.; Kleynen, C.E.; Heesters, M.A.A.M.; Verhoeff, J.J.C. Patterns of Re-Irradiation for Recurrent Gliomas and Validation of a Prognostic Score. Radiother. Oncol. 2019, 130, 156–163.
- Christ, S.M.; Youssef, G.; Tanguturi, S.K.; Cagney, D.; Shi, D.; McFaline-Figueroa, J.R.; Chukwueke, U.; Lee, E.Q.; Hertler, C.; Andratschke, N.; et al. Re-Irradiation of Recurrent IDH-Wildtype Glioblastoma in the Bevacizumab and Immunotherapy Era: Target Delineation, Outcomes and Patterns of Recurrence. Clin. Transl. Radiat. Oncol. 2023, 44, 100697.
- Pineda, E.; Domenech, M.; Hernández, A.; Comas, S.; Balaña, C. Recurrent Glioblastoma: Ongoing Clinical Challenges and Future Prospects. Onco Targets Ther. 2023, 16, 71–86.
- Norden, A.D.; Drappatz, J.; Wen, P.Y. Antiangiogenic Therapies for High-Grade Glioma. Nat. Rev. Neurol. 2009, 5, 610–620.
- Hegde, P.S.; Wallin, J.J.; Mancao, C. Predictive Markers of Anti-VEGF and Emerging Role of Angiogenesis Inhibitors as Immunotherapeutics. Semin. Cancer Biol. 2018, 52, 117–124.
- Tamura, R.; Tanaka, T.; Ohara, K.; Miyake, K.; Morimoto, Y.; Yamamoto, Y.; Kanai, R.; Akasaki, Y.; Murayama, Y.; Tamiya, T.; et al. Persistent Restoration to the Immunosupportive Tumor Microenvironment in Glioblastoma by Bevacizumab. Cancer Sci. 2019, 110, 499–508.
- Fu, M.; Zhou, Z.; Huang, X.; Chen, Z.; Zhang, L.; Zhang, J.; Hua, W.; Mao, Y. Use of Bevacizumab in Recurrent Glioblastoma: A Scoping Review and Evidence Map. BMC Cancer 2023, 23, 544.
- Cohen, M.H.; Shen, Y.L.; Keegan, P.; Pazdur, R. FDA Drug Approval Summary: Bevacizumab (Avastin) as Treatment of Recurrent Glioblastoma Multiforme. Oncologist 2009, 14, 1131–1138.
- Balañá, C.; Etxaniz, O.; Bugés, C.; Martínez, A. Approval Denied by the European Medicines Agency (EMA) for Bevacizumab in the Treatment of High-Grade Glioma Recurrence: A Good Idea or a Grave Error? Clin. Transl. Oncol. 2011, 13, 209–210.
- Taal, W.; Oosterkamp, H.M.; Walenkamp, A.M.E.; Dubbink, H.J.; Beerepoot, L.V.; Hanse, M.C.J.; Buter, J.; Honkoop, A.H.; Boerman, D.; Vos, F.Y.F.; et al. Single-Agent Bevacizumab or Lomustine versus a Combination of Bevacizumab plus Lomustine in Patients with Recurrent Glioblastoma (BELOB Trial): A Randomised Controlled Phase 2 Trial. Lancet Oncol. 2014, 15, 943–953.
- Franceschi, E.; Brandes, A.A. The Role of Bevacizumab in Recurrent Glioblastoma: New Insights from Randomized Trials. CNS Oncol. 2015, 4, 117–119.
- Field, K.M.; Simes, J.; Nowak, A.K.; Cher, L.; Wheeler, H.; Hovey, E.J.; Brown, C.S.B.; Barnes, E.H.; Sawkins, K.; Livingstone, A.; et al. Randomized Phase 2 Study of Carboplatin and Bevacizumab in Recurrent Glioblastoma. Neuro Oncol. 2015, 17, 1504–1513.
- Hovey, E.J.; Field, K.M.; Rosenthal, M.A.; Barnes, E.H.; Cher, L.; Nowak, A.K.; Wheeler, H.; Sawkins, K.; Livingstone, A.; Phal, P.; et al. Continuing or Ceasing Bevacizumab beyond Progression in Recurrent Glioblastoma: An Exploratory Randomized Phase II Trial. Neurooncol. Pract. 2017, 4, 171–181.
- Brandes, A.A.; Gil-Gil, M.; Saran, F.; Carpentier, A.F.; Nowak, A.K.; Mason, W.; Zagonel, V.; Dubois, F.; Finocchiaro, G.; Fountzilas, G.; et al. A Randomized Phase II Trial (TAMIGA) Evaluating the Efficacy and Safety of Continuous Bevacizumab Through Multiple Lines of Treatment for Recurrent Glioblastoma. Oncologist 2019, 24, 521–528.
- Vredenburgh, J.J.; Cloughesy, T.; Samant, M.; Prados, M.; Wen, P.Y.; Mikkelsen, T.; Schiff, D.; Abrey, L.E.; Yung, W.K.A.; Paleologos, N.; et al. Corticosteroid Use in Patients with Glioblastoma at First or Second Relapse Treated with Bevacizumab in the BRAIN Study. Oncologist 2010, 15, 1329–1334.
- Dirven, L.; van den Bent, M.J.; Bottomley, A.; van der Meer, N.; van der Holt, B.; Vos, M.J.; Walenkamp, A.M.E.; Beerepoot, L.V.; Hanse, M.C.J.; Reijneveld, J.C.; et al. The Impact of Bevacizumab on Health-Related Quality of Life in Patients Treated for Recurrent Glioblastoma: Results of the Randomised Controlled Phase 2 BELOB Trial. Eur. J. Cancer 2015, 51, 1321–1330.
- Tsien, C.; Pugh, S.; Dicker, A.P.; Raizer, J.J.; Matuszak, M.M.; Lallana, E.; Huang, J.; Algan, O.; Taylor, N.; Portelance, L.; et al. Randomized Phase II Trial of Re-Irradiation and Concurrent Bevacizumab versus Bevacizumab Alone as Treatment for Recurrent Glioblastoma (NRG Oncology/RTOG 1205): Initial Outcomes and RT Plan Quality Report. Int. J. Radiat. Oncol. Biol. Phys. 2019, 105, S78.
- Fleischmann, D.F.; Jenn, J.; Corradini, S.; Ruf, V.; Herms, J.; Forbrig, R.; Unterrainer, M.; Thon, N.; Kreth, F.W.; Belka, C.; et al. Bevacizumab Reduces Toxicity of Reirradiation in Recurrent High-Grade Glioma. Radiother. Oncol. 2019, 138, 99–105.
- Marwah, R.; Xing, D.; Squire, T.; Soon, Y.Y.; Gan, H.K.; Ng, S.P. Reirradiation versus Systemic Therapy versus Combination Therapy for Recurrent High-Grade Glioma: A Systematic Review and Meta-Analysis of Survival and Toxicity. J. Neurooncol. 2023, 164, 505–524.
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963.
- Fallah, J.; Chaudhary, R.T.; Rogers, L.R.; Wei, W.; Brewer, C.J.; Peereboom, D.M.; Ahluwalia, M.S. Clinical Outcomes of the Combination of Bevacizumab and Ttfields in Patients with Recurrent Glioblastoma: Results of a Phase II Clinical Trial. JCO 2020, 38, 2537.
- Lu, Q.R.; Qian, L.; Zhou, X. Convergence of Developmental Origins and Oncogenic Pathways in Malignant Brain Tumors. Wiley Interdiscip. Rev. Dev. Biol. 2019, 8, e342.
- Diplas, B.H.; He, X.; Brosnan-Cashman, J.A.; Liu, H.; Chen, L.H.; Wang, Z.; Moure, C.J.; Killela, P.J.; Loriaux, D.B.; Lipp, E.S.; et al. The Genomic Landscape of TERT Promoter Wildtype-IDH Wildtype Glioblastoma. Nat. Commun. 2018, 9, 2087.
- Yeo, A.T.; Shah, R.; Aliazis, K.; Pal, R.; Xu, T.; Zhang, P.; Rawal, S.; Rose, C.M.; Varn, F.S.; Appleman, V.A.; et al. Driver Mutations Dictate the Immunologic Landscape and Response to Checkpoint Immunotherapy of Glioblastoma. Cancer Immunol. Res. 2023, 11, 629–645.
- Scherm, A.; Ippen, F.M.; Hau, P.; Baurecht, H.; Wick, W.; Gempt, J.; Knüttel, H.; Leitzmann, M.F.; Seliger, C. Targeted Therapies in Patients with Newly Diagnosed Glioblastoma—A Systematic Meta-Analysis of Randomized Clinical Trials. Int. J. Cancer 2023, 152, 2373–2382.
- Zhang, A.B.; Mozaffari, K.; Aguirre, B.; Li, V.; Kubba, R.; Desai, N.C.; Wei, D.; Yang, I.; Wadehra, M. Exploring the Past, Present, and Future of Anti-Angiogenic Therapy in Glioblastoma. Cancers 2023, 15, 830.
- Andersen, R.S.; Anand, A.; Harwood, D.S.L.; Kristensen, B.W. Tumor-Associated Microglia and Macrophages in the Glioblastoma Microenvironment and Their Implications for Therapy. Cancers 2021, 13, 4255.
- Wang, G.; Zhong, K.; Wang, Z.; Zhang, Z.; Tang, X.; Tong, A.; Zhou, L. Tumor-Associated Microglia and Macrophages in Glioblastoma: From Basic Insights to Therapeutic Opportunities. Front. Immunol. 2022, 13, 964898.
- Fanelli, G.N.; Grassini, D.; Ortenzi, V.; Pasqualetti, F.; Montemurro, N.; Perrini, P.; Naccarato, A.G.; Scatena, C. Decipher the Glioblastoma Microenvironment: The First Milestone for New Groundbreaking Therapeutic Strategies. Genes 2021, 12, 445.
- Chistiakov, D.A.; Chekhonin, I.V.; Chekhonin, V.P. The EGFR Variant III Mutant as a Target for Immunotherapy of Glioblastoma Multiforme. Eur. J. Pharmacol. 2017, 810, 70–82.
- Choi, B.D.; Maus, M.V.; June, C.H.; Sampson, J.H. Immunotherapy for Glioblastoma: Adoptive T-Cell Strategies. Clin. Cancer Res. 2019, 25, 2042–2048.
- Nam, L.; Coll, C.; Erthal, L.C.S.; de la Torre, C.; Serrano, D.; Martínez-Máñez, R.; Santos-Martínez, M.J.; Ruiz-Hernández, E. Drug Delivery Nanosystems for the Localized Treatment of Glioblastoma Multiforme. Materials 2018, 11, 779.
- Di Filippo, L.D.; Duarte, J.L.; Luiz, M.T.; de Araújo, J.T.C.; Chorilli, M. Drug Delivery Nanosystems in Glioblastoma Multiforme Treatment: Current State of the Art. Curr. Neuropharmacol. 2021, 19, 787–812.
- Hsu, J.-F.; Chu, S.-M.; Liao, C.-C.; Wang, C.-J.; Wang, Y.-S.; Lai, M.-Y.; Wang, H.-C.; Huang, H.-R.; Tsai, M.-H. Nanotechnology and Nanocarrier-Based Drug Delivery as the Potential Therapeutic Strategy for Glioblastoma Multiforme: An Update. Cancers 2021, 13, 195.
- Marei, H.E. Multimodal Targeting of Glioma with Functionalized Nanoparticles. Cancer Cell Int. 2022, 22, 265.
- Wadajkar, A.S.; Dancy, J.G.; Hersh, D.S.; Anastasiadis, P.; Tran, N.L.; Woodworth, G.F.; Winkles, J.A.; Kim, A.J. Tumor-Targeted Nanotherapeutics: Overcoming Treatment Barriers for Glioblastoma. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol 2017, 9.
- Alphandéry, E. Nano-Therapies for Glioblastoma Treatment. Cancers 2020, 12, 242.
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and Challenges towards Targeted Delivery of Cancer Therapeutics. Nat. Commun. 2018, 9, 1410.
- Qiu, Z.; Yu, Z.; Xu, T.; Wang, L.; Meng, N.; Jin, H.; Xu, B. Novel Nano-Drug Delivery System for Brain Tumor Treatment. Cells 2022, 11, 3761.
- Wei, D.; Zhang, N.; Qu, S.; Wang, H.; Li, J. Advances in Nanotechnology for the Treatment of GBM. Front. Neurosci. 2023, 17.
- Tang, L.; Feng, Y.; Gao, S.; Mu, Q.; Liu, C. Nanotherapeutics Overcoming the Blood-Brain Barrier for Glioblastoma Treatment. Front. Pharmacol. 2021, 12.
- Aravind, A.; Veeranarayanan, S.; Poulose, A.C.; Nair, R.; Nagaoka, Y.; Yoshida, Y.; Maekawa, T.; Kumar, D.S. Aptamer-Functionalized Silica Nanoparticles for Targeted Cancer Therapy. BioNanoSci. 2012, 2, 1–8.
- Kuo, Y.-C.; Lee, C.-H. Dual Targeting of Solid Lipid Nanoparticles Grafted with 83-14 MAb and Anti-EGF Receptor for Malignant Brain Tumor Therapy. Life Sci. 2016, 146, 222–231.
- Song, P.; Zhao, X.; Xiao, S. Application Prospect of Peptide-Modified Nano Targeting Drug Delivery System Combined with PD-1/PD-L1 Based Immune Checkpoint Blockade in Glioblastoma. Int. J. Pharm. 2020, 589, 119865.
- Fang, C.; Wang, K.; Stephen, Z.R.; Mu, Q.; Kievit, F.M.; Chiu, D.T.; Press, O.W.; Zhang, M. Temozolomide Nanoparticles for Targeted Glioblastoma Therapy. ACS Appl. Mater. Interfaces 2015, 7, 6674–6682.
- Gonawala, S.; Ali, M.M. Application of Dendrimer-Based Nanoparticles in Glioma Imaging. J. Nanomed. Nanotechnol. 2017, 8, 444.
- Ruiz-Molina, D.; Mao, X.; Alfonso-Triguero, P.; Lorenzo, J.; Bruna, J.; Yuste, V.J.; Candiota, A.P.; Novio, F. Advances in Preclinical/Clinical Glioblastoma Treatment: Can Nanoparticles Be of Help? Cancers 2022, 14, 4960.
- Melnick, K.; Shin, D.; Dastmalchi, F.; Kabeer, Z.; Rahman, M.; Tran, D.; Ghiaseddin, A. Role of Laser Interstitial Thermal Therapy in the Management of Primary and Metastatic Brain Tumors. Curr. Treat. Options Oncol. 2021, 22, 108.
- Lerner, E.C.; Edwards, R.M.; Wilkinson, D.S.; Fecci, P.E. Laser Ablation: Heating up the Anti-Tumor Response in the Intracranial Compartment. Adv. Drug Deliv. Rev. 2022, 185, 114311.
- Traylor, J.I.; Patel, R.; Muir, M.; de Almeida Bastos, D.C.; Ravikumar, V.; Kamiya-Matsuoka, C.; Rao, G.; Thomas, J.G.; Kew, Y.; Prabhu, S.S. Laser Interstitial Thermal Therapy for Glioblastoma: A Single-Center Experience. World Neurosurg. 2021, 149, e244–e252.
- Alkazemi, M.; Lo, Y.T.; Hussein, H.; Mammi, M.; Saleh, S.; Araujo-Lama, L.; Mommsen, S.; Pisano, A.; Lamba, N.; Bunevicius, A.; et al. Laser Interstitial Thermal Therapy for the Treatment of Primary and Metastatic Brain Tumors: A Systematic Review and Meta-Analysis. World Neurosurg. 2023, 171, e654–e671.
- de Groot, J.F.; Kim, A.H.; Prabhu, S.; Rao, G.; Laxton, A.W.; Fecci, P.E.; O’Brien, B.J.; Sloan, A.; Chiang, V.; Tatter, S.B.; et al. Efficacy of Laser Interstitial Thermal Therapy (LITT) for Newly Diagnosed and Recurrent IDH Wild-Type Glioblastoma. Neurooncol. Adv. 2022, 4, vdac040.
- Roberts, J.W.; Powlovich, L.; Sheybani, N.; LeBlang, S. Focused Ultrasound for the Treatment of Glioblastoma. J. Neurooncol. 2022, 157, 237–247.
- Johansen, P.M.; Hansen, P.Y.; Mohamed, A.A.; Girshfeld, S.J.; Feldmann, M.; Lucke-Wold, B. Focused Ultrasound for Treatment of Peripheral Brain Tumors. Explor. Drug Sci. 2023, 1, 107–125.
- Mungur, R.; Zheng, J.; Wang, B.; Chen, X.; Zhan, R.; Tong, Y. Low-Intensity Focused Ultrasound Technique in Glioblastoma Multiforme Treatment. Front. Oncol. 2022, 12.
- Kong, C.; Chang, W.S. Preclinical Research on Focused Ultrasound-Mediated Blood–Brain Barrier Opening for Neurological Disorders: A Review. Neurol. Int. 2023, 15, 285–300.
- Elhelf, I.A.S.; Albahar, H.; Shah, U.; Oto, A.; Cressman, E.; Almekkawy, M. High Intensity Focused Ultrasound: The Fundamentals, Clinical Applications and Research Trends. Diagn. Interv. Imaging 2018, 99, 349–359.
- Quadri, S.A.; Waqas, M.; Khan, I.; Khan, M.A.; Suriya, S.S.; Farooqui, M.; Fiani, B. High-Intensity Focused Ultrasound: Past, Present, and Future in Neurosurgery. Neurosurg. Focus 2018, 44, E16.
- Coluccia, D.; Fandino, J.; Schwyzer, L.; O’Gorman, R.; Remonda, L.; Anon, J.; Martin, E.; Werner, B. First Noninvasive Thermal Ablation of a Brain Tumor with MR-Guided Focused Ultrasound. J. Ther. Ultrasound 2014, 2, 17.
- MacDonell, J.; Patel, N.; Rubino, S.; Ghoshal, G.; Fischer, G.; Burdette, E.C.; Hwang, R.; Pilitsis, J.G. Magnetic Resonance-Guided Interstitial High-Intensity Focused Ultrasound for Brain Tumor Ablation. Neurosurg. Focus 2018, 44, E11.
- Hersh, A.M.; Bhimreddy, M.; Weber-Levine, C.; Jiang, K.; Alomari, S.; Theodore, N.; Manbachi, A.; Tyler, B.M. Applications of Focused Ultrasound for the Treatment of Glioblastoma: A New Frontier. Cancers 2022, 14, 4920.
- Arsiwala, T.A.; Sprowls, S.A.; Blethen, K.E.; Adkins, C.E.; Saralkar, P.A.; Fladeland, R.A.; Pentz, W.; Gabriele, A.; Kielkowski, B.; Mehta, R.I.; et al. Ultrasound-Mediated Disruption of the Blood Tumor Barrier for Improved Therapeutic Delivery. Neoplasia 2021, 23, 676–691.
- Hersh, D.S.; Wadajkar, A.S.; Roberts, N.; Perez, J.G.; Connolly, N.P.; Frenkel, V.; Winkles, J.A.; Woodworth, G.F.; Kim, A.J. Evolving Drug Delivery Strategies to Overcome the Blood Brain Barrier. Curr. Pharm. Des. 2016, 22, 1177–1193.
- Liu, H.-L.; Hua, M.-Y.; Chen, P.-Y.; Chu, P.-C.; Pan, C.-H.; Yang, H.-W.; Huang, C.-Y.; Wang, J.-J.; Yen, T.-C.; Wei, K.-C. Blood-Brain Barrier Disruption with Focused Ultrasound Enhances Delivery of Chemotherapeutic Drugs for Glioblastoma Treatment. Radiology 2010, 255, 415–425.
- Wei, H.-J.; Upadhyayula, P.S.; Pouliopoulos, A.N.; Englander, Z.K.; Zhang, X.; Jan, C.-I.; Guo, J.; Mela, A.; Zhang, Z.; Wang, T.J.C.; et al. Focused Ultrasound-Mediated Blood-Brain Barrier Opening Increases Delivery and Efficacy of Etoposide for Glioblastoma Treatment. Int. J. Radiat. Oncol. Biol. Phys. 2021, 110, 539–550.
- Liu, H.-L.; Huang, C.-Y.; Chen, J.-Y.; Wang, H.-Y.J.; Chen, P.-Y.; Wei, K.-C. Pharmacodynamic and Therapeutic Investigation of Focused Ultrasound-Induced Blood-Brain Barrier Opening for Enhanced Temozolomide Delivery in Glioma Treatment. PLoS ONE 2014, 9, e114311.
- Noroozian, Z.; Xhima, K.; Huang, Y.; Kaspar, B.K.; Kügler, S.; Hynynen, K.; Aubert, I. MRI-Guided Focused Ultrasound for Targeted Delivery of rAAV to the Brain. Methods Mol. Biol. 2019, 1950, 177–197.
- Alkins, R.; Burgess, A.; Ganguly, M.; Francia, G.; Kerbel, R.; Wels, W.S.; Hynynen, K. Focused Ultrasound Delivers Targeted Immune Cells to Metastatic Brain Tumors. Cancer Res. 2013, 73, 1892–1899.
- Nance, E.; Timbie, K.; Miller, G.W.; Song, J.; Louttit, C.; Klibanov, A.L.; Shih, T.-Y.; Swaminathan, G.; Tamargo, R.J.; Woodworth, G.F.; et al. Non-Invasive Delivery of Stealth, Brain-Penetrating Nanoparticles across the Blood-Brain Barrier Using MRI-Guided Focused Ultrasound. J. Control Release 2014, 189, 123–132.
- Coluccia, D.; Figueiredo, C.A.; Wu, M.Y.; Riemenschneider, A.N.; Diaz, R.; Luck, A.; Smith, C.; Das, S.; Ackerley, C.; O’Reilly, M.; et al. Enhancing Glioblastoma Treatment Using Cisplatin-Gold-Nanoparticle Conjugates and Targeted Delivery with Magnetic Resonance-Guided Focused Ultrasound. Nanomedicine 2018, 14, 1137–1148.
- Janjua, T.I.; Cao, Y.; Ahmed-Cox, A.; Raza, A.; Moniruzzaman, M.; Akhter, D.T.; Fletcher, N.L.; Kavallaris, M.; Thurecht, K.J.; Popat, A. Efficient Delivery of Temozolomide Using Ultrasmall Large-Pore Silica Nanoparticles for Glioblastoma. J. Control Release 2023, 357, 161–174.
- Liu, H.-L.; Hsieh, H.-Y.; Lu, L.-A.; Kang, C.-W.; Wu, M.-F.; Lin, C.-Y. Low-Pressure Pulsed Focused Ultrasound with Microbubbles Promotes an Anticancer Immunological Response. J. Transl. Med. 2012, 10, 221.
- Unga, J.; Hashida, M. Ultrasound Induced Cancer Immunotherapy. Adv. Drug Deliv. Rev. 2014, 72, 144–153.
- Liu, S.; Zhang, Y.; Liu, Y.; Wang, W.; Gao, S.; Yuan, W.; Sun, Z.; Liu, L.; Wang, C. Ultrasound-Targeted Microbubble Destruction Remodels Tumour Microenvironment to Improve Immunotherapeutic Effect. Br. J. Cancer 2023, 128, 715–725.
- Cohen-Inbar, O.; Xu, Z.; Sheehan, J.P. Focused Ultrasound-Aided Immunomodulation in Glioblastoma Multiforme: A Therapeutic Concept. J. Ther. Ultrasound 2016, 4, 2.
- Chen, K.-T.; Chai, W.-Y.; Lin, Y.-J.; Lin, C.-J.; Chen, P.-Y.; Tsai, H.-C.; Huang, C.-Y.; Kuo, J.S.; Liu, H.-L.; Wei, K.-C. Neuronavigation-Guided Focused Ultrasound for Transcranial Blood-Brain Barrier Opening and Immunostimulation in Brain Tumors. Sci. Adv. 2021, 7, eabd0772.
- Bathini, P.; Sun, T.; Schenk, M.; Schilling, S.; McDannold, N.J.; Lemere, C.A. Acute Effects of Focused Ultrasound-Induced Blood-Brain Barrier Opening on Anti-Pyroglu3 Abeta Antibody Delivery and Immune Responses. Biomolecules 2022, 12, 951.
- Idbaih, A.; Canney, M.; Belin, L.; Desseaux, C.; Vignot, A.; Bouchoux, G.; Asquier, N.; Law-Ye, B.; Leclercq, D.; Bissery, A.; et al. Safety and Feasibility of Repeated and Transient Blood-Brain Barrier Disruption by Pulsed Ultrasound in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2019, 25, 3793–3801.
- Park, S.H.; Kim, M.J.; Jung, H.H.; Chang, W.S.; Choi, H.S.; Rachmilevitch, I.; Zadicario, E.; Chang, J.W. One-Year Outcome of Multiple Blood-Brain Barrier Disruptions With Temozolomide for the Treatment of Glioblastoma. Front. Oncol. 2020, 10, 1663.
- Sonabend, A.M.; Gould, A.; Amidei, C.; Ward, R.; Schmidt, K.A.; Zhang, D.Y.; Gomez, C.; Bebawy, J.F.; Liu, B.P.; Bouchoux, G.; et al. Repeated Blood-Brain Barrier Opening with an Implantable Ultrasound Device for Delivery of Albumin-Bound Paclitaxel in Patients with Recurrent Glioblastoma: A Phase 1 Trial. Lancet Oncol. 2023, 24, 509–522.
- Kim, E.; Van Reet, J.; Kim, H.-C.; Kowsari, K.; Yoo, S.-S. High Incidence of Intracerebral Hemorrhaging Associated with the Application of Low-Intensity Focused Ultrasound Following Acute Cerebrovascular Injury by Intracortical Injection. Pharmaceutics 2022, 14, 2120.
- McHale, A.P.; Callan, J.F.; Nomikou, N.; Fowley, C.; Callan, B. Sonodynamic Therapy: Concept, Mechanism and Application to Cancer Treatment. Adv. Exp. Med. Biol. 2016, 880, 429–450.
- Cramer, S.W.; Chen, C.C. Photodynamic Therapy for the Treatment of Glioblastoma. Front. Surg. 2020, 6.
- Zheng, Y.; Ye, J.; Li, Z.; Chen, H.; Gao, Y. Recent Progress in Sono-Photodynamic Cancer Therapy: From Developed New Sensitizers to Nanotechnology-Based Efficacy-Enhancing Strategies. Acta Pharm. Sin. B 2021, 11, 2197–2219.
- Bhanja, D.; Wilding, H.; Baroz, A.; Trifoi, M.; Shenoy, G.; Slagle-Webb, B.; Hayes, D.; Soudagar, Y.; Connor, J.; Mansouri, A. Photodynamic Therapy for Glioblastoma: Illuminating the Path toward Clinical Applicability. Cancers 2023, 15, 3427.
- Song, D.; Yue, W.; Li, Z.; Li, J.; Zhao, J.; Zhang, N. Study of the Mechanism of Sonodynamic Therapy in a Rat Glioma Model. Onco Targets Ther. 2014, 7, 1801–1810.
- Mroz, P.; Hashmi, J.T.; Huang, Y.-Y.; Lange, N.; Hamblin, M.R. Stimulation of Anti-Tumor Immunity by Photodynamic Therapy. Expert Rev. Clin. Immunol. 2011, 7, 75–91.
- Falk-Mahapatra, R.; Gollnick, S.O. Photodynamic Therapy and Immunity: An Update. Photochem. Photobiol. 2020, 96, 550–559.
- Bunevicius, A.; Pikis, S.; Padilla, F.; Prada, F.; Sheehan, J. Sonodynamic Therapy for Gliomas. J. Neurooncol. 2022, 156, 1–10.
- Mahmoudi, K.; Garvey, K.; Bouras, A.; Cramer, G.; Stepp, H.; Jesu Raj, J.; Bozec, D.; Busch, T.; Hadjipanayis, C. 5-Aminolevulinic Acid Photodynamic Therapy for the Treatment of High-Grade Gliomas. J. Neurooncol. 2019, 141, 595–607.
- Hsia, T.; Small, J.L.; Yekula, A.; Batool, S.M.; Escobedo, A.K.; Ekanayake, E.; You, D.G.; Lee, H.; Carter, B.S.; Balaj, L. Systematic Review of Photodynamic Therapy in Gliomas. Cancers 2023, 15, 3918.
- Wang, W.; Tabu, K.; Hagiya, Y.; Sugiyama, Y.; Kokubu, Y.; Murota, Y.; Ogura, S.-I.; Taga, T. Enhancement of 5-Aminolevulinic Acid-Based Fluorescence Detection of Side Population-Defined Glioma Stem Cells by Iron Chelation. Sci. Rep. 2017, 7, 42070.
- Müller, P.; Abdel Gaber, S.A.; Zimmermann, W.; Wittig, R.; Stepp, H. ABCG2 Influence on the Efficiency of Photodynamic Therapy in Glioblastoma Cells. J. Photochem. Photobiol. B 2020, 210, 111963.
- Leroy, H.-A.; Guérin, L.; Lecomte, F.; Baert, G.; Vignion, A.-S.; Mordon, S.; Reyns, N. Is Interstitial Photodynamic Therapy for Brain Tumors Ready for Clinical Practice? A Systematic Review. Photodiagnosis Photodyn. Ther. 2021, 36, 102492.
- Schwartz, C.; Rühm, A.; Tonn, J.-C.; Kreth, S.; Kreth, F.-W. SURG-25INTERSTITIAL PHOTODYNAMIC THERAPY OF DE-NOVO GLIOBLASTOMA MULTIFORME WHO IV. Neuro Oncol. 2015, 17, v219–v220.
- Nitta, M.; Muragaki, Y.; Maruyama, T.; Iseki, H.; Komori, T.; Ikuta, S.; Saito, T.; Yasuda, T.; Hosono, J.; Okamoto, S.; et al. Role of Photodynamic Therapy Using Talaporfin Sodium and a Semiconductor Laser in Patients with Newly Diagnosed Glioblastoma. J. Neurosurg. 2018, 131, 1361–1368.
- Lietke, S.; Schmutzer, M.; Schwartz, C.; Weller, J.; Siller, S.; Aumiller, M.; Heckl, C.; Forbrig, R.; Niyazi, M.; Egensperger, R.; et al. Interstitial Photodynamic Therapy Using 5-ALA for Malignant Glioma Recurrences. Cancers 2021, 13, 1767.
- Foglar, M.; Aumiller, M.; Bochmann, K.; Buchner, A.; El Fahim, M.; Quach, S.; Sroka, R.; Stepp, H.; Thon, N.; Forbrig, R.; et al. Interstitial Photodynamic Therapy of Glioblastomas: A Long-Term Follow-up Analysis of Survival and Volumetric MRI Data. Cancers 2023, 15, 2603.
- Henderson, T.A.; Morries, L.D. Near-Infrared Photonic Energy Penetration: Can Infrared Phototherapy Effectively Reach the Human Brain? Neuropsychiatr. Dis. Treat. 2015, 11, 2191–2208.
- Lee, S.Y.; Lee, R.; Kim, E.; Lee, S.; Park, Y.I. Near-Infrared Light-Triggered Photodynamic Therapy and Apoptosis Using Upconversion Nanoparticles With Dual Photosensitizers. Front. Bioeng. Biotechnol. 2020, 8, 275.
- Burley, T.A.; Mączyńska, J.; Shah, A.; Szopa, W.; Harrington, K.J.; Boult, J.K.R.; Mrozek-Wilczkiewicz, A.; Vinci, M.; Bamber, J.C.; Kaspera, W.; et al. Near-Infrared Photoimmunotherapy Targeting EGFR-Shedding New Light on Glioblastoma Treatment. Int. J. Cancer 2018, 142, 2363–2374.
- Mączyńska, J.; Raes, F.; Da Pieve, C.; Turnock, S.; Boult, J.K.R.; Hoebart, J.; Niedbala, M.; Robinson, S.P.; Harrington, K.J.; Kaspera, W.; et al. Triggering Anti-GBM Immune Response with EGFR-Mediated Photoimmunotherapy. BMC Med. 2022, 20, 16.
- Mohiuddin, T.M.; Zhang, C.; Sheng, W.; Al-Rawe, M.; Zeppernick, F.; Meinhold-Heerlein, I.; Hussain, A.F. Near Infrared Photoimmunotherapy: A Review of Recent Progress and Their Target Molecules for Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 2655.
- Zhang, Y.; He, L.; Wu, J.; Wang, K.; Wang, J.; Dai, W.; Yuan, A.; Wu, J.; Hu, Y. Switchable PDT for Reducing Skin Photosensitization by a NIR Dye Inducing Self-Assembled and Photo-Disassembled Nanoparticles. Biomaterials 2016, 107, 23–32.
- Kang, R.H.; Kim, Y.; Um, H.J.; Kim, J.; Bang, E.-K.; Yeo, S.G.; Kim, D. Glioblastoma Homing Photodynamic Therapy Based on Multifunctionalized Porous Silicon Nanoparticles. ACS Appl. Nano Mater. 2022, 5, 5387–5397.
- Pellosi, D.S.; Paula, L.B.; de Melo, M.T.; Tedesco, A.C. Targeted and Synergic Glioblastoma Treatment: Multifunctional Nanoparticles Delivering Verteporfin as Adjuvant Therapy for Temozolomide Chemotherapy. Mol. Pharm. 2019, 16, 1009–1024.
- Ihata, T.; Nonoguchi, N.; Fujishiro, T.; Omura, N.; Kawabata, S.; Kajimoto, Y.; Wanibuchi, M. The Effect of Hypoxia on Photodynamic Therapy with 5-Aminolevulinic Acid in Malignant Gliomas. Photodiagnosis Photodyn. Ther. 2022, 40, 103056.
- Ma, S.; Wang, F.; Dong, J.; Wang, N.; Tao, S.; Du, J.; Hu, S. Inhibition of Hypoxia-Inducible Factor 1 by Acriflavine Renders Glioblastoma Sensitive for Photodynamic Therapy. J. Photochem. Photobiol. B 2022, 234, 112537.
- Caverzán, M.D.; Oliveda, P.M.; Beaugé, L.; Palacios, R.E.; Chesta, C.A.; Ibarra, L.E. Metronomic Photodynamic Therapy with Conjugated Polymer Nanoparticles in Glioblastoma Tumor Microenvironment. Cells 2023, 12, 1541.
- Omura, N.; Nonoguchi, N.; Fujishiro, T.; Park, Y.; Ikeda, N.; Kajimoto, Y.; Hosomi, R.; Yagi, R.; Hiramatsu, R.; Furuse, M.; et al. Ablation Efficacy of 5-Aminolevulinic Acid-Mediated Photodynamic Therapy on Human Glioma Stem Cells. Photodiagnosis Photodyn. Ther. 2023, 41, 103119.
- Borah, B.M.; Cacaccio, J.; Durrani, F.A.; Bshara, W.; Turowski, S.G.; Spernyak, J.A.; Pandey, R.K. Sonodynamic Therapy in Combination with Photodynamic Therapy Shows Enhanced Long-Term Cure of Brain Tumor. Sci. Rep. 2020, 10, 21791.
- Park, J.; Kong, C.; Shin, J.; Park, J.Y.; Na, Y.C.; Han, S.H.; Chang, J.W.; Song, S.H.; Chang, W.S. Combined Effects of Focused Ultrasound and Photodynamic Treatment for Malignant Brain Tumors Using C6 Glioma Rat Model. Yonsei Med. J. 2023, 64, 233–242.
- Ohmura, T.; Fukushima, T.; Shibaguchi, H.; Yoshizawa, S.; Inoue, T.; Kuroki, M.; Sasaki, K.; Umemura, S.-I. Sonodynamic Therapy with 5-Aminolevulinic Acid and Focused Ultrasound for Deep-Seated Intracranial Glioma in Rat. Anticancer. Res. 2011, 31, 2527–2533.
- Suehiro, S.; Ohnishi, T.; Yamashita, D.; Kohno, S.; Inoue, A.; Nishikawa, M.; Ohue, S.; Tanaka, J.; Kunieda, T. Enhancement of Antitumor Activity by Using 5-ALA-Mediated Sonodynamic Therapy to Induce Apoptosis in Malignant Gliomas: Significance of High-Intensity Focused Ultrasound on 5-ALA-SDT in a Mouse Glioma Model. J. Neurosurg. 2018, 129, 1416–1428.
- Wu, S.-K.; Santos, M.A.; Marcus, S.L.; Hynynen, K. MR-Guided Focused Ultrasound Facilitates Sonodynamic Therapy with 5-Aminolevulinic Acid in a Rat Glioma Model. Sci. Rep. 2019, 9, 10465.
- Raspagliesi, L.; D’Ammando, A.; Gionso, M.; Sheybani, N.D.; Lopes, M.-B.; Moore, D.; Allen, S.; Gatesman, J.; Porto, E.; Timbie, K.; et al. Intracranial Sonodynamic Therapy With 5-Aminolevulinic Acid and Sodium Fluorescein: Safety Study in a Porcine Model. Front. Oncol. 2021, 11, 679989.
- Bonosi, L.; Marino, S.; Benigno, U.E.; Musso, S.; Buscemi, F.; Giardina, K.; Gerardi, R.; Brunasso, L.; Costanzo, R.; Iacopino, D.G.; et al. Sonodynamic Therapy and Magnetic Resonance-Guided Focused Ultrasound: New Therapeutic Strategy in Glioblastoma. J. Neurooncol. 2023, 163, 219–238.
- Mumtaz, S.; Rana, J.N.; Choi, E.H.; Han, I. Microwave Radiation and the Brain: Mechanisms, Current Status, and Future Prospects. Int. J. Mol. Sci. 2022, 23, 9288.
- Chandra, R.; Zhou, H.; Balasingham, I.; Narayanan, R.M. On the Opportunities and Challenges in Microwave Medical Sensing and Imaging. IEEE Trans. Biomed. Eng. 2015, 62, 1667–1682.
- Ryan, T.P.; Brace, C.L. Interstitial Microwave Treatment for Cancer: Historical Basis and Current Techniques in Antenna Design and Performance. Int. J. Hyperth. 2017, 33, 3–14.
- Izzo, F.; Granata, V.; Grassi, R.; Fusco, R.; Palaia, R.; Delrio, P.; Carrafiello, G.; Azoulay, D.; Petrillo, A.; Curley, S.A. Radiofrequency Ablation and Microwave Ablation in Liver Tumors: An Update. Oncologist 2019, 24, e990–e1005.
- Hu, C.; Zuo, H.; Li, Y. Effects of Radiofrequency Electromagnetic Radiation on Neurotransmitters in the Brain. Front. Public Health 2021, 9.
- Rana, J.N.; Mumtaz, S.; Choi, E.H.; Han, I. ROS Production in Response to High-Power Microwave Pulses Induces P53 Activation and DNA Damage in Brain Cells: Radiosensitivity and Biological Dosimetry Evaluation. Front. Cell Dev. Biol. 2023, 11.
- Li, W.; Zhang, S.; Xing, D.; Qin, H. Pulsed Microwave-Induced Thermoacoustic Shockwave for Precise Glioblastoma Therapy with the Skin and Skull Intact. Small 2022, 18, e2201342.
- Jenkins, E.P.W.; Finch, A.; Gerigk, M.; Triantis, I.F.; Watts, C.; Malliaras, G.G. Electrotherapies for Glioblastoma. Adv. Sci. 2021, 8, 2100978.
- Lorenzo, M.F.; Arena, C.B.; Davalos, R.V. Maximizing Local Access to Therapeutic Deliveries in Glioblastoma. Part III: Irreversible Electroporation and High-Frequency Irreversible Electroporation for the Eradication of Glioblastoma. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017; ISBN 978-0-9944381-2-6.
- Schoenbach, K.H.; Beebe, S.J.; Buescher, E.S. Intracellular Effect of Ultrashort Electrical Pulses. Bioelectromagnetics 2001, 22, 440–448.
- Vernier, P.T.; Sun, Y.; Marcu, L.; Craft, C.M.; Gundersen, M.A. Nanoelectropulse-Induced Phosphatidylserine Translocation. Biophys. J. 2004, 86, 4040–4048.
- Pakhomov, A.G.; Shevin, R.; White, J.A.; Kolb, J.F.; Pakhomova, O.N.; Joshi, R.P.; Schoenbach, K.H. Membrane Permeabilization and Cell Damage by Ultrashort Electric Field Shocks. Arch. Biochem. Biophys. 2007, 465, 109–118.
- Pakhomov, A.G.; Bowman, A.M.; Ibey, B.L.; Andre, F.M.; Pakhomova, O.N.; Schoenbach, K.H. Lipid Nanopores Can Form a Stable, Ion Channel-like Conduction Pathway in Cell Membrane. Biochem. Biophys. Res. Commun. 2009, 385, 181–186.
- Kotnik, T.; Rems, L.; Tarek, M.; Miklavčič, D. Membrane Electroporation and Electropermeabilization: Mechanisms and Models. Annu. Rev. Biophys. 2019, 48, 63–91.
- Vernier, P.T.; Li, A.; Marcu, L.; Craft, C.M.; Gundersen, M.A. Ultrashort Pulsed Electric Fields Induce Membrane Phospholipid Translocation and Caspase Activation: Differential Sensitivities of Jurkat T Lymphoblasts and Rat Glioma C6 Cells. IEEE Trans. Dielectr. Electr. Insul. 2003, 10, 795–809.
- White, J.A.; Blackmore, P.F.; Schoenbach, K.H.; Beebe, S.J. Stimulation of Capacitative Calcium Entry in HL-60 Cells by Nanosecond Pulsed Electric Fields *. J. Biol. Chem. 2004, 279, 22964–22972.
- Ren, W.; Beebe, S.J. An Apoptosis Targeted Stimulus with Nanosecond Pulsed Electric Fields (nsPEFs) in E4 Squamous Cell Carcinoma. Apoptosis 2011, 16, 382–393.
- Stacey, M.; Stickley, J.; Fox, P.; Statler, V.; Schoenbach, K.; Beebe, S.J.; Buescher, S. Differential Effects in Cells Exposed to Ultra-Short, High Intensity Electric Fields: Cell Survival, DNA Damage, and Cell Cycle Analysis. Mutat. Res. /Genet. Toxicol. Environ. Mutagen. 2003, 542, 65–75.
- Nuccitelli, R. Application of Pulsed Electric Fields to Cancer Therapy. Bioelectricity 2019, 1, 30–34.
- Garcia, P.A.; Pancotto, T.; Rossmeisl, J.H.; Henao-Guerrero, N.; Gustafson, N.R.; Daniel, G.B.; Robertson, J.L.; Ellis, T.L.; Davalos, R.V. Non-Thermal Irreversible Electroporation (N-TIRE) and Adjuvant Fractionated Radiotherapeutic Multimodal Therapy for Intracranial Malignant Glioma in a Canine Patient. Technol. Cancer Res. Treat. 2011, 10, 73–83.
- Herranz, C.; Fernández, F.; Martín-Ibáñez, R.; Blasco, E.; Crespo, E.; De la Fuente, C.; Añor, S.; Rabanal, R.M.; Canals, J.M.; Pumarola, M. Spontaneously Arising Canine Glioma as a Potential Model for Human Glioma. J. Comp. Pathol. 2016, 154, 169–179.
- Rossmeisl, J.H.; Garcia, P.A.; Pancotto, T.E.; Robertson, J.L.; Henao-Guerrero, N.; Neal, R.E.; Ellis, T.L.; Davalos, R.V. Safety and Feasibility of the NanoKnife System for Irreversible Electroporation Ablative Treatment of Canine Spontaneous Intracranial Gliomas. J. Neurosurg. 2015, 123, 1008–1025.
- Lefevre, M.C.; Dijk, G.; Kaszas, A.; Baca, M.; Moreau, D.; O’Connor, R.P. Integrating Flexible Electronics for Pulsed Electric Field Delivery in a Vascularized 3D Glioblastoma Model. npj Flex. Electron. 2021, 5, 1–9.
- Sano, M.B.; Arena, C.B.; Bittleman, K.R.; DeWitt, M.R.; Cho, H.J.; Szot, C.S.; Saur, D.; Cissell, J.M.; Robertson, J.; Lee, Y.W.; et al. Bursts of Bipolar Microsecond Pulses Inhibit Tumor Growth. Sci. Rep. 2015, 5, 14999.
- Sweeney, D.C.; Reberšek, M.; Dermol, J.; Rems, L.; Miklavčič, D.; Davalos, R.V. Quantification of Cell Membrane Permeability Induced by Monopolar and High-Frequency Bipolar Bursts of Electrical Pulses. Biochim. Biophys. Acta (BBA) Biomembr. 2016, 1858, 2689–2698.
- Latouche, E.L.; Arena, C.B.; Ivey, J.W.; Garcia, P.A.; Pancotto, T.E.; Pavlisko, N.; Verbridge, S.S.; Davalos, R.V.; Rossmeisl, J.H. High-Frequency Irreversible Electroporation for Intracranial Meningioma: A Feasibility Study in a Spontaneous Canine Tumor Model. Technol. Cancer Res. Treat. 2018, 17.
- Campelo, S.N.; Lorenzo, M.F.; Partridge, B.; Alinezhadbalalami, N.; Kani, Y.; Garcia, J.; Saunier, S.; Thomas, S.C.; Hinckley, J.; Verbridge, S.S.; et al. High-Frequency Irreversible Electroporation Improves Survival and Immune Cell Infiltration in Rodents with Malignant Gliomas. Front. Oncol. 2023, 13.
- Lepareur, N.; Ramée, B.; Mougin-Degraef, M.; Bourgeois, M. Clinical Advances and Perspectives in Targeted Radionuclide Therapy. Pharmaceutics 2023, 15, 1733.
- Kunikowska, J.; Morgenstern, A.; Pełka, K.; Bruchertseifer, F.; Królicki, L. Targeted Alpha Therapy for Glioblastoma. Front. Med. 2022, 9, 1085245.
- Cimini, A.; Ricci, M.; Russo, F.; Egidi, M.; Calabria, F.; Bagnato, A.; Schillaci, O.; Chiaravalloti, A. Peptide Receptor Radionuclide Therapy and Primary Brain Tumors: An Overview. Pharmaceuticals 2021, 14, 872.
- Li, Y.; Marcu, L.G.; Hull, A.; Bezak, E. Radioimmunotherapy of Glioblastoma Multiforme—Current Status and Future Prospects. Crit. Rev. Oncol. /Hematol. 2021, 163, 103395.
- Bailly, C.; Vidal, A.; Bonnemaire, C.; Kraeber-Bodéré, F.; Chérel, M.; Pallardy, A.; Rousseau, C.; Garcion, E.; Lacoeuille, F.; Hindré, F.; et al. Potential for Nuclear Medicine Therapy for Glioblastoma Treatment. Front. Pharmacol. 2019, 10.
- Casaco, A.; López, G.; García, I.; Arsenio Rodríguez, J.; Fernández, R.; Figueredo, J.; Torres, L.; Perera Pintado, A.; Batista, J.; Leyva, R.; et al. Phase I Single-Dose Study of Intracavitary-Administered Nimotuzumab Labeled with 188-Re in Adult Recurrent High-Grade Glioma. Cancer Biol. Ther. 2008, 7, 333–339.
- Li, L.; Quang, T.S.; Gracely, E.J.; Kim, J.H.; Emrich, J.G.; Yaeger, T.E.; Jenrette, J.M.; Cohen, S.C.; Black, P.; Brady, L.W. A Phase II Study of Anti–Epidermal Growth Factor Receptor Radioimmunotherapy in the Treatment of Glioblastoma Multiforme: Clinical Article. J. Neurosurg. 2010, 113, 192–198.
- Riva, P.; Franceschi, G.; Frattarelli, M.; Riva, N.; Guiducci, G.; Cremonini, A.M.; Giuliani, G.; Casi, M.; Gentile, R.; Jekunen, A.A.; et al. 131I Radioconjugated Antibodies for the Locoregional Radioimmunotherapy of High-Grade Malignant Glioma--Phase I and II Study. Acta Oncol. 1999, 38, 351–359.
- Reardon, D.A.; Quinn, J.A.; Akabani, G.; Coleman, R.E.; Friedman, A.H.; Friedman, H.S.; Herndon, J.E.; McLendon, R.E.; Pegram, C.N.; Provenzale, J.M.; et al. Novel Human IgG2b/Murine Chimeric Antitenascin Monoclonal Antibody Construct Radiolabeled with 131I and Administered into the Surgically Created Resection Cavity of Patients with Malignant Glioma: Phase I Trial Results. J. Nucl. Med. 2006, 47, 912–918.
- Zalutsky, M.R.; Reardon, D.A.; Akabani, G.; Coleman, R.E.; Friedman, A.H.; Friedman, H.S.; McLendon, R.E.; Wong, T.Z.; Bigner, D.D. Clinical Experience with α-Particle–Emitting 211At: Treatment of Recurrent Brain Tumor Patients with 211At-Labeled Chimeric Antitenascin Monoclonal Antibody 81C6. J. Nucl. Med. 2008, 49, 30–38.
- Reardon, D.A.; Zalutsky, M.R.; Akabani, G.; Coleman, R.E.; Friedman, A.H.; Herndon, J.E.; McLendon, R.E.; Pegram, C.N.; Quinn, J.A.; Rich, J.N.; et al. A Pilot Study: 131I-Antitenascin Monoclonal Antibody 81c6 to Deliver a 44-Gy Resection Cavity Boost. Neuro Oncol. 2008, 10, 182–189.
- Hdeib, A.; Sloan, A. Targeted Radioimmunotherapy: The Role of 131I-chTNT-1/B mAb (Cotara®) for Treatment of High-Grade Gliomas. Future Oncol. 2012, 8, 659–669.
- Reulen, H.-J.; Poepperl, G.; Goetz, C.; Gildehaus, F.J.; Schmidt, M.; Tatsch, K.; Pietsch, T.; Kraus, T.; Rachinger, W. Long-Term Outcome of Patients with WHO Grade III and IV Gliomas Treated by Fractionated Intracavitary Radioimmunotherapy. J. Neurosurg. 2015, 123, 760–770.
- Heute, D.; Kostron, H.; von Guggenberg, E.; Ingorokva, S.; Gabriel, M.; Dobrozemsky, G.; Stockhammer, G.; Virgolini, I.J. Response of Recurrent High-Grade Glioma to Treatment with (90)Y-DOTATOC. J. Nucl. Med. 2010, 51, 397–400.
- Krolicki, L.; Bruchertseifer, F.; Kunikowska, J.; Koziara, H.; Królicki, B.; Jakuciński, M.; Pawlak, D.; Apostolidis, C.; Mirzadeh, S.; Rola, R.; et al. Prolonged Survival in Secondary Glioblastoma Following Local Injection of Targeted Alpha Therapy with 213Bi-Substance P Analogue. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1636–1644.
- Królicki, L.; Bruchertseifer, F.; Kunikowska, J.; Koziara, H.; Królicki, B.; Jakuciński, M.; Pawlak, D.; Apostolidis, C.; Mirzadeh, S.; Rola, R.; et al. Safety and Efficacy of Targeted Alpha Therapy with 213Bi-DOTA-Substance P in Recurrent Glioblastoma. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 614–622.
- Królicki, L.; Bruchertseifer, F.; Kunikowska, J.; Koziara, H.; Pawlak, D.; Kuliński, R.; Rola, R.; Merlo, A.; Morgenstern, A. Dose Escalation Study of Targeted Alpha Therapy with Ac-DOTA-Substance P in Recurrence Glioblastoma—Safety and Efficacy. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 3595–3605.
- Mamelak, A.N.; Rosenfeld, S.; Bucholz, R.; Raubitschek, A.; Nabors, L.B.; Fiveash, J.B.; Shen, S.; Khazaeli, M.B.; Colcher, D.; Liu, A.; et al. Phase I Single-Dose Study of Intracavitary-Administered Iodine-131-TM-601 in Adults with Recurrent High-Grade Glioma. J. Clin. Oncol. 2006, 24, 3644–3650.
- Cordier, D.; Forrer, F.; Bruchertseifer, F.; Morgenstern, A.; Apostolidis, C.; Good, S.; Müller-Brand, J.; Mäcke, H.; Reubi, J.C.; Merlo, A. Targeted Alpha-Radionuclide Therapy of Functionally Critically Located Gliomas with 213Bi-DOTA--Substance P: A Pilot Trial. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 1335–1344.
- Gill, M.R.; Falzone, N.; Du, Y.; Vallis, K.A. Targeted Radionuclide Therapy in Combined-Modality Regimens. Lancet Oncol. 2017, 18, e414–e423.
- Bolcaen, J.; Kleynhans, J.; Nair, S.; Verhoeven, J.; Goethals, I.; Sathekge, M.; Vandevoorde, C.; Ebenhan, T. A Perspective on the Radiopharmaceutical Requirements for Imaging and Therapy of Glioblastoma. Theranostics 2021, 11, 7911–7947.
- Dadgar, H.; Jokar, N.; Nemati, R.; Larvie, M.; Assadi, M. PET Tracers in Glioblastoma: Toward Neurotheranostics as an Individualized Medicine Approach. Front. Nucl. Med. 2023, 3.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.5K
Revisions:
2 times
(View History)
Update Date:
15 Mar 2024
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No