Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Luis German Gonzalez-Bonet and Version 2 by Peter Tang.
Glioblastoma (GB) stands out as the most prevalent and lethal form of brain cancer. Despite multimodality treatments, recurrence is almost universal with survival rates under 2 years after diagnosis.
- glioblastoma
- targeted therapy
- immunotherapy
- nanotherapy
- non-ionizing radiation
1. Introduction
1.1. The Nature and Prognosis of Glioblastoma
Glioblastoma (GB), categorized as a grade IV astrocytoma, is the most prevalent, aggressive, and lethal primary brain tumor in adults. In 2021, the World Health Organization (WHO) introduced significant changes in the criteria for the diagnosis of gliomas, focusing on the importance of genetic and molecular alterations. According to these new criteria (Figure 1), GB should be diagnosed in adults as an isocitrate dehydrogenase wild-type (IDHwt) diffuse astrocytic glioma if there is microvascular proliferation or necrosis (the conventional criteria), and/or at least one of the following three criteria: concurrent gain of whole chromosome 7 and loss of whole chromosome 10 (+7/−10), telomerase reverse transcriptase (TERT) promoter mutations, and epidermal growth factor receptor (EGFR) amplification [1][2][1,2]. The primary driver behind the change in diagnosis criteria is the IDH mutation status, which results in the following modifications: restricting the diagnosis of GB to tumors that do not have IDH mutations (IDHwt); reclassifying tumors previously identified as IDH-mutated GBs as astrocytomas with IDH mutations (grade IV); and establishing the presence of IDH mutations as a requirement for classifying tumors as astrocytomas or oligodendrogliomas Consequently, due to its more favorable prognosis, the previously designated IDH-mutant GB is now categorized within the astrocytomas group, which covers grades II–IV, thus eliminating the term IDH-mutant GB [3]. Moreover, in IDHwt diffuse astrocytomas occurring in younger people, diagnostic consideration should be given to the different types of diffuse pediatric-type gliomas [1]. Gliosarcoma, epithelioid cell GB and giant cell GB are still registered subtypes of GBs, and the term “glioblastoma multiforme” should not be used [4][5][4,5]. New clinical trials will need to be designed with these new distinctions in mind [6].
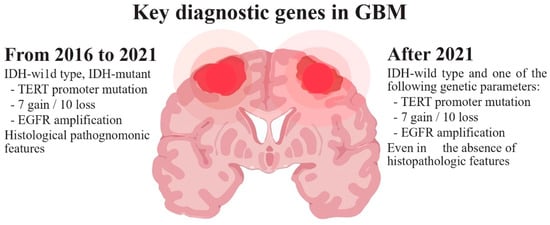
Figure 1.
WHO diagnostic criteria for glioblastoma.
GB typically appears in the cerebral hemispheres, with 95% of these tumors arising in the supratentorial region, especially in the frontal and temporal globes. It infiltrates inside the brain parenchyma and usually does not spread to other parts of the body [7][8][9][7,8,9]. Histologically, GBs are characterized by prominent cellular and nuclear atypia, increased mitotic activity, areas of necrosis, and microvascular proliferation. At least one of these two features must be present for a histologic diagnosis of GB [5]. GB causes death in less than 6 months if untreated [10]. Despite advances in neurosurgery, chemotherapy and radiotherapy, GB remains one of the most treatment-resistant malignancies and its relapse is, in practice, inevitable [7][11][12][7,11,12]. Recurrence often implies a more aggressive form and a median survival of less than 18 months in treated patients [13][14][13,14]. Survival beyond 5 years is observed in less than 5.8% of patients [7]. Patients with recurrent GB (rGB) show an approx. 6-month progression-free survival (PFS6) in only 15% of the cases, and overall survival (OS) ranging between 24 and 40 weeks. Survival rate decreases with age [11][15][11,15].
As suggested by the moniker “multiforme”, GB has a widespread tumoral heterogeneity and plasticity at the cytopathological, transcriptional, and genomic levels [16][17][18][19][20][21][16,17,18,19,20,21]. Moreover, its highly infiltrative nature and the protection by the blood–brain barrier (BBB) have posed significant treatment challenges [9][22][23][9,22,23]. Glioma stem cells (GSCs) are a small subpopulation of cells within the GB, with genomic instability, self-renewal and tumor-initiating capacity, and the ability to differentiate into different GB subpopulations, being responsible for tumor heterogeneity [24][25][26][27][24,25,26,27]. Moreover, GSCs are resistant to apoptosis [28][29][30][31][28,29,30,31], can modulate the components of the tumor microenvironment (TME), are involved in angiogenesis activation and immunosuppression and drive radio/chemoresistance [22][32][33][22,32,33]. The inability of current therapies to eliminate specific GSC subpopulations has been considered a major factor contributing to the inevitable recurrence after treatment [33][34][33,34].
Verhaak et al. proposed a four-subtype classification of GB (classical, mesenchymal, proneural, and neural) based on the analysis of mutational changes in 601 genes in the context of The Cancer Genome Atlas (TCGA) [35]. Verhaak’s latest update removed the neural subtype attributing its origin to a peripheral contamination of the tumor samples [20]. The proneural subtype is associated with a more favorable outcome with respect to the mesenchymal, but this difference is relative to the more favorable outcome of IDH-mutant GBs which were consistently classified as proneural GBs [20][36][20,36]. Moreover, mesenchymal GSCs are enriched with genes associated with angiogenesis, inflammation, and cell migration/invasion. They tend to develop immunosuppression and exhibit increased radio/chemoresistance, all of which are features linked to a worse prognosis [36][37][38][39][36,37,38,39]. In any of the cases, the survival difference is minimal because both subtypes can coexist in the same tumor, and dynamic transitions from a proneural to a mesenchymal phenotype can be induced by TNF-α, temozolomide (TMZ), or radiation through an NF-κB-dependent mechanism [20][36][40][41][20,36,40,41]. Meanwhile, multiplatform analyses of the genetic, epigenetic, and transcriptional profiles have proven useful in refining the classification of gliomas and predicting patient outcomes [42][43][44][45][42,43,44,45].
1.2. Incidence and Risk Factors
GB is the most common (50.1%) among all malignant brain tumors [15]. The annual incidence is low (≈3.19 per 100,000 people in developed countries) but seems to be increasing in some countries owing to aging populations and improvements in diagnosis, among other factors [46]. The median age of diagnosis is approx. 64 and the incidence increases with age reaching its maximum value (15 per 100,000 people) between 75 and 84 years [11][15][47][11,15,47]. It is extremely rare in a pediatric population (0.15 per 100,000), which usually shows longer survivals. The occurrence of GB is 1.6 times more common in males than females and in Caucasians relative to other ethnicities [15][48][15,48].
Beyond rare cases of genetic susceptibility and high-dose radiation exposure, there are no known GB risk factors. An increased risk is seen in some specific genetic diseases, such as hereditary retinoblastoma or Cowden, Turcot, Lynch, Li-Fraumeni and Maffucci syndromes. However, less than 1% of GB patients have a known hereditary disease. Radiation-induced GB can be diagnosed several years after radiation therapy for another tumor or condition in children [49], but no increased risk was observed in adults exposed to IR [50]. Patients diagnosed with previous non-neurological cancers may have an overall elevated incidence of GB compared to the general population [51].
1.3. Criteria to Evaluate Treatment Response and Progression
The MacDonald criteria [52] have traditionally been used to determine treatment response and progression by assessing contrast-enhancing tumor size [by computed tomography (CT) or magnetic resonance imaging (MRI)] along with clinical evaluation and corticosteroid dosage. These criteria categorized the response into four groups: complete response (CR), partial response (PR), stable disease (SD), and progressive disease (PD). Nevertheless, these criteria have several limitations. One is the temporary increase in tumor enhancement (known as pseudo-progression), which occurs in 20–30% of the patients treated with chemo/radiotherapy and challenges differentiation with a real tumor progression. Another limitation is the high radiographic response rates seen with anti-angiogenic agents and other treatments [53]. To address these issues in 2010, the Response Assessment in Neuro-Oncology (RANO) criteria were developed to address these limitations. Although the RANO criteria improved therapy evaluation in high-grade glioma, the assessment of treatment-related side effects can hinder accurate response evaluation. The appearance of new lesions is considered a criterion for disease progression according to both RANO and MacDonald criteria. However, neuro-oncology patients receiving immunotherapies may experience the transient appearance of new enhancing lesions, either locally or in distant sites. In such cases, it is advisable to evaluate imaging findings within 6 months of starting immunotherapy, including the development of new lesions or radiographic progression, as long as there is no significant clinical deterioration [54].
To tackle the challenges in assessing immunotherapy response for neuro-oncology, the immunotherapy RANO (iRANO) criteria were introduced. The iRANO criteria combine the response assessment framework of RANO with guidelines for confirming disease progression, as originally proposed by the Immunotherapy Response Criteria in Solid Tumors to assist in clinical decision-making. The aim is to minimize premature discontinuation of potentially beneficial therapies while ensuring patient safety [54]. However, in most recurrence cases, there is a mixture of tumor cells and tissue affected by radiation injury. Radiologists strive to identify the predominant component of the lesion to determine prognostic factors and categorize the findings according to the RANO criteria, thus providing the most appropriate treatment for the patient. To overcome the aforementioned limitations in the follow-up, incorporating changes measured by advanced MRI and positron emission tomography (PET) imaging, which may precede anatomical changes in tumor volume, shows promise [55][56][55,56]. PET may also help to differentiate actual progression from pseudo progression [57]. Additionally, 18F-FMISO-PET can localize regions of hypoxia that are thought to drive radio/chemoresistance in GBs and promote immune suppression [58].
2. Lessons Learned in the Pathophysiology of Glioblastoma
2.1. Glioma Stem Cells and Tumor Microenvironment
The two prevailing hypotheses for the origin of GB are the GSC and the astrocyte de-differentiation theories [25][26][37][25,26,37]. Neural stem cells (NSCs), as unique stem cell type in the brain, have the ability to self-renew and can differentiate into neurons, astrocytes, and oligodendrocytes (Figure 2) [25][59][25,59]. NSCs are most active during development, but small populations remain functional in specific stem-cell niches in the adult brain. Compelling evidence suggests that GSCs may arise from NSCs located in the adult subventricular zone (SVZ) [60][61][62][63][64][65][60,61,62,63,64,65], and a recent article provided molecular genetic confirmation of this hypothesis in a preclinical model [60]. GSCs express the mutated genes TERT, PTEN, EGFR, TP53, and PDGF present in NSCs. In addition, there is an evident functional overlap and similarity between both types of stem cells, reflected in numerous shared gene expression patterns such as CD133, Sox10, nestin, vimentin, musashi, GFAP, and Olig1/2 [65][66][67][65,66,67]. Due to the migration ability of GSCs and the unique environment of SVZ (the vascular system of SVZ is richer than that of other brain regions), treatment-resistant GSCs easily migrate to and colonize the SVZ [68]. Consequently, numerous retrospective studies have confirmed that GBs in close contact with the SVZ possess more aggressive patterns of recurrence and worse clinical outcomes [67][69][70][67,69,70]. Therefore, new therapy strategies are being assayed with the aim of targeting SVZ to eradicate NSCs or GSCs [71].
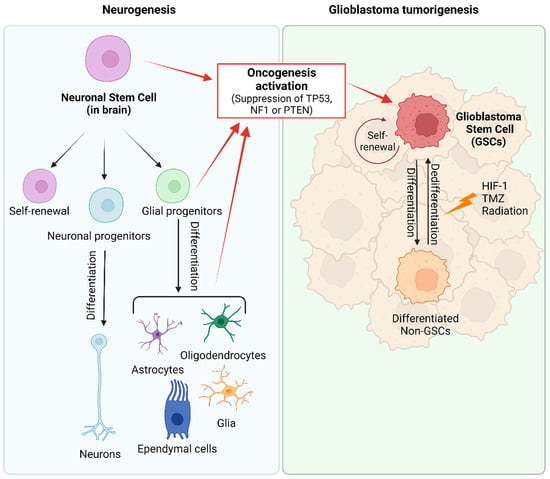
Figure 2. Origin of glioblastoma. During normal embryonic development and in the adult brain, neural stem cells (NSCs) generate glial and neuronal cells. Glioma stem cells (GSCs) arise from NSCs, astrocytes, oligodendrocytes, or glial precursor cells through the activation of oncogenic pathways (inactivation of TP53, NF1 or PTEN). GSCs are described as slow-dividing or quiescent cells, with multilineage differentiation capacity that allows them to differentiate into GB cells and cells with astrocytic, neuronal, and endothelial features and even trans-differentiation abilities. In GB tumors, there exists a dynamic equilibrium between quiescent and proliferative GSCs, and between GSC populations and their lineage-committed counterparts (differentiated non-GSC) that can also dedifferentiate into stem-lineage GSCs. Created with BioRender.com, accessed on 24 January 2024.
The origin of GB, based on the stem cell theory, explains the versatility and plasticity of heterogeneous GB tumor populations. However, several studies provide evidence suggesting that partially differentiated glial cells, including oligodendrocyte and astrocyte precursors, may play a role in or be responsible for tumorigenesis [60][72][73][60,72,73]. The astrocyte de-differentiation theory is supported by experiments demonstrating the formation of tumors that are histologically similar to GB after activation of oncogenes and/or suppression of tumor suppressor genes in astrocytes [24][72][74][75][24,72,74,75]. Nevertheless, this manipulation in astrocytes results in their acquisition of stem-cell-like characteristics. Consequently, both hypotheses are not mutually exclusive and explain the presence of cancer stem cells within the tumor [64][76][77][64,76,77]. Moreover, the dedifferentiation of non-GSCs to GSCs further complicates the GSC-targeted therapy [25][78][25,78].
GSCs represent a very low percentage of cells within GBs, and are functionally defined and distinguished from their differentiated tumor progeny at central transcriptional, epigenetic, and metabolic regulatory levels [79][80][79,80]. Recognized markers of GSCs include CD133 (PROM1) [81], CD44 [82], SOX2 and nestin [76][83][76,83], but none of them are specific markers of GSC. Other putative biomarkers are CD15 (FUT4), A2B5 antigen, CD90 (THY1), integrin ITGA6, CD171 (L1CAM), S100A4, ATP-binding cassette transporters and the combination of CD44 and ID1 (reviewed in [84]). GSCs develop genetic variability and possess self-renewal capacity and specific characteristics that support tumor development, heterogeneity, recurrence, immunosuppression and radio and chemotherapeutic resistance [85]. Therefore, the heterogeneity of GB tumor cells can be attributed to the clonal evolution and differentiation/dedifferentiation capacity of GSC [25][66][72][77][86][87][25,66,72,77,86,87]. The GSCs’ ability to adapt to different niches implies that they can dynamically restructure their transcriptional program, inducing the transient expression of genes with specific functions for each cell state [18][36][18,36]. Furthermore, microglia and endothelial cells of the perivascular niche produce numerous growth factors that contribute to the support of proliferation, migration, and differentiation of NSCs and GSCs [27][88][89][27,88,89]. In turn, GSCs release transforming growth factor β (TGFβ) that enhances the tumor vasculature and can even transdifferentiate and generate endothelial cells or pericytes to form new tumor vascular niches [90][91][90,91]. GSCs exhibit elevated migratory and invasive potential, eliciting infiltration into healthy tissue, thus limiting the effect of total surgical resection and radiotherapy [92]. Residual cells have the ability to regenerate GB in brain regions distant from the initial tumor by acquiring new and different driver mutations that make them resistant to treatments [41]. Consequently, GSCs are more radioresistant than GB cells [93], can be resilient to TMZ-mediated cell death [94], and have mutations that facilitate recurrence after therapy [95]. DNA damage repair mechanisms, such as ATM, ATR, CHK1, and PARP1, are upregulated in GSCs, and CHK1 is preferentially activated following irradiation [96][97][96,97].Consequently, GSCs exhibit rapid G2-M cell cycle checkpoint activation and enhanced DNA repair [98]. The preferential activation of DNA damage checkpoint responses [34] and the increased expression of drug efflux pumps and antiapoptotic proteins [99] contribute to GSC recruitment after treatment. Interestingly, the inhibition of DNA repair protein RAD51 homolog 1 has been found to delay G2 cell cycle arrest, thereby sensitizing GSCs to radiation [100].
Ionizing radiation also enhances the motility, invasiveness and aggressiveness of GSCs. The increased motility and invasiveness result from the activation of the HIF(hypoxia-inducible factor)-1α, whereas aggressiveness is attributable to a pro-neural-to-mesenchymal transition associated with the activation of the STAT3 transcriptional factor [101]. STAT3 is overexpressed in GSCs [87] and plays a crucial role in sustaining stem-like characteristics [102]. Moreover, it enhances the expression of pro-tumorigenic genes related to cell cycle progression, extracellular matrix remodeling, as well as the secretion of cytokines and growth factors [103]. Consequently, STAT3 deletion or inhibition in GB cell lines markedly decreases tumorigeneses in vitro and in vivo [103][104][103,104] and has a radiosensitizing effect [93]. WP1066, one of the most promising STAT3 inhibitors, will be investigated in a phase II clinical trial for patients with recurrent malignant glioma [105].
GB cells have the ability to manipulate the TME to favor immunosuppression and to develop a niche sustaining tumor growth, invasion, migration, and survival [28][106][28,106]. GB cells can evade immune surveillance through the release of various soluble mediators such as TGFβ, IL-10, and PGE-2. In the presence of TGFβ, CD4+ T cells upregulate FoxP3 and differentiate into Treg cells with potent immunosuppressive potential. This cytokine inhibits the expression of cytolytic gene products (perforin, granzyme A, granzyme B, Fas ligand, and IFN-γ) which are co-responsible for CD8+ T-cell-mediated tumor cytotoxicity. Increased secretion of IL-10 is associated with enhanced expression of anti-inflammatory cytokines, such as IL-4, CCL2, and TGFβ. In the presence of IL-10, TAMs downregulate the expression of antigen-presenting molecules, thereby impairing CD4+ T cell activation. In turn, PGE-2 has been shown as a key mediator of immunosuppressive activity through the expansion of myeloid-derived suppressor cells (MDSCs) [107]. In fact, GSCs and GB cells play the role in recruiting and activating MDSCs [108] and M2 macrophages to drive immune suppression [109][110][111][109,110,111]. Simultaneously, GSCs protect themselves from T-cell-mediated killing by secreting extracellular vesicles containing programmed death ligand 1 (PD-L1) [112][113][112,113].
Consequently, new therapies that effectively target this important population may help to prevent recurrence and improve patient survival, and for sure, no single therapeutic modality will be effective against such a heterogeneous population of cells.
2.2. Metabolic Features Favoring Growth and Resistance
Metabolic reprogramming plays a crucial role in enabling GB invasive cells to generate the energy required for colonizing the surrounding brain tissue and adapting to hypoxic microenvironments [114][115][114,115]. The metabolism of GB is characterized by the upregulation of the PI3K/Akt/mTOR signaling pathway, a high rate of glycolysis, and increased lipid storage [116][117][116,117]. Aerobic glycolysis along with glucose consumption and lactate production supports rapid GB growth and correlates with a lower survival rate [118]. Nevertheless, GB cells adapt their metabolism according to glucose availability, which gives them extra resistance to hypoxia or altered redox situations. Selective pressure on GB cells makes them overexpress glucose transporters (GLUT1 and, particularly, GLUT3). GLUT3 has a five-fold higher affinity for glucose compared to GLUT1, thus facilitating glucose uptake in environments with lower glucose concentrations. Additionally, the acquisition of a stem cell state is associated with a significant increase in GLUT3 expression in induced pluripotent cells, and this overexpression correlates with poor glioma patient survival [119][120][119,120]. When glucose levels are low, HIF-1α guarantees the upregulation of GLUT3 and hexokinase-2, increasing the glycolytic pathway [121][122][121,122].
The activation of sterol regulatory element-binding protein-1, a crucial transcription factor controlling fatty acid and cholesterol synthesis, as well as cholesterol uptake, enables GB to obtain significant quantities of lipids essential for its rapid growth [123]. GSCs exhibit high expression of mediators of lipid metabolism, such as brain-fatty-acid-binding protein (FABP7), which leads to an increase in lipid contents that are specifically metabolized under glucose-deprived conditions [116]. GB cells direct significant amounts of lipids into specialized storage organelles known as lipid droplets, thus avoiding lipotoxicity. This process involves the overexpression of diacylglycerol acyltransferase-1 and sterol-O-acyltransferase-1, which convert surplus fatty acids and cholesterol into triacylglycerol and cholesteryl esters, respectively, increasing the storage as neutral lipids within lipid droplets [123].
Amino acids play a crucial role as important fuels for GB growth. Gene expression profiling has shown an upregulation of the L-Gln importer ASCT2 in GB compared to low-grade gliomas, and L-Gln deprivation has slowed tumor growth in some in vitro studies [124]. The L-Gln-derived glutamate and glucose-derived pyruvate are substrates for the glutamate-pyruvate transaminase 2 (GPT2), which synthetizes α-ketoglutarate. Through GPT2 upregulation, the anaplerotic replenishment of the TCA cycle is possible; otherwise, it is impaired by augmented pyruvate conversion to lactate. In other words, the Warburg effect, manifested as increased lactate release, drives L-Gln addiction in order to maintain the TCA cycle function [124]. Moreover, L-Gln has been shown to promote the mTOR-dependent signaling pathway, a potent driver of GB growth and progression [125][126][125,126]. Other amino acids are also utilized to fuel bioenergetic reactions and the synthesis of macromolecules in GBs [114]. L-Asp has been shown to be a limiting metabolite for GB cellular proliferation in hypoxic conditions [127]. L-Arg is involved in GB cell adhesion, and thereby in tumor cell migration and invasion [128]. L-Trp and L-Arg metabolism have also been linked to decreased detection by neighboring immune cells, creating a favorable environment [129].
In gliomas, autocrine glutamatergic signaling has been identified as a promoter of invasion [130]. GB cells release high levels of glutamate, which not only enhances tumor invasiveness but also promotes the turnover of GSCs [131]. In other words, GB cells create a positive feedback system whereby an excess of glutamate promotes their own growth and secondarily causes excitotoxicity-induced cell death in surrounding brain tissue [132]. It is probable that such tissue damage contributes to cerebral edema and the neurotoxicity associated with a growing GB. Consequently, the inhibition of glutamatergic signaling has been proposed as a strategy to mitigate GB-induced neurotoxicity [133].
Moreover, the invasive nature of GB is modulated by cell-to-cell crosstalk within the TME and altered expression of specific genes, such as ANXA2 (encoding the protein annexin A2, a Ca2+-dependent phospholipid-binding protein that helps to organize exocytosis of intracellular proteins to the extracellular domain) [134], GBP2 (encoding the guanylate-binding protein 2, which binds to guanine nucleotides and works in intracellular signaling) [135], FN1 (encoding fibronectin, which binds to integrins and facilitates adhesion, growth, migration, and differentiation) [136], PHIP (encoding the pleckstrin homology domain interacting protein, which regulates growth and survival of GB cells) [137], and SLC2A3 (encoding the glucose transporter 3) [114][138][114,138].
2.3. Ion Channels
Different studies have demonstrated the upregulation of Ca2+ selective ion channels in GB, contributing to invasion, proliferation and resistance to apoptosis [139]. By blocking L-type voltage-gated Ca2+ channels, cell invasion is inhibited as filopodia (also known as tumor microtubes, TMs) formation is blocked [140]. Indeed, GB cells possess the ability to extract specific signals from healthy neurons using TMs [141]. Furthermore, inhibition of T-type Ca2+ channels has been shown to induce apoptosis in GB cells [142]. Therefore, blocking Ca2+ could prevent tumorigenesis through several mechanisms, i.e., cell cycle progression, induction of apoptosis and inhibition of cell migration.
K+ ion channels play a crucial role in the proliferation and the resistance to apoptosis in GB. Specifically, certain voltage-gated K+ channels are overexpressed in GBs participating in signaling pathways that promote proliferation and inhibit apoptosis [143]. Some of these effects are due to the role of K+ channels in establishing the resting membrane potential, and therefore affecting the cell cycle. Different studies have shown that inhibition of K+ channels improves survival in GB patients, which emphasizes their role in GB development and progression [144]. Consequently, blocking of ion channels could represent an interesting therapeutic approach against GB progression.
2.4. Epigenetics of Glioblastoma
GB progression is associated with different types of epigenetic alterations, including histone modifications, DNA methylation, chromatin remodeling, and aberrant microRNA (miRNA) [145][146][145,146], a group of small non-coding RNA (19–22 nucleotide long) molecules that regulate the post-transcriptional degradation of mRNA [147]. Ciafré et al. performed the first experiment related to miRNAs in GB, investigating the expression of 245 miRNAs using microarrays [148]. The most interesting results came from miR-221 upregulation, and a set of brain-enriched miRNAs (miR-128, miR-181a, miR-181b, and miR-181c) that are down-regulated in GB. At the same time, miRNAs have been shown to be important regulators of gene expression and may also regulate cellular processes, including apoptosis, proliferation, invasion, angiogenesis, and chemoresistance [149][150][149,150]. Therefore, microRNAs can be classified according to their role in tumorigenesis (i.e., tumor suppressor or oncogenic).
CircRNAs exert their biological effects through four different mechanisms: serving as sponges of RNA binding proteins, modulating parental gene transcription, encoding functional proteins and, most importantly, serving as sponges of miRNAs [151][189]. As is thoroughly reviewed in [151][152][189,190], circRNAs regulate GB proliferation and invasion and are also involved in angiogenesis activation. Good stability, broad distribution and high specificity make circRNAs promising biomarkers for GB prognosis and/or diagnosis, although their clinical implementation still has a long way to go.
Long-noncoding RNAs (lncRNAs) are a class of regulatory noncoding RNAs (>200 nt) that interact with DNA, RNA, and proteins to regulate various biological processes. As reviewed in [153][154][155][156][191,192,193,194], numerous studies have shown that lncRNA regulates the expression of genes involved in GB tumorigenesis (CHRM3-AS2, DLGAP1-AS1, DGCR10, LINC01057, LINC-PINT, MIR31HG, MIR210HG, NEAT1, NONHSAT079852.2, PVT1, SEMA3B, RBPMS-AS1), progression (ASLNC22381, ASLNC20819, CRNDE, DGCR10, HNF1A-AS1, HOXD-AS2, HRA1B, HOTAIRM1, LINC-PINT, PRADX, NEAT1, OXCT1-AS, TCONS-00004099) and therapeutic resistance (H19, MALAT1, MUF, DANCR, HOTAIR, HOTAIRM1, LINC00511, UCA1, OIP5-AS1, DANCR, FOXO3, HERC2P2) of GB cells. Moreover, lncRNAs exhibit stable secondary structure; thus, some of them (HOTAIR, GAS5, HOXA11-AS, HOTAIRM1, AGAP2-AS1, and AC002456.1) have been proposed as prognostic and diagnostic GB biomarkers [157][158][159][160][195,196,197,198]. More specifically, SBF2-AS1, MALAT1, CRNDE, TP73-AS1 and LINC00511 have been suggested as biomarkers of TMZ resistance in GB [160][198]. Lately, some evidence has indicated that lncRNAs also take part in GB cell metabolism. For instance, the lncRNA TP53TG1, under glucose deprivation, may promote cell proliferation and migration by influencing the expression of glucose-metabolism-related genes in glioma cells [161][199], and the lncRNA LEF1-AS1 facilitates the multiplication of GB cells and impedes apoptosis via the Akt/mTOR pathway [162][200]. Clinical trials involving the use of lncRNA as biomarkers for GB detection and prognosis are only in the recruitment phase but look promising.
Other epigenetic alterations, such as DNA methylation, histone modifications, and chromatin remodeling, are mechanisms involved in transcriptional activation of critical genes for GB development, lethality and resistance [145][163][164][165][145,201,202,203]. Thus, several epigenetic agents, including histone methyltransferase inhibitors, DNA methyltransferase inhibitors, histone deacetylase (HDAC) inhibitors, and other agents, are currently being tested for GB treatment in preclinical and clinical trials [146][165][146,203]. Protein arginine methyltransferase 5 (PRMT5) is a member of the PRMT family of proteins that plays a key role in the regulation of cellular signaling and gene expression by methylating histones as well as nonhistone proteins [166][204]. Nuclear expression of PRMT5 negatively correlates with glioma patient survival [167][205]. Engineered loss of PRMT5 or treatment with CMP5 (a PRMT5 inhibitor) results in apoptosis or loss of self-renewal for differentiated or undifferentiated GB cells, respectively, [168][206]. CMP5 derails the negative regulation of PTEN by PRMT5, which, in turn, decreases Akt activity in patient-derived GB neurospheres [169][207].
HDACs have been widely studied in GBM cells due to their relationship with therapeutic resistance, cell proliferation and invasion, angiogenesis and apoptosis [170][171][172][173][208,209,210,211]. In preclinical studies, HDAC inhibitors (HDACi) have proven to be effective anti-GB agents via multiple mechanisms, such as upregulating the expression of tumor suppressor genes, inhibiting oncogenes, inducing cell cycle arrest, promoting cell apoptosis and differentiation, inhibiting motility/migration, abolishing autophagy and tumor angiogenesis, and upregulating natural killer (NK)-cell-mediated tumor immunity [174][175][176][177][212,213,214,215]. Additionally, HDACis have demonstrated the capability to reduce cancer stem cell burden in GB tumors by modulating stemness, proliferation, differentiation, cell cycle arrest, apoptosis, autophagy and vasculogenic mimicry of GSCs [171][172][178][209,210,216]. Several HDACis (i.e., valproic acid, voristonat, panobinostat) have been assayed in clinical trials due to their capacity to act as chemo/radio-sensitizers and target GSCs [178][216]. Voristonat combination regimens with TMZ/radiotherapy and/or bevacizumab (BEV, recombinant humanized monoclonal antibody that blocks VEGFR-A) have proven to be tolerable (NCT01738646), but no statistical improvement in OS and/or PFS was noted [179][217]. Similar results were obtained with panobinostat in combination with BEV (NCT00859222) [180][218]. Valproic acid is a potent anticonvulsant that promotes apoptosis and impairs glioma cell proliferation and invasiveness and sensitizes GB cells to several anticancer drugs, such as TMZ, etoposide, gefitinib, nitrosoureas, and radiation therapy [181][182][183][219,220,221]. A meta-analysis [184][222] and a recent open-label phase II study [185][223] results seem to confirm that GB patients may experience prolonged survival due to valproic acid administration, providing further justification for a phase III trial of valproic acid/SOC. Levetiracetam, a relatively new antiepileptic drug, modulates HDAC levels ultimately silencing MGMT, thus increasing TMZ effectiveness in GCSs [186][224]. Retrospective analyses and an open-label phase II study (NCT02815410) seem to evidence that LEV improves GB patients’ PFS and OS [187][188][225,226]. So, it is perhaps time to reconsider the results performed in 2016, where a pooled analysis of a large series of cases treated with valproic acid or levetiracetam failed to find an association with patients’ survival [189][227]. A double-blind randomized clinical trial (ChiCTR2100049941) focusing on the clinical benefits of LEV + TMZ in the treatment of GB is ongoing in China. Nuclear imaging of HDAC expression in GB can be useful to improve the understanding and role of HDAC enzymes in gliomagenesis and identify patients likely to benefit from HDACi-targeted therapy [177][190][215,228].
2.5. The Angiogenetic Capacity of Glioblastoma
Aberrant vascular proliferation, necrosis, and infiltration of surrounding brain tissues are considered “hallmarks” of GB. Neo-vessels form from preexisting blood vessels due to VEGF expression by tumor and stromal cells under hypoxic conditions. The combination of VEGF with FGF (fibroblast growth factor)-2 or PDGF (platelet-derived growth factor) is known to synergistically enhance angiogenesis [191][229]. Vasculogenic mimicry (VM) is a new mechanism of tumor neovascularization in which highly invasive and genetically dysregulated tumor cells acquire vascular cell function, forming de novo vascular-like structures [192][230]. The involvement of GSCs in VM has been reported by several studies [193][194][231,232]. The disruption of GB vasculature through radiation or anti-angiogenic therapies induces a hypoxic microenvironment that promotes VM as an adaptative strategy to assist GB cells in surviving and progressing even when angiogenesis is blocked [195][196][233,234]. In keeping with this idea, the inhibition of vasculogenesis, but not sprouting angiogenesis, prevents the recurrence of GB after irradiation in mice [197][235].
SCs play a crucial role in VM, mainly due to their high plasticity and potential differentiation into endothelial-like cells [198][236]. The vascular niche is very important for the maintenance of GSCs as it promotes their survival and proliferation [192][199][230,237]. Additionally, communication between endothelial and tumor cells allows tumor vasculature formation and tumor cell dissemination [194][200][232,238]. Tumor vasculature has been considered a contributor to treatment resistance and relapse [201][239]. GSCs seem to be attached to the arterioles but not to the capillaries [202][240]. Arterioles transport, but do not exchange, gasses and nutrients, and promote a peri-hypoxic area. Integrin ligation causes an activation of the integrin-linked kinase leading to increased HIF-1α, as well as increased VEGF production [203][241]. HIF-1α acts as a potent activator of angiogenesis by stimulating the production of VEGF-A, PDGF and many other factors that initiate endothelial cell proliferation, invasion, and migration [204][242]. In GB, HIF-1α is not only influenced by oxygen but also by oncogenic signaling pathways, such as MAPK/ERK, p53, and PI3K/Akt/mTOR [205][243]. Although many approaches have been tried to inhibit HIF-1α, drugs that only target specific components of the hypoxia signaling pathway have generally failed to produce an enduring clinical response in GB. It is thought that the complete inhibition of HIF-1α is necessary to show potent antitumor activity and to promote the activation of the immune system [205][243]. Inhibition HIF-2α, which can also block the hypoxia pathway, is an alternative attractive strategy for GB treatment. HIF-2α is specifically overexpressed in GB cells and GSCs, but not in normal tissues [206][244]. Although the HIF-2α inhibitor PT2385 had limited activity in rGB (phase II, NCT03216499) [207][245], other HIF-2α inhibitors that are currently under research may help in blocking GB progression. It is also important to mention that recent findings suggest that GB hypoxia regulates gene expression in an HIF-independent way. In that sense, Srivastava et al. demonstrated that FAT1 (a FAT atypical cadherin) modulates the epithelial-mesenchymal transition and stemness gene expression in hypoxic GB [208][246], and hypoxia induces epigenetic regulation of the transmembrane protein odd Oz, altering DNA methylation status and activating the ODZ1-mediated migration of GB cells [164][202].
Importantly, Aderetti et al. demonstrated the existence of hypoxic peri-arteriolar GSC niches in GB tumor samples [209][247]. Apparently, GSCs remain attached to peri-arteriolar niches by the same receptor–ligand interactions as hematopoietic stem cells in the bone marrow. GSCs’ infiltration can be promoted via VEGF secreted by endothelial cells, which may induce the trans-differentiation of GSCs into endothelial cells, promoting angiogenesis and invasiveness [86]. Furthermore, a phenomenon of metabolic zonation has been described depending on the relative distance between the tumor cell and the blood vessel [210][248]. Proximity to the blood vessels promotes the mammalian target of rapamycin mTOR-derived anabolic metabolism and enhances tumor aggressiveness and therapy resistance [210][248]. Indeed, GB cells located in the perivascular tier exhibit robust anabolic metabolism and deviate from the Warburg principle by extensively engaging in oxidative phosphorylation. These perivascular cancer cells acquire specific functional characteristics, such as heightened tumorigenicity, enhanced migratory and invasive abilities, and surprisingly, remarkable resistance to chemotherapy and radiation; most of these traits are dependent on the mTOR pathway [210][248].
The BBB is a major obstacle to drug penetration within the brain parenchyma. Only 20% of small molecules/therapeutics agents cross the BBB and reach tumor cells at an effective concentration. GSC or GB cells protected against therapeutic agents by an intact BBB are the source of tumor recurrence [23]. P-glycoprotein, multidrug resistance proteins, organic anion transporters and breast cancer resistance proteins are especially important efflux pumps within the BBB that limit the accumulation of small-molecule-targeted therapies [211][249]. To make it more difficult, GSCs overexpress ABC transporters, further hindering drug delivery. ABC transporters promote therapy resistance by promoting the efflux of exogenous compounds, such as TMZ, at the cellular and BBB levels [22]. Infiltrating tumor cells are known to compromise the integrity of the BBB, resulting in a vasculature known as the blood–tumor barrier (BTB), which is highly heterogeneous and characterized by numerous distinct features, i.e., non-uniform permeability and active efflux of molecules [211][249]. Therefore, delivering therapeutic agents across the BBB and BTB, but avoiding their accumulation in the healthy parenchyma, is essential to making significant progress in GB treatment.
3. Present Therapy and Challenges
3.1. Standard of Care in Newly Diagnosed GB Patients
The Stupp protocol became the standard of care (SOC) for newly diagnosed GB (ndGB) patients since a randomized phase III trial evidenced an improved mOS from 12.1 to 14.6 months and an increase in the 2-year survival rate from 10% to 27% [7]. This SOC includes maximal safe resection, radiotherapy with concurrent (75 mg/m2/day × 6 weeks) and adjuvant TMZ (150–200 mg/m2/day × 5 days for six 28-day cycles). Since then, similar results (mOS 15–18 months) have been observed in other clinical studies [212][213][214][250,251,252]. Despite significant advances in the understanding of the molecular biology and pathophysiology of the GB, SOC has remained unchanged, excepting the possibility of adding or not tumor treating fields (TTFields) [13][215][216][13,253,254].
GB mostly recurs within 2–3 cm from the borders of the initial lesion and with multiple lesions, thus, maximal surgical resection improves survival irrespective of the age of the patient or the molecular status of the tumor [214][217][252,268]. Preoperative brain mapping techniques such as navigated transcranial magnetic stimulation (nTMS), magnetoencephalography, functional MRI, and diffusion tract imaging (DTI) are used to facilitate safe resections and minimize surgical complications [218][269]. Compared to non-nTMS techniques, nTMS has been associated in GB patients with smaller craniotomy size, less residual tumor tissue, shorter hospital stays, and improved survival at 3, 6, and 9 months, with no significant difference in surgery-induced neurological deficits [219][270].
During surgery, various tools are employed to optimize the extent of resection and minimize residual tumor volume. These include functional monitoring, fluorescence-based visualization of the tumor using 5-aminolevulinic acid (5-ALA), ultrasonography, and intraoperative MRI (ioMRI) [217][218][220][268,269,271]. Additionally, techniques like evoked potentials, electromyography, and brain mapping in awake patients, under local anesthesia, are used to monitor and preserve language and cognition during resections in critical brain areas [221][272]. The use of the amino acid 5-Ala helps to identify tumor volume and areas of neoplastic infiltration through fluorescent visualization, improves PFS, OS, and reduces postoperative neurological damages [218][222][223][224][225][269,273,274,275,276]. 5-Ala has also been effectively used in rGB resection, but the risk of false-positive fluorescence for reactive non-tumor tissue is more remarkable in relapse forms, likely due to an altered BBB [226][277]. Nevertheless, recently, an off-label fluorophore (sodium fluorescein) has become popular due to numerous benefits compared to 5-Ala, including lower cost, non-toxicity, easy administration and a wide indication for other brain tumors [227][278]. Microsurgical resection of GB using sodium fluorescein has been associated with an increased GTR rate and OS [228][279], although it is still considered inferior compared to 5-Ala [229][280].
Intraoperative ultrasound (ioUS) involves the use of sonography to locate tumor tissue during surgery and to delineate it from healthy brain tissue. As opposed to 5-Ala, which can only identify high-grade gliomas, ioUS is able to identify both low- and high-grade gliomas. In practice, 5-Ala and ioUS are considered complementary techniques [218][269]. Intraoperative magnetic resonance imaging (IoMRI) improves the accuracy and definition of the tumor and provides near real-time information about the dynamic changes occurring during surgery [230][281]. Analysis of residual GB volumes and neurological outcomes demonstrates that ioMRI is significantly superior to 5-Ala and white-light surgery at comparable peri- and post-operative morbidities [231][282]. The combination of ioMRI and 5-Ala facilitated achievement of the highest extent of resection (95%), followed by ioMRI alone (94%), 5-Ala alone (74%), and no imaging (73%), and this was associated with fewer post-chirurgic neurological deficits [232][233][283,284]. However, the lack of evidence regarding the cost-effectiveness compared to less advanced techniques raises uncertainty [217][234][268,285]. Regardless of the technique used, a postoperative contrast-enhanced MRI should be carried out within 48 h to assess the extent of resection and serve as a baseline for further treatments. Additionally, MRIs are performed every 2–3 cycles of TMZ treatment to monitor the tumor’s response [235][286].
After surgery, the smallest amount of residual tumor correlates with higher survivals [236][237][287,288]. However, radical surgical resection is limited by the highly invasive nature of GB cells [238][289]. Additionally, postoperative complications are a negative prognostic factor, and in this it is essential to prevent permanent neurologic deficits to safeguard the quality of life of the patients [14][47][238][14,47,289]. Carmustine (BCNU) wafers placed in the tumor resection cavity at the time of surgery provide a modest survival advantage (≈2 months) [239][290]. Wafer implants have been approved by the FDA and the EMA, but are not included in the SOC mainly due to their limited brain penetration, safety and tolerability, and because the treatment may preclude patients from enrolling into clinical trials [240][291]. Where surgical resection is not possible, stereotactic biopsy or open biopsy are alternative options for histological diagnosis and further molecular testing, which can determine an optional therapy [241][242][292,293]. However, this recommendation is not exempt from criticisms since in GB patients with low-performance status and/or advanced age, biopsies imply very little clinical gain [243][294].
Compared with surgery alone, postoperative radiotherapy is used to control microscopic unresectable disease, delay neurological deterioration and increase survival [7][244][7,295]. Radiotherapy is less efficacious in hypoxic TME due to a lower oxidative stress and because cancer cells develop mechanisms to repair DNA [115].
TMZ is a mono-alkylating agent that induces cytotoxic lesions including N7-methylguanine, N3-methyladenine and O6-methylguanine. N7-methylguanine and N3-methyladenine are repaired by base-excision repair (BER) and contribute minimally to the overall cytotoxicity of TMZ, while O6-methylguanine is repaired by O6-methylguanine-DNA-methyl transferase (MGMT) [245][296]. Methylation of the MGMT gene promoter (40% of GB patients) causes a reduction in MGMT protein expression and activity that results in persistent O6MeG lesions that trigger replicative stress and cytotoxicity via futile cycles of mismatch repair (MMR) [246][297]. Therefore, MGMT promoter methylation confers a better prognosis and overall survival (OS) associated with a positive response to alkylating agents in GB patients aged <70 years [7][13][247][7,13,298]. Radiotherapy has been shown to upregulate MGMT, whereas prolonged exposure to alkylating agents may suppress MGMT activity making the cells more susceptible to TMZ [248][257]. Nevertheless, several trials evidence that extending post-radiation TMZ from 6 to 12 months does not improve PFS6 and is associated with greater toxicity, functional deterioration, and poorer quality of life [248][249][257,258].
TMZ is a cornerstone of GB treatment, but its effectiveness is limited by the blood–brain and blood–tumor barriers, and the inherently or acquired GB resistance [19][250][251][252][19,299,300,301]. Upon TMZ treatment, GB and GSC cells induce DNA repair mechanisms, NF-kB signaling mediated antiapoptotic pathways, the expression of anti-apoptotic Bcl-2 family members, EGFR activity, drug efflux by ATP-binding cassette (ABC) transporters, autophagy-mediated resistance, expression of STAT3 and miRNAs, and overexpression of antioxidant proteins [83][250][252][253][254][255][256][83,299,301,302,303,304,305]. Nitrosoureas, i.e., lomustine (CCNU), carmustine and procarbazine, were widely used before the availability of TMZ, but their use is now limited to the treatment of rGB. Patients in good physical condition with hypermethylated MTMG promoters (NCT01149109) slightly increase their OS survival by the addition of lomustine to SOC (48.1 vs. 31.4 months) [257][258][259,306]. Nevertheless, the benefit of this regimen remains unclear since the sample size was small and few patients were able to complete all six cycles of adjuvant treatment due to the greater hematologic toxicity in the lomustine-TMZ arm [259][307]. The addition of BEV to the SOC improved PFS but not OS in both AVAglio and RTOG 0825 trials (NCT00943826 and NCT00884741) [260][261][260,262].
Up to now, TMZ is still commonly used for GBs with unmethylated MGMT promoters, due to the lack of significant benefits of alternative options (BEV plus irinotecan, dose-dense TMZ, BEV+SOC) [247][248][260][261][257,260,262,298]. Several preclinical studies demonstrated that O6-benzylguanine (O6BG) or O6-bromothenylguanine inactivate MGMT, but the addition of O6BG to radiation and BCNU treatment did not provide further benefit and instead increased toxicity [262][264].
3.2. Tumor-Treating Fields (TTFields)
TTFields is a non-invasive cancer treatment modality that applies low-intensity (0.7–3 V/cm), intermediate-frequency (100–500 kHz), and alternating electric fields over regions of the body where tumors are localized [263][264][308,309]. In growing GB cells, TTFields cause chromosome missegregation, disrupt DNA repair, inhibit mitosis and the cell cycle, and induce apoptosis and autophagy [265][266][267][268][269][270][271][272][310,311,312,313,314,315,316,317]. TTFields also interfere with the directionality of cancer migration by inducing changes in the organization and dynamics of microtubules and actin and ablate the primary cilia on GB cells that contribute to tumor growth and chemoresistance to TMZ [273][318]. TTFields also downregulate the expression levels of VEGF, HIF-1α, and matrix metalloproteinases (MMP2 and MMP9), which are necessary for tumor growth, invasion and metastasis [274][319]. TTFields also increase the membrane permeability of cancer cells and the BBB [275][276][320,321], which can help to increase the uptake and bioefficacy of chemotherapeutic drugs. Although this treatment modality reduces the viability of proliferating T cells [277][322], it also stimulates maturation and phagocytosis by dendritic cells (DCs) [278][323], increases CD8 T infiltration in TME [270][315], promotes the production of type I IFNs in GB cells in a cGAS/STING- and AIM2 inflammasome-dependent mechanism [279][324] and, thereby, facilitates the immune system response. Interestingly, the combination of hyperthermia and TTFields has shown synergistic effects in GB [280][325].
Over the past decade, TTFields have emerged as a complementary treatment strategy, which is now part of the SOC in GB treatment [238][281][282][283][289,326,327,328]. The FDA’s approval of rGB was based on the results from the EF-11 trial (NCT00379470) showing that TTFields monotherapy provided similar efficacy compared to the best physician’s choice chemotherapy in patients with rGB, albeit with better quality of life, less toxicity and a lower incidence of serious adverse events [284][329]. A randomized clinical trial in ndGB patients (NCT00916409) previously treated with chemoradiotherapy showed that patients treated with the TTFields and TMZ had a median free survival (mPFS) of 6.7 months compared to 4.0 months with TMZ alone. The addition of TTFields to the SOC therapy improved median OS (mOS) from 15.6 to 20.5 months without a negative influence on the health-related quality of life [13][285][286][13,330,331]. In ndGB, TTFields are applied within 6 weeks after the end of the radio-chemotherapy, ideally simultaneously with TMZ monotherapy [13][283][13,328]. Patients with compliance > 90% showed extended median and 5-year survival rates [287][256]. The most common adverse effect is skin irritation, occurring in 43% of patients (2% grade 3 or higher) [13][264][282][288][13,309,327,332], which is generally managed with array relocation and topical treatments including antibiotics and steroids. The frequency of systemic adverse events was 48% in the TTFields-TMZ group and 44% in the TMZ-alone group. Several limitations should be noted in the NCT00916409 trial: (a) only PF patients after the completion of chemoradiation were enrolled, which excluded those who were more likely to have a poor prognosis; (b) randomization in the EF-14 trial occurred over 2 months after diagnosis, which suggests a selection bias of patients who did not have progression after the initial treatment and would therefore likely have a better survival rate; (c) a “sham” device—to better discern a potential placebo-effect of wearing the device—was not used; (d) second-line therapies (chemotherapies, salvage radiation, radiosurgeries or craniotomies) after tumor progression in both groups were not reported while the TTFields plus TMZ group allowed patients to continue TTFields for up to 24 months or after the second GB progression; (e) molecular markers, such as the IDH1/2 status, were not performed [263][289][308,333]. Recently, recognized brain cancer experts concluded that TTFields plus TMZ represents a major advance in the field of GB therapy, though other experts maintain their skepticism regarding the use of the TTFields because of the lack of effect in some patients and because the time lengths required to reach (modest) benefits (at least 18 h per day) limit its utility [263][264][290][308,309,334].
Dexamethasone is administered to GB patients to alleviate cerebral edema and provide symptomatic relief. However, the corticoid-induced immunosuppressive effects may also increase infections and decrease survival [291][292][335,336]. In fact, a recent meta-analysis revealed that dexamethasone interferes with the therapeutic effects of TTFields [279][324]. The threshold dose at which dexamethasone can be used with minimal interactions with the TTFields was 4.1 mg per day or lower [293][337]. Several ongoing clinical trials are studying the optimal timing for TTFields administration (e.g., NCT04471844, NCT04492163, NCT03705351) and the safety and efficacy of the combination of TTFields with other cancer modalities [263][270][271][294][295][308,315,316,338,339]. For instance, PriCoTTFields is a phase I/II clinical trial that evaluates the safety and efficacy of TTFields initiated prior and concomitant to combined radiation and TMZ therapy in ndGB patients [296][340].
TTFields can reduce the DNA double-strand repair by downregulating the activity of the breast cancer type 1 susceptibility (BRCA1) signaling pathway, thereby increasing the sensitivity to the blockade of DNA repair caused by PARP inhibition [297][341]. Consequently, an ongoing phase II trial (NCT04221503) will try to determine whether niraparib (a PARP inhibitor) can enhance the effect of TTFields in GB patients (NCT04221503). In addition, the combination of TTFields, TMZ and lomustine has shown benefits in ndGB patients [298][342] and the triple combination of BEV, irinotecan, and TMZ plus TTFields improved the OS of patients with rGB [299][343]. Mechanisms involved in the acquisition of TTField resistance include activation of voltage-gated Ca2+ channels linked to cell migration [300][344]; CDK2NA deletion, mTOR (V2006I) mutations [301][345], and the upregulation of autophagy which can be reversed by combining TTFields with an autophagy inhibitor [267][312].
In clinical practice, TTFields are mainly used at a frequency of 200 kHz, but preclinical studies show that different GB cell lines respond to other optimal electric frequencies, as is the case of SF188 (400 kHz) or U87 (100 kHz) [302][346]. This phenomenon highlights the need for further investigation to individualize “TTFields prescription”. Despite the advances associated with the incorporation of TTFields to GB treatment, its clinical use is still quite restricted. EANO guidelines argue that the clinical benefit of TTFields has not been established yet, which contradicts the ASCO-NSO recommendations [215][303][253,347]. Certainly, price, regulation, the increase in the efficacy of combined treatments, and likely the development of novel intracranial electrodes, may assist in increasing the utilization and acceptance of TTFields [271][316].
3.3. Treatment in Special Patient Populations
Elderly patients (>65 years) or patients with a poor functional status have worse prognosis and are less tolerant to toxicities. Surgical resection is not associated with improved survival [304][348], but according to a recent retrospective single-center study, BCNU wafer implantation during the surgical resection is safe and improves mOS 39.0 months (≥12 implanted wafers) vs. 16.5 months (<12 implanted wafers) in patients in “extreme” neurosurgical conditions (>80 years and patients with preoperative Karnofsky Performance Status score < 50) [305][349]. Although just a few patients (6/49) reached that number of implants, these results are impressive, since mOS in the “extreme” conditions subgroup was 10.0 months, and there was a significant improvement in the postoperative KPS score compared to the preoperative KPS score.
The combination of TTFields with maintenance TMZ resulted in improved PFS and OS in ≥ 65-year-old patients with ndGB in the phase III EF-14 trial, without affecting patient quality of life [306][350]. Nevertheless, clinical trials have shown that standard radiotherapy is associated with poor outcomes, especially in patients older than 70 years [307][266]. Here, abbreviated courses of radiation therapy must be considered [307][308][266,267], although age alone should not represent the sole determining factor for the duration and intensity of the therapy [309][351]. Hypofractionated radiotherapy schedule (40 Gy delivered in 15) fractions and the addition of concurrent and adjuvant TMZ (NCT00482677) significantly increase survival (9.3 vs. 7.6 months, respectively) without impairing the quality of life [308][267]. Consequently, partial-brain fractionated radiotherapy with concurrent and adjuvant TMZ is the SOC for elderly patients with good performance status [310][311][352,353]. The addition of BEV to radiotherapy had no benefits in elderly patients [312][354].
A single modality therapy can be considered for patients with poor functional status. RT was more effective than TMZ for unmethylated MGMT-promoter tumors, whereas TMZ was more effective than RT for methylated MGMT-promoter tumors [307][308][266,267].
3.4. Options of Treatment in rGB patients
Regardless of the use of multimodality treatments, GB invariably returns after a median interval of less than 10 months, and typically even sooner (≈6 months) in older patients [308][267]. The genetic and biological changes induced by radiotherapy and/or cytotoxic chemotherapy differentiate rGB from primary tumors. These changes empower GB tumors to navigate the host microenvironment, evade the immune system, and foster intrinsic and acquired resistance to further administration of radiation and/or alkylating agents. Upon recurrence, patients typically exhibit a poor performance status and compromised overall health, with GB tumors often being unresectable, thus requiring substantial use of corticosteroids to manage cerebral edema [313][355]. This makes rGB prognosis much worse than that of the primary GB.
Actually, although there is no clear SOC salvage therapy for rGB [238][289], patients who received no salvage treatment had poorer survival than those who received radiation and/or chemotherapy [314][356]. Therefore, re-resection, re-irradiation and systemic chemotherapy with TMZ rechallenge, nitrosoureas, BEV, and TTFields or clinical trial enrolment to test experimental drugs are considered for all recurrent patients [244][315][316][317][318][295,357,358,359,360]. Unfortunately, fewer than 43% of rGB patients were fit enough to be included in clinical trials [319][361].
Consensus guidelines for selecting candidates for second surgery recommend that patients need to have a good performance status, particularly if more than 6 months have elapsed since the initial surgery [315][320][357,362]. According to a retrospective review of the brain tumor database (1997–2016), stereotactic radiosurgery is associated with longer OS and/or PFS in rGB patients with good performance status and small-volume tumor recurrences [321][363]. In practice, not more than 20–30% of relapsed patients are eligible and only complete resections have any survival benefit (11–17 months) [322][323][364,365]. Toxicity to normal brain parenchyma limits re-irradiation in rGB [324][366]. Radiosurgery or hypofractionated radiotherapy (30–35 Gy in 5–15 fractions) is considered a potentially effective option and is increasingly used for younger patients with good performance status [314][325][356,367]. Data from a few prospective studies in rGB suggest that re-irradiation modestly improves PFS compared with systemic treatment alone [314][356]. OS after re-irradiation (9.7 months) was sufficient to justify this treatment [325][326][367,368], but marginal recurrence is significantly more frequent in patients who had prior BEV exposure [327][369].
Lomustine has become the SOC at relapse in Europe, with thrombocytopenia being the most frequent limiting toxicity [316][358]. Lomustine is generally preferred to other nitrosoureas given its oral formulation, schedule of administration, and better safety profile. However, lomustine activity is largely restricted to patients with tumors with MGMT promoter methylation and its survival benefit has been found limited: objective response rate was around 10%, mPFS < 2 months, PFS6 was 20%, and OS was 6–9 months [316][328][358,370].
One of the most significant features of GB is its hypervascularization, mainly promoted by the hypoxia-facilitated VEGF overexpression in tumor and stromal cells [329][371]. BEV is an anti-VEGF humanized monoclonal antibody that inhibits tumor-driven angiogenesis and may help in reducing patients’ immune suppression [196][330][331][234,372,373]. rGB with a low apparent diffusion coefficient, large tumor burden, or IDH mutation is more likely to benefit from BEV treatment [332][374]. BEV gained approval in 2009 for rGB treatment in the US and later in other countries, but BEV is not approved by the EMA as an SOC for rGB [333][334][375,376]. BEV has shown promise in extending PFS treating GB, but there is no evidence for its ability to prolong OS [335][336][337][338][339][377,378,379,380,381]. The anti-angiogenic effect of BEV decreases contrast uptake during MRI, which can lead to false negatives in recurrences [328][370]. Despite this, BEV is almost used due to the lack of alternative treatment options, and because it also serves to control brain vasogenic edema [238][333][289,375] and to avoid the need for corticoid treatment [340][341][382,383]. BEV combined with re-irradiation was found to be safe and tolerable and showed a significant reduction in the incidence of radiation necrosis, patient dependence on corticosteroids and improvement in the Karnofsky score during disease progression-free periods. Survival benefits (10.1 months) have been reported following fractionated stereotactic radiotherapy (35 Gy/10 fractions) and concurrent BEV in a prospective randomized phase II trial [342][343][384,385]. A recent retrospective study showed that BEV combined with re-irradiation improved mPFS and mOS to 8 and 13.6 months, respectively [317][359]. Nevertheless, the validity of these results is constrained by the inclusion of a small number of patients, the heterogeneity of treatment options, and the absence of a control group. Despite these limitations, recent conclusions drawn from a meta-analysis endorse the benefits of this therapeutic option [344][386]. Lomustine plus BEV for rGB (phase II, NCT01290939) somewhat prolonged PFS but did not confer a survival advantage over treatment with lomustine alone [345][387]. Although earlier reports suggested that BEV had glucocorticoid-sparing effects, in this trial, the addition of BEV did not reduce the use of glucocorticoids [345][387].
TTFields did not increase OS (phase III, NCT00379470) but showed efficacy equivalent to chemotherapy commonly used for rGB, with lower toxicity and improved quality of life [284][329]. According to a recent phase II trial (NCT01894061), the combination of BEV and TTFields is safe and has clinical efficacy in rGB [346][388].
4. Targeted Therapies
Genetic changes that have been well recognized in GB cells include alterations in the Rb/p16 pathway (> 90%), loss of heterozygosity of 10q (70%), EGFR amplification or mutation (≈50%), TP53 mutations (31%), PDGF receptor gain/amplification (≈25%), mouse double minute homolog 2 (MDM2) gene mutations (10–15%) and the phosphatase and tensin homolog (PTEN) gene mutations (20–34%) [16][347][16,389]. Analysis of the large-scale molecular and genomic information present in the Cancer Genome Atlas Program (TCGA) database indicated that p53 pathway (TP53/MDM2/P14arfç), the PI3K/Akt/mTOR pathway, and the RB pathway (CDK4/RB1/P16ink4) are the main signaling pathways involved in GB tumorigenesis, pathophysiology and acquisition of resistance to treatment [16][348][349][16,390,391]. Intrinsically targeting these altered molecules and pathways was seen as a novel avenue in GB treatment. Unfortunately, despite research efforts and clinical trials, except for prolonged PFS afforded by the BEV, no pharmacological intervention has been demonstrated to alter the course of disease [350][351][392,393].
5. Immunotherapies
Recent studies show the presence of a variety of immune cell types within the GB TME with a dominance of immunosuppressive cells, i.e., MDSCs, microglia, M2 macrophages, FoxP3+ regulatory T cells (Tregs), and antigen-presenting cells (APCs) (including DCs and bone-marrow-derived macrophages). The presence of M2 macrophages is linked to an increased GB aggressiveness and plays a pivotal role in the acquisition of chemo and radioresistance of GB cells [352][353][653,654]. In addition, frequently, CD4+ and CD8+ T cells are functionally deficient, inactivated, or exhausted, often co-expressing immune checkpoint molecules, i.e., programmed cell death receptor 1 (PD-1), lymphocyte activation gene 3 (LAG3) and T cell immunoglobulin mucin 3 (TIM-3) [354][655]. GB cells secrete immunosuppressive factors such as TGFβ2, PGE-2, IL-1, IL-10 and indoleamine 2,3-dioxygenase (IDO), which work cooperatively to suppress the activity of effector cells and to evading the anti-tumor immune response [355][356][516,656]. As described here below, a plethora of novel immunotherapies, i.e., checkpoint inhibitors (ICIs), vaccines, T-cell-based immunotherapies, NK-cell-based therapies, viral therapies, and combined treatments, have been attempted in order to control GB expansion and/or recurrence.