 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yingbin Fu | -- | 2891 | 2024-03-11 16:43:27 | | | |
| 2 | Mona Zou | + 3 word(s) | 2894 | 2024-03-12 09:25:34 | | | | |
| 3 | Mona Zou | + 1 word(s) | 2895 | 2024-03-18 09:53:00 | | |
Video Upload Options
Despite extensive use of intravitreal anti-vascular endothelial growth factor (anti-VEGF) biologics for over a decade, neovascular age-related macular degeneration (nAMD) or choroidal neovascularization (CNV) continues to be a major cause of irreversible vision loss in developed countries. Many nAMD patients demonstrate persistent disease activity or experience declining responses over time despite anti-VEGF treatment. The underlying mechanisms of anti-VEGF resistance are poorly understood, and no effective treatment strategies are available to date. Emerging strong evidence from animal models and clinical studies supports the roles of neovascular remodeling and arteriolar CNV formation in anti-VEGF resistance. Cholesterol dysregulation, inflammation, and ensuing macrophage activation are critically involved in arteriolar CNV formation and anti-VEGF resistance. Combination therapy by neutralizing VEGF and enhancing cholesterol removal from macrophages is a promising strategy to combat anti-VEGF resistance in CNV.
1. Limitation of Anti-VEGF Therapies
2. Animal models of anti-VEGF resistance.
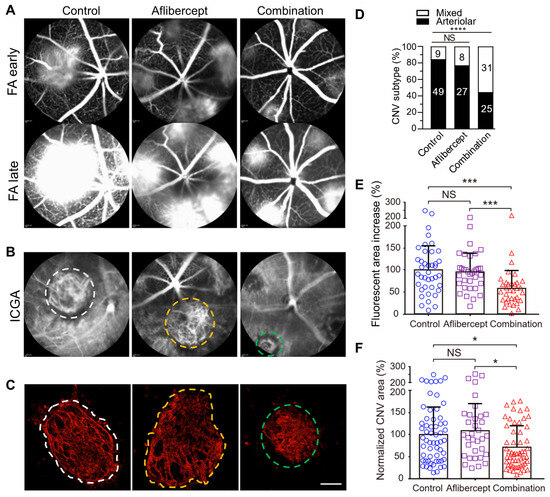
Multiple pivotal clinical trials (ANCHOR, MARINA, CATT) have shown that patients of advanced age with larger baseline CNV lesions are less responsive to anti-VEGF treatment and have worse outcomes [13][35][36][37]. Importantly, anti-VEGF resistance in CNV patients is frequently associated with arteriolar CNV, characterized by large-caliber branching arterioles, vascular loops, and anastomotic connections. The persistence of fluid leakage in arteriolar CNV likely results from increased exudation through poorly formed tight junctions at arteriovenous anastomotic loops, particularly during periods of elevated blood flow. In contrast, individuals responding well to anti-VEGF treatment typically exhibit capillary CNV, where VEGF-mediated permeability is the primary cause of leakage. Moreover, recurrent anti-VEGF treatment can induce vessel abnormalization, arteriolar CNV formation, and ultimately contribute to anti-VEGF resistance [14][38], suggesting a mechanism for acquired anti-VEGF resistance.
The researchers found that laser photocoagulation produces larger CNV lesions in aged mice that are markedly more resistant to anti-VEGF treatment compared with young mice [39][40][41]. Importantly, laser-induced CNV in young and old mice, respectively, mimics capillary and arteriolar CNV [9][40]. The researchers propose that laser-induced CNV in aged mice is a clinically relevant model of anti-VEGF resistance [39][40].
3. Capillary CNV versus Arteriolar CNV
4. Role of Macrophages in Anti-VEGF Resistance
5. Treatment Strategies for Anti-VEGF Resistance by Simultaneously Targeting Capillary and Arteriolar CNV
6. How does the combination therapy compare with anti-VEGF gene therapy and higher dose anti-VEGF regimen currently in development?
AMD is a complex multi-factorial disease. It is unrealistic to expect that targeting one factor or one pathway will solve all the problems. The anti-VEGF gene therapy and higher dose regimen that are currently in development target VEGF-dependent angiogenesis without targeting arteriogenesis, which are unlikely to resolve resistance (see Discussion regarding high dose regimen in 1. Limitation of anti-VEGF therapies). In the HARBOR trial, high dose ranibizumab (2.0 mg) did not increase efficacy in treatment-naïve patients [97]. In the recently completed PULSAR trial, 8 mg aflibercept sustained improvements of visual acuity and retinal anatomy at 22 months with 36% fewer injections relative to the standard 2-mg dose, suggesting the potential to reduce treatment burdens. However, there is no evidence that the high-dose aflibercept eliminates anti-VEGF resistance. Rather, there is evidence that unbalanced treatments targeting VEGF-dependent angiogenesis alone can cause vessel abnormalization, arteriolar CNV formation, and anti-VEGF resistance [14][38] (Figure 2). Combination therapy has an advantage by targeting both angiogenesis and arteriogenesis.
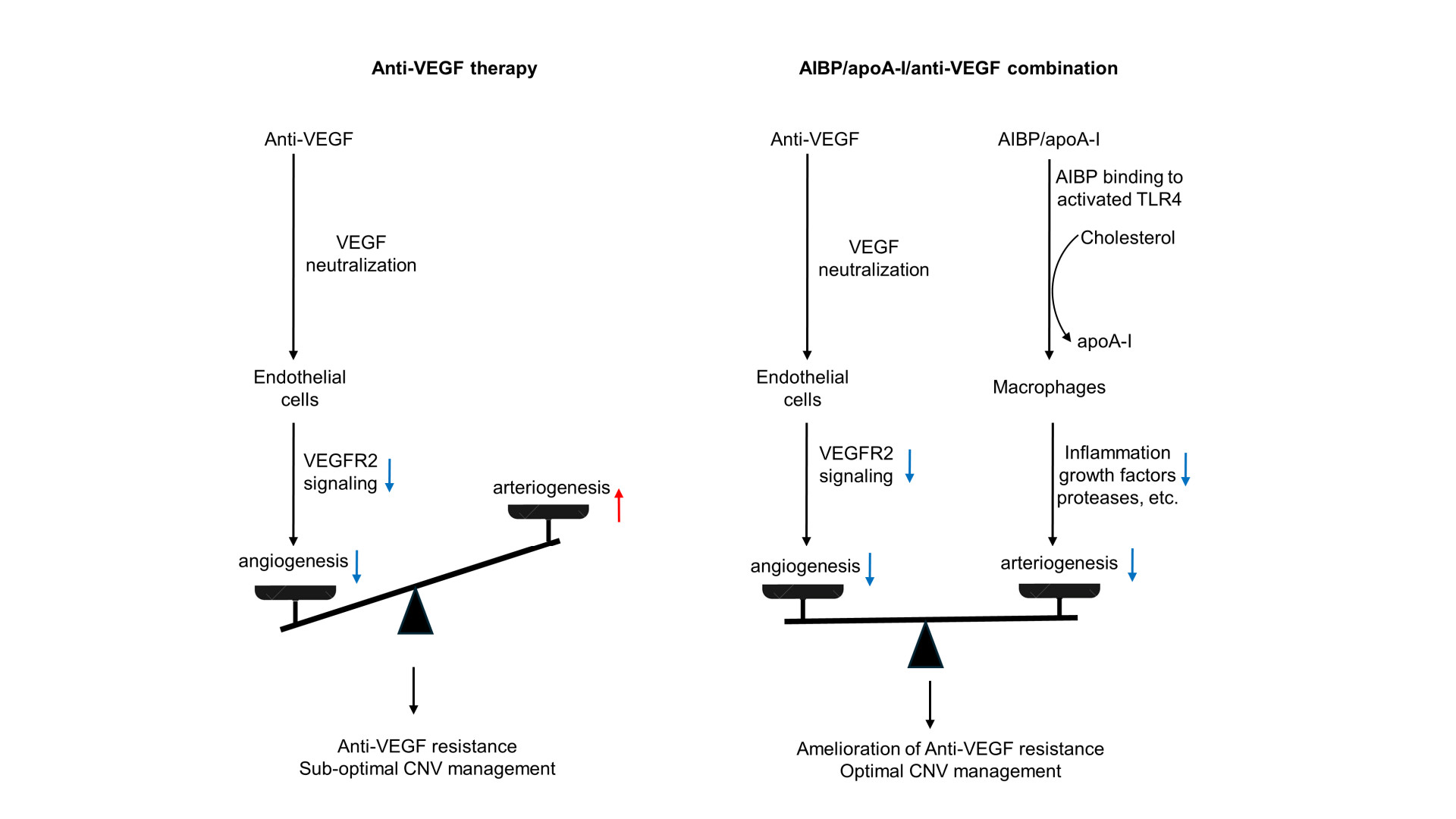
Figure 2. Comparison of anti-VEGF monotherapy with AIBP/apoA-I/anti-VEGF combination therapy in the treatment of CNV. Anti-VEGF therapies neutralize VEGF, inhibit VEGFR2 signaling in endothelial cells, and thereby inhibit angiogenesis and capillary CNV. However, this treatment results in unchecked arteriogenesis, vessel abnormalization, and arteriolar CNV formation, leading to anti-VEGF resistance and sub-optimal CNV management. In AIBP/apoA-I/anti-VEGF combination therapy, AIBP binds to activated TLR4 and augments cholesterol efflux from macrophages and microglia to apoA-I, normalizing plasma lipid rafts and suppressing inflammation, which inhibits arteriolar CNV. Simultaneously, anti-VEGF therapies inhibit VEGFR2 signaling in endothelial cells, thereby suppressing angiogenesis and capillary CNV. Thus, the combination therapy leads to the amelioration of anti-VEGF resistance and optimal CNV management.
7. Perspectives
Because long-term efficacy of anti-VEGF therapy is suboptimal and repeated anti-VEGF treatment can lead to arteriolar CNV and anti-VEGF resistance [14][38], The researchers predict that combination therapy with AIBP/apoA-I/anti-VEGF not only overcomes anti-VEGF resistance for monotherapy non-responders, but also improves therapeutic efficacy at all levels of anti-VEGF response in the treatment of nAMD. To our knowledge, there is no treatment available for arteriolar CNV. Combination therapy has the potential to replace current anti-VEGF monotherapies and become a new first-line therapy. The global anti-VEGF therapeutics market size was valued at USD 12.3 billion in 2022 and is estimated to reach USD 13.7 billion by 2031, representing a significant portion of global healthcare cost. The researchers' objective is to generate preclinical efficacy and safety data to support an Investigational New Drug (IND) application for AIBP/apoA-I/aflibercept therapy and advance to a first-in-human Phase I clinical trial that will ultimately benefit a wide range of nAMD patients including anti-VEGF non-responders and responders with sub-optimal long-term efficacy.
References
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.G.; Klein, R.; Cheng, C.-Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116.
- Bressler, N.M.; Bressler, S.B.; Fine, S.L. Age-related macular degeneration. Surv. Ophthalmol. 1988, 32, 375–413.
- Comparison of Age-related Macular Degeneration Treatments Trials (CATT) Research Group; Maguire, M.G.; Martin, D.F.; Ying, G.-S.; Jaffe, G.J.; Daniel, E.; Grunwald, J.E.; Toth, C.A.; Ferris, F.L.; Fine, S.L. Five-Year Outcomes with Anti-Vascular Endothelial Growth Factor Treatment of Neovascular Age-Related Macular Degeneration: The Comparison of Age-Related Macular Degeneration Treatments Trials. Ophthalmology 2016, 123, 1751–1761.
- Yang, S.; Zhao, J.; Sun, X. Resistance to anti-VEGF therapy in neovascular age-related macular degeneration: A comprehensive review. Drug Des. Dev. Ther. 2016, 10, 1857–1867.
- Ehlken, C.; Jungmann, S.; Böhringer, D.; Agostini, H.T.; Junker, B.; Pielen, A. Switch of anti-VEGF agents is an option for nonresponders in the treatment of AMD. Eye 2014, 28, 538–545.
- Heier, J.S.; Brown, D.M.; Chong, V.; Korobelnik, J.-F.; Kaiser, P.K.; Nguyen, Q.D.; Kirchhof, B.; Ho, A.; Ogura, Y.; Yancopoulos, G.D.; et al. Intravitreal aflibercept (VEGF trap-eye) in wet age-related macular degeneration. Ophthalmology 2012, 119, 2537–2548.
- Rofagha, S.; Bhisitkul, R.B.; Boyer, D.S.; Sadda, S.R.; Zhang, K. Seven-year outcomes in ranibizumab-treated patients in ANCHOR, MARINA, and HORIZON: A multicenter cohort study (SEVEN-UP). Ophthalmology 2013, 120, 2292–2299.
- Krebs, I.; Glittenberg, C.; Ansari-Shahrezaei, S.; Hagen, S.; Steiner, I.; Binder, S. Non-responders to treatment with antagonists of vascular endothelial growth factor in age-related macular degeneration. Br. J. Ophthalmol. 2013, 97, 1443–1446.
- Mettu, P.S.; Allingham, M.J.; Cousins, S.W. Incomplete response to Anti-VEGF therapy in neovascular AMD: Exploring disease mechanisms and therapeutic opportunities. Prog. Retin. Eye Res. 2021, 82, 100906.
- Otsuji, T.; Nagai, Y.; Sho, K.; Tsumura, A.; Koike, N.; Tsuda, M.; Nishimura, T.; Takahashi, K. Initial non-responders to ranibizumab in the treatment of age-related macular degeneration (AMD). Clin. Ophthalmol. 2013, 7, 1487–1490.
- Cobos, E.; Recalde, S.; Anter, J.; Hernandez-Sanchez, M.; Barreales, C.; Olavarrieta, L.; Valverde, A.; Suarez-Figueroa, M.; Cruz, F.; Abraldes, M.; et al. Association between CFH, CFB, ARMS2, SERPINF1, VEGFR1 and VEGF polymorphisms and anatomical and functional response to ranibizumab treatment in neovascular age-related macular degeneration. Acta Ophthalmol. 2018, 96, e201–e212.
- Kitchens, J.W.; Kassem, N.; Wood, W.; Stone, T.W.; Isernhagen, R.; Wood, E.; Hancock, B.A.; Radovich, M.; Waymire, J.; Li, L.; et al. A pharmacogenetics study to predict outcome in patients receiving anti-VEGF therapy in age related macular degeneration. Clin. Ophthalmol. 2013, 7, 1987–1993.
- Rosenfeld, P.J.; Shapiro, H.; Tuomi, L.; Webster, M.; Elledge, J.; Blodi, B. Characteristics of Patients Losing Vision after 2 Years of Monthly Dosing in the Phase III Ranibizumab Clinical Trials. Ophthalmology 2011, 118, 523–530.
- Spaide, R.F. Optical Coherence Tomography Angiography Signs of Vascular Abnormalization With Antiangiogenic Therapy for Choroidal Neovascularization. Am. J. Ophthalmol. 2015, 160, 6–16.
- Sharma, D.; Zachary, I.; Jia, H. Mechanisms of Acquired Resistance to Anti-VEGF Therapy for Neovascular Eye Diseases. Investig. Ophthalmol. Vis. Sci. 2023, 64, 28.
- Zuber-Laskawiec, K.; Kubicka-Trzaska, A.; Karska-Basta, I.; Pociej-Marciak, W.; Romanowska-Dixon, B. Non-responsiveness and tachyphylaxis to anti-vascular endothelial growth factor treatment in naive patients with exudative age-related macular degeneration. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2019, 70, 779–785.
- Hara, C.; Wakabayashi, T.; Fukushima, Y.; Sayanagi, K.; Kawasaki, R.; Sato, S.; Sakaguchi, H.; Nishida, K. Tachyphylaxis during treatment of exudative age-related macular degeneration with aflibercept. Graefe’s Arch. Clin. Exp. Ophthalmol. 2019, 257, 2559–2569.
- Schaal, S.; Kaplan, H.J.; Tezel, T.H. Is there tachyphylaxis to intravitreal anti-vascular endothelial growth factor pharmacotherapy in age-related macular degeneration? Ophthalmology 2008, 115, 2199–2205.
- Forooghian, F.; Cukras, C.; Meyerle, C.B.; Chew, E.Y.; Wong, W.T. Tachyphylaxis after intravitreal bevacizumab for exudative age-related macular degeneration. Retina 2009, 29, 723–731.
- Hwang, R.Y.; Santos, D.; Oliver, S.C.N. Rates of exudative recurrence for eyes with inactivated wet age-related macular degeneration on 12-week interval dosing with bevacizumab therapy. Retina 2020, 40, 679–685.
- Kuroda, Y.; Yamashiro, K.; Miyake, M.; Yoshikawa, M.; Nakanishi, H.; Oishi, A.; Tamura, H.; Ooto, S.; Tsujikawa, A.; Yoshimura, N. Factors Associated with Recurrence of Age-Related Macular Degeneration after Anti-Vascular Endothelial Growth Factor Treatment: A Retrospective Cohort Study. Ophthalmology 2015, 122, 2303–2310.
- Kim, J.H.; Chang, Y.S.; Kim, J.W.; Kim, C.G.; Lee, D.W. recurrence in patients with type 3 neovascularization (retinal angiomatous proliferation) after intravitreal ranibizumab. Retina 2017, 37, 1508–1515.
- You, Q.S.; Gaber, R.; Meshi, A.; Ramkumar, H.L.; Alam, M.; Muftuoglu, I.K.; Freeman, W.R. high-dose high-frequency aflibercept for recalcitrant neovascular age-related macular degeneration. Retina 2018, 38, 1156–1165.
- Brown, D.M.; Chen, E.; Mariani, A.; Major, J.C.; SAVE Study Group. Super-dose anti-VEGF (SAVE) trial: 2.0 mg intravitreal ranibizumab for recalcitrant neovascular macular degeneration-primary end point. Ophthalmology 2013, 120, 349–354.
- Fung, A.T.; Kumar, N.; Vance, S.K.; Slakter, J.S.; Klancnik, J.M.; Spaide, R.S.; Freund, K.B. Pilot study to evaluate the role of high-dose ranibizumab 2.0 mg in the management of neovascular age-related macular degeneration in patients with persistent/recurrent macular fluid <30 days following treatment with intravitreal anti-VEGF therapy (the LAST Study). Eye 2012, 26, 1181–1187.
- Chang, A.A.; Li, H.; Broadhead, G.K.; Hong, T.; Schlub, T.E.; Wijeyakumar, W.; Zhu, M. Intravitreal aflibercept for treatment-resistant neovascular age-related macular degeneration. Ophthalmology 2014, 121, 188–192.
- Marquis, L.-M.; Mantel, I. Beneficial switch from aflibercept to ranibizumab for the treatment of refractory neovascular age-related macular degeneration. Graefe’s Arch. Clin. Exp. Ophthalmol. 2020, 258, 1591–1596.
- Spooner, K.; Hong, T.; Nair, R.; Chow, N.C.C.; Broadhead, G.K.; Wijeyakumar, W.; Chang, A.A. Long-term outcomes of switching to aflibercept for treatment-resistant neovascular age-related macular degeneration. Acta Ophthalmol. 2019, 97, e706–e712.
- Broadhead, G.K.; Keenan, T.D.L.; Chew, E.Y.; Wiley, H.E.; Cukras, C.A. Comparison of agents using higher dose anti-VEGF therapy for treatment-resistant neovascular age-related macular degeneration. Graefe’s Arch. Clin. Exp. Ophthalmol. 2022, 260, 2239–2247.
- Dunn, E.N.; Hariprasad, S.M.; Sheth, V.S. An Overview of the Fovista and Rinucumab Trials and the Fate of Anti-PDGF Medications. Ophthalmic Surg. Lasers Imaging Retina 2017, 48, 100–104.
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264.
- Heier, J.S.; Khanani, A.M.; Ruiz, C.Q.; Basu, K.; Ferrone, P.J.; Brittain, C.; Figueroa, M.S.; Lin, H.; Holz, F.G.; Patel, V.; et al. Efficacy, durability, and safety of intravitreal faricimab up to every 16 weeks for neovascular age-related macular degeneration (TENAYA and LUCERNE): Two randomised, double-masked, phase 3, non-inferiority trials. Lancet 2022, 399, 729–740.
- Lange, C.; Tetzner, R.; Strunz, T.; Rittenhouse, K.D. Aflibercept Suppression of Angiopoietin-2 in a Rabbit Retinal Vascular Hyperpermeability Model. Transl. Vis. Sci. Technol. 2023, 12, 17.
- Regula, J.T.; Lundh von Leithner, P.; Foxton, R.; Barathi, V.A.; Gemmy Cheung, C.M.; Bo Tun, S.B.; Wey, Y.S.; Iwata, D.; Dostalek, M.; Moelleken, J.; et al. Targeting key angiogenic pathways with a bispecific CrossMAb optimized for neovascular eye diseases. EMBO Mol. Med. 2017, 9, 985.
- Kaiser, P.K.; Brown, D.M.; Zhang, K.; Hudson, H.L.; Holz, F.G.; Shapiro, H.; Schneider, S.; Acharya, N.R. Ranibizumab for predominantly classic neovascular age-related macular degeneration: Subgroup analysis of first-year ANCHOR results. Am. J. Ophthalmol. 2007, 144, 850–857.
- Finger, R.P.; Wickremasinghe, S.S.; Baird, P.N.; Guymer, R.H. Predictors of anti-VEGF treatment response in neovascular age-related macular degeneration. Surv. Ophthalmol. 2014, 59, 1–18.
- Boyer, D.S.; Antoszyk, A.N.; Awh, C.C.; Bhisitkul, R.B.; Shapiro, H.; Acharya, N.R. Subgroup analysis of the MARINA study of ranibizumab in neovascular age-related macular degeneration. Ophthalmology 2007, 114, 246–252.
- Lumbroso, B.; Rispoli, M.; Savastano, M.C.; Jia, Y.; Tan, O.; Huang, D. Optical Coherence Tomography Angiography Study of Choroidal Neovascularization Early Response after Treatment. Dev. Ophthalmol. 2016, 56, 77–85.
- Zhu, L.; Parker, M.; Enemchukwu, N.; Shen, M.; Zhang, G.; Yan, Q.; Handa, J.T.; Fang, L.; Fu, Y. Combination of apolipoprotein-A-I/apolipoprotein-A-I binding protein and anti-VEGF treatment overcomes anti-VEGF resistance in choroidal neovascularization in mice. Commun. Biol. 2020, 3, 386.
- Zhang, Z.; Shen, M.M.; Fu, Y. Combination of AIBP, apoA-I, and Aflibercept Overcomes Anti-VEGF Resistance in Neovascular AMD by Inhibiting Arteriolar Choroidal Neovascularization. Investig. Ophthalmol. Vis. Sci. 2022, 63, 2.
- Attarde, A.; Riad, T.S.; Zhang, Z.; Ahir, M.; Fu, Y. Characterization of Vascular Morphology of Neovascular Age-Related Macular Degeneration by Indocyanine Green Angiography. JoVE J. Vis. Exp. 2023, 2023, e65682.
- Adams, R.H.; Alitalo, K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 2007, 8, 464–478.
- Patel-Hett, S.; D’Amore, P.A. Signal transduction in vasculogenesis and developmental angiogenesis. Int. J. Dev. Biol. 2011, 55, 353–363.
- Grossniklaus, H.E.; Kang, S.J.; Berglin, L. Animal models of choroidal and retinal neovascularization. Prog Retin Eye Res 2010, 29, 500–519.
- Campochiaro, P.A. Molecular pathogenesis of retinal and choroidal vascular diseases. Prog. Retin. Eye Res. 2015, 49, 67–81.
- Carmeliet, P. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 2000, 6, 389–395.
- Buschmann, I.; Schaper, W. Arteriogenesis Versus Angiogenesis: Two Mechanisms of Vessel Growth. Physiology 1999, 14, 121–125.
- Deindl, E.; Buschmann, I.; Hoefer, I.E.; Podzuweit, T.; Boengler, K.; Vogel, S.; van Royen, N.; Fernandez, B.; Schaper, W. Role of ischemia and of hypoxia-inducible genes in arteriogenesis after femoral artery occlusion in the rabbit. Circ. Res. 2001, 89, 779–786.
- Schierling, W.; Troidl, K.; Troidl, C.; Schmitz-Rixen, T.; Schaper, W.; Eitenmüller, I.K. The role of angiogenic growth factors in arteriogenesis. J. Vasc. Res. 2009, 46, 365–374.
- Wu, S.; Wu, X.; Zhu, W.; Cai, W.-J.; Schaper, J.; Schaper, W. Immunohistochemical study of the growth factors, aFGF, bFGF, PDGF-AB, VEGF-A and its receptor (Flk-1) during arteriogenesis. Mol. Cell. Biochem. 2010, 343, 223–229.
- Helisch, A.; Schaper, W. Arteriogenesis: The development and growth of collateral arteries. Microcirculation 2003, 10, 83–97.
- Heil, M.; Ziegelhoeffer, T.; Pipp, F.; Kostin, S.; Martin, S.; Clauss, M.; Schaper, W. Blood monocyte concentration is critical for enhancement of collateral artery growth. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H2411–H2419.
- la Sala, A.; Pontecorvo, L.; Agresta, A.; Rosano, G.; Stabile, E. Regulation of collateral blood vessel development by the innate and adaptive immune system. Trends Mol. Med. 2012, 18, 494–501.
- Tatar, O.; Yoeruek, E.; Szurman, P.; Bartz-Schmidt, K.U.; Tübingen Bevacizumab Study Group; Adam, A.; Shinoda, K.; Eckardt, C.; Boeyden, V.; Claes, C.; et al. Effect of bevacizumab on inflammation and proliferation in human choroidal neovascularization. Arch. Ophthalmol. 2008, 126, 782–790.
- Subhi, Y.; Krogh Nielsen, M.; Molbech, C.R.; Krüger Falk, M.; Singh, A.; Hviid, T.V.F.; Nissen, M.H.; Sørensen, T.L. Association of CD11b+ Monocytes and Anti-Vascular Endothelial Growth Factor Injections in Treatment of Neovascular Age-Related Macular Degeneration and Polypoidal Choroidal Vasculopathy. JAMA Ophthalmol. 2019, 137, 515–522.
- Heil, M.; Eitenmüller, I.; Schmitz-Rixen, T.; Schaper, W. Arteriogenesis versus angiogenesis: Similarities and differences. J. Cell. Mol. Med. 2006, 10, 45–55.
- Arras, M.; Ito, W.D.; Scholz, D.; Winkler, B.; Schaper, J.; Schaper, W. Monocyte activation in angiogenesis and collateral growth in the rabbit hindlimb. J. Clin. Investig. 1998, 101, 40–50.
- McLeod, D.S.; Bhutto, I.; Edwards, M.M.; Silver, R.E.; Seddon, J.M.; Lutty, G.A. Distribution and Quantification of Choroidal Macrophages in Human Eyes With Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5843–5855.
- Li, M.; Dolz-Marco, R.; Messinger, J.D.; Wang, L.; Feist, R.M.; Girkin, C.A.; Gattoussi, S.; Ferrara, D.; Curcio, C.A.; Freund, K.B. Clinicopathologic Correlation of Anti-Vascular Endothelial Growth Factor-Treated Type 3 Neovascularization in Age-Related Macular Degeneration. Ophthalmology 2018, 125, 276–287.
- Pang, C.E.; Messinger, J.D.; Zanzottera, E.C.; Freund, K.B.; Curcio, C.A. The Onion Sign in Neovascular Age-Related Macular Degeneration Represents Cholesterol Crystals. Ophthalmology 2015, 122, 2316–2326.
- Kamei, M.; Yoneda, K.; Kume, N.; Suzuki, M.; Itabe, H.; Matsuda, K.-I.; Shimaoka, T.; Minami, M.; Yonehara, S.; Kita, T.; et al. Scavenger receptors for oxidized lipoprotein in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1801–1807.
- Lin, J.B.; Sene, A.; Santeford, A.; Fujiwara, H.; Sidhu, R.; Ligon, M.M.; Shankar, V.A.; Ban, N.; Mysorekar, I.U.; Ory, D.S.; et al. Oxysterol Signatures Distinguish Age-Related Macular Degeneration from Physiologic Aging. EBioMedicine 2018, 32, 9–20.
- Sene, A.; Khan, A.A.; Cox, D.; Nakamura, R.E.I.; Santeford, A.; Kim, B.M.; Sidhu, R.; Onken, M.D.; Harbour, J.W.; Hagbi-Levi, S.; et al. Impaired cholesterol efflux in senescent macrophages promotes age-related macular degeneration. Cell Metab. 2013, 17, 549–561.
- Neale, B.M.; Fagerness, J.; Reynolds, R.; Sobrin, L.; Parker, M.; Raychaudhuri, S.; Tan, P.L.; Oh, E.C.; Merriam, J.E.; Souied, E.; et al. Genome-wide association study of advanced age-related macular degeneration identifies a role of the hepatic lipase gene (LIPC). Proc. Natl. Acad. Sci. USA 2010, 107, 7395–7400.
- Sarks, J.P.; Sarks, S.H.; Killingsworth, M.C. Morphology of early choroidal neovascularisation in age-related macular degeneration: Correlation with activity. Eye 1997, 11 Pt 4, 515–522.
- Espinosa-Heidmann, D.G.; Suner, I.J.; Hernandez, E.P.; Monroy, D.; Csaky, K.G.; Cousins, S.W. Macrophage depletion diminishes lesion size and severity in experimental choroidal neovascularization. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3586–3592.
- Sakurai, E.; Anand, A.; Ambati, B.K.; van Rooijen, N.; Ambati, J. Macrophage depletion inhibits experimental choroidal neovascularization. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3578–3585.
- Ishida, S.; Usui, T.; Yamashiro, K.; Kaji, Y.; Amano, S.; Ogura, Y.; Hida, T.; Oguchi, Y.; Ambati, J.; Miller, J.W.; et al. VEGF164-mediated inflammation is required for pathological, but not physiological, ischemia-induced retinal neovascularization. J. Exp. Med. 2003, 198, 483–489.
- Nagineni, C.N.; Kommineni, V.K.; William, A.; Detrick, B.; Hooks, J.J. Regulation of VEGF expression in human retinal cells by cytokines: Implications for the role of inflammation in age-related macular degeneration. J. Cell. Physiol. 2012, 227, 116–126.
- Apte, R.S.; Richter, J.; Herndon, J.; Ferguson, T.A. Macrophages inhibit neovascularization in a murine model of age-related macular degeneration. PLoS Med. 2006, 3, e310.
- Cherepanoff, S.; McMenamin, P.; Gillies, M.C.; Kettle, E.; Sarks, S.H. Bruch’s membrane and choroidal macrophages in early and advanced age-related macular degeneration. Br. J. Ophthalmol. 2010, 94, 918–925.
- Grossniklaus, H.E.; Ling, J.X.; Wallace, T.M.; Dithmar, S.; Lawson, D.H.; Cohen, C.; Elner, V.M.; Elner, S.G.; Sternberg, P. Macrophage and retinal pigment epithelium expression of angiogenic cytokines in choroidal neovascularization. Mol. Vis. 2002, 8, 119–126.
- Hagbi-Levi, S.; Grunin, M.; Jaouni, T.; Tiosano, L.; Rinsky, B.; Elbaz-Hayoun, S.; Peled, A.; Chowers, I. Proangiogenic characteristics of activated macrophages from patients with age-related macular degeneration. Neurobiol. Aging 2017, 51, 71–82.
- Killingsworth, M.C.; Sarks, J.P.; Sarks, S.H. Macrophages related to Bruch’s membrane in age-related macular degeneration. Eye Lond. Engl. 1990, 4 Pt 4, 613–621.
- Lopez, P.F.; Lambert, H.M.; Grossniklaus, H.E.; Sternberg, P. Well-defined subfoveal choroidal neovascular membranes in age-related macular degeneration. Ophthalmology 1993, 100, 415–422.
- Campa, C.; Costagliola, C.; Incorvaia, C.; Sheridan, C.; Semeraro, F.; De Nadai, K.; Sebastiani, A.; Parmeggiani, F. Inflammatory mediators and angiogenic factors in choroidal neovascularization: Pathogenetic interactions and therapeutic implications. Mediat. Inflamm. 2010, 2010, 546826.
- Chen, M.; Chan, C.-C.; Xu, H. Cholesterol homeostasis, macrophage malfunction and age-related macular degeneration. Ann. Transl. Med. 2018, 6, S55.
- Kang, E.Y.-C.; Chen, T.-H.; Garg, S.J.; Sun, C.-C.; Kang, J.-H.; Wu, W.-C.; Hung, M.-J.; Lai, C.-C.; Cherng, W.-J.; Hwang, Y.-S. Association of Statin Therapy With Prevention of Vision-Threatening Diabetic Retinopathy. JAMA Ophthalmol. 2019, 137, 363–371.
- Chung, Y.-R.; Park, S.W.; Choi, S.-Y.; Kim, S.W.; Moon, K.Y.; Kim, J.H.; Lee, K. Association of statin use and hypertriglyceridemia with diabetic macular edema in patients with type 2 diabetes and diabetic retinopathy. Cardiovasc. Diabetol. 2017, 16, 4.
- Vail, D.; Callaway, N.F.; Ludwig, C.A.; Saroj, N.; Moshfeghi, D.M. Lipid-Lowering Medications Are Associated with Lower Risk of Retinopathy and Ophthalmic Interventions among United States Patients with Diabetes. Am. J. Ophthalmol. 2019, 207, 378–384.
- Vavvas, D.G.; Daniels, A.B.; Kapsala, Z.G.; Goldfarb, J.W.; Ganotakis, E.; Loewenstein, J.I.; Young, L.H.; Gragoudas, E.S.; Eliott, D.; Kim, I.K.; et al. Regression of Some High-risk Features of Age-related Macular Degeneration (AMD) in Patients Receiving Intensive Statin Treatment. EBioMedicine 2016, 5, 198–203.
- Miller, Y.I.; Navia-Pelaez, J.M.; Corr, M.; Yaksh, T.L. Lipid rafts in glial cells: Role in neuroinflammation and pain processing. J. Lipid Res. 2020, 61, 655–666.
- Labrecque, L.; Royal, I.; Surprenant, D.S.; Patterson, C.; Gingras, D.; Béliveau, R. Regulation of Vascular Endothelial Growth Factor Receptor-2 Activity by Caveolin-1 and Plasma Membrane Cholesterol. Mol. Biol. Cell 2003, 14, 334–347.
- Laurenzana, A.; Fibbi, G.; Chillà, A.; Margheri, G.; Del Rosso, T.; Rovida, E.; Del Rosso, M.; Margheri, F. Lipid rafts: Integrated platforms for vascular organization offering therapeutic opportunities. Cell. Mol. Life Sci. CMLS 2015, 72, 1537–1557.
- Pilarczyk, M.; Mateuszuk, L.; Rygula, A.; Kepczynski, M.; Chlopicki, S.; Baranska, M.; Kaczor, A. Endothelium in Spots—High-Content Imaging of Lipid Rafts Clusters in db/db Mice. PLoS ONE 2014, 9, e106065.
- Fang, L.; Choi, S.-H.; Baek, J.S.; Liu, C.; Almazan, F.; Ulrich, F.; Wiesner, P.; Taleb, A.; Deer, E.; Pattison, J.; et al. Control of angiogenesis by AIBP-mediated cholesterol efflux. Nature 2013, 498, 118–122.
- Schneider, D.A.; Choi, S.-H.; Agatisa-Boyle, C.; Zhu, L.; Kim, J.; Pattison, J.; Sears, D.D.; Gordts, P.L.S.M.; Fang, L.; Miller, Y.I. AIBP protects against metabolic abnormalities and atherosclerosis. J. Lipid Res. 2018, 59, 854–863.
- Zhang, M.; Zhao, G.-J.; Yao, F.; Xia, X.-D.; Gong, D.; Zhao, Z.-W.; Chen, L.-Y.; Zheng, X.-L.; Tang, X.-E.; Tang, C.-K. AIBP reduces atherosclerosis by promoting reverse cholesterol transport and ameliorating inflammation in apoE-/-mice. Atherosclerosis 2018, 273, 122–130.
- Zhang, M.; Li, L.; Xie, W.; Wu, J.-F.; Yao, F.; Tan, Y.-L.; Xia, X.-D.; Liu, X.-Y.; Liu, D.; Lan, G.; et al. Apolipoprotein A-1 binding protein promotes macrophage cholesterol efflux by facilitating apolipoprotein A-1 binding to ABCA1 and preventing ABCA1 degradation. Atherosclerosis 2016, 248, 149–159.
- Dubrovsky, L.; Ward, A.; Choi, S.-H.; Pushkarsky, T.; Brichacek, B.; Vanpouille, C.; Adzhubei, A.A.; Mukhamedova, N.; Sviridov, D.; Margolis, L.; et al. Inhibition of HIV Replication by Apolipoprotein A-I Binding Protein Targeting the Lipid Rafts. mBio 2020, 11, 10–1128.
- Gu, Q.; Yang, X.; Lv, J.; Zhang, J.; Xia, B.; Kim, J.-D.; Wang, R.; Xiong, F.; Meng, S.; Clements, T.P.; et al. AIBP-mediated cholesterol efflux instructs hematopoietic stem and progenitor cell fate. Science 2019, 363, 1085–1088.
- Choi, S.-H.; Wallace, A.M.; Schneider, D.A.; Burg, E.; Kim, J.; Alekseeva, E.; Ubags, N.D.; Cool, C.D.; Fang, L.; Benjamin, T.S.; et al. AIBP augments cholesterol efflux from alveolar macrophages to surfactant and reduces acute lung inflammation. JCI Insight 2018, 3, 120519.
- Qiu, X.; Luo, J.; Fang, L. AIBP, Angiogenesis, Hematopoiesis, and Atherogenesis. Curr. Atheroscler. Rep. 2020, 23, 1.
- Navia-Pelaez, J.M.; Choi, S.-H.; Dos Santos Aggum Capettini, L.; Xia, Y.; Gonen, A.; Agatisa-Boyle, C.; Delay, L.; Gonçalves Dos Santos, G.; Catroli, G.F.; Kim, J.; et al. Normalization of cholesterol metabolism in spinal microglia alleviates neuropathic pain. J. Exp. Med. 2021, 218, e20202059.
- Woller, S.A.; Choi, S.-H.; An, E.J.; Low, H.; Schneider, D.A.; Ramachandran, R.; Kim, J.; Bae, Y.S.; Sviridov, D.; Corr, M.; et al. Inhibition of Neuroinflammation by AIBP: Spinal Effects upon Facilitated Pain States. Cell Rep. 2018, 23, 2667–2677.
- Corliss, B.A.; Azimi, M.S.; Munson, J.M.; Peirce, S.M.; Murfee, W.L. Macrophages: An Inflammatory Link Between Angiogenesis and Lymphangiogenesis. Microcirculation 2016, 23, 95–121.
- Busbee, B.G.; Ho, A.C.; Brown, D.M.; Heier, J.S.; Suñer, I.J.; Li, Z.; Rubio, R.G.; Lai, P.; HARBOR Study Group. Twelve-month efficacy and safety of 0.5 mg or 2.0 mg ranibizumab in patients with subfoveal neovascular age-related macular degeneration. Ophthalmology 2013, 120, 1046–1056.

