Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Natascha Vidovic | -- | 3007 | 2024-03-11 09:52:52 | | | |
| 2 | Jessie Wu | + 17 word(s) | 3024 | 2024-03-12 03:36:54 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Vidovic, N.; Spittau, B. Microglial Transforming Growth Factor-β Signaling in Alzheimer’s Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/56093 (accessed on 24 June 2026).
Vidovic N, Spittau B. Microglial Transforming Growth Factor-β Signaling in Alzheimer’s Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/56093. Accessed June 24, 2026.
Vidovic, Natascha, Björn Spittau. "Microglial Transforming Growth Factor-β Signaling in Alzheimer’s Disease" Encyclopedia, https://encyclopedia.pub/entry/56093 (accessed June 24, 2026).
Vidovic, N., & Spittau, B. (2024, March 11). Microglial Transforming Growth Factor-β Signaling in Alzheimer’s Disease. In Encyclopedia. https://encyclopedia.pub/entry/56093
Vidovic, Natascha and Björn Spittau. "Microglial Transforming Growth Factor-β Signaling in Alzheimer’s Disease." Encyclopedia. Web. 11 March, 2024.
Copy Citation
Alzheimer’s disease is the most common form of dementia (60–70%) affecting the elderly. To date, the major risk factor of Alzheimer’s disease (AD) is aging. In these times of demographic change, the numbers of patients diagnosed with AD are rising.
neurodegenerative diseases
Alzheimer’s disease
amyloid beta
microglia
TGFβ
hippocampus
APOE
1. Microglia Origin, Maturation, and Activation
During embryonic development, primitive macrophages from the yolk sac migrate into the developing central nervous system (CNS), where they undergo maturation processes and form specialized immune cells called microglia [1][2]. They contribute to the brain’s cellular content by about 5–12% [3]. Maturation and differentiation are highly regulated processes that rely on several signaling molecules such as PU.1, IRF8, IL34, CSF1, and transforming growth factor-beta 1 (TGFβ1) [2][4].
Comparable to macrophages in the periphery, microglia are capable of phagocytosis and display morphological plasticity. Under homeostatic conditions, microglia appear in to have a “ramified” shape and function as the brain’s controlling unit—scanning the surroundings for invaders, pathogens, or aggregated proteins [5][6]. In reaction to environmental changes or acute damage, they adopt different “(re-)activation” states. For a long time, the activation states of microglia have been divided into M1- and M2-like phenotypes, with differences in their morphology and cytokine profiles [7]. Reactive microglia shift towards an ameboid shape, which allows them to move freely through neural tissues and phagocytose cellular debris. Moreover, M1-like microglia tend towards a pro-inflammatory phenotype, while M2-like cells act in immunosuppression and tissue repair tasks [3]. This polarization is not final and can be altered during pathogenic progress [8]. Recently, a more diverse activation pattern of microglia that react to various “input signals” was introduced, and, at present, microglia are characterized not only on the basis of their morphology or cytokine profile but also based on their genetic (transcriptional) profile [9].
The input signals can have a broad spectrum and, among others, include pathogen-associated molecular patterns (PAMP), danger-associated molecular patterns (DAMP), cytokines, and growth and stimulating factors [10]. Even without DAMP or PAMP stimulation, microglia undergo morphological (hypertrophy, de-ramification, and thickening), biochemical, and transcriptional changes during aging processes, with differences across grey and white matter [11]. The transcriptome of microglia throughout the CNS is heterogenous and varies across anatomical regions, as does their distribution. Greater numbers of microglia are found in the grey compared to white matter, with the highest density being found in the hippocampus and substantia nigra [12]. Moreover, microglia expression profiles have been found to be different in the cortex and striatum compared to the cerebellum and hippocampus, with greater age-related phenotypic changes in the white matter of the cerebellum [11]. There are also regional differences in response to injury and inflammation [13].
Reactive microglia have been found in close proximity to amyloid plaques, indicating an interaction with amyloid oligomers and fibrils. Supporting evidence for an involvement of amyloid oligomers in the activation of microglia has been derived from receptors that are capable of binding Aβ42 [14]. Among others, the cluster of differentiation (CD) 14, which has gained importance as the lipopolysaccharide (LPS) receptor involved in innate immunity, is also expressed on the microglial cell surface, has been found to assist in the phagocytosis of Aβ fibrils [15][16]. Additionally, the receptor for advanced glycation end products (RAGE), CD36, and various scavenger receptors are able to stimulate the phagocytosis of Aβ species [17][18][19][20]. Binding to these receptors activates a signaling cascade resulting in the expression of cytokines of either pro- or anti-inflammatory origins or leads to the internalization of Aβ molecules via micropinocytosis or endocytosis. Additionally, an involvement of CD14 long time-activation by Aβ fibrils in the chronic neuroinflammatory process in Alzheimer’s disease (AD) has been hypothesized [15]. Moreover, pattern recognition receptors belonging to the group of Toll-like receptors (TLRs) are also expressed by microglia [21]. Upon activation, these receptors act via signaling induction through interferon (IFN)-γ and nuclear factor-κB [19]. TLR4 and CD14 are both involved in the recognition of bacterial LPS [22]. Like TLR2 and 4, TLR6 is also known for its Aβ-binding properties and the provoking of neuroinflammation [23][24]. CD36 acts as a coreceptor for TLR4 and TLR6, which is triggered by Aβ.
The microglial receptor TREM2 assists in the interaction with apoptotic cells, lipoproteins, and accumulated proteins such as Aβ. TREM2 is a member of immunoglobulin superfamily receptors and is involved in proliferation, survival, and regulating inflammatory processes by enhancing microglial phagocytosis. Consequently, in AD, TREM2 is increased in the microglia surrounding amyloid deposits [25].
2. Transforming Growth Factor-Beta and Alzheimer’s Disease
Recent studies on the reactomes from human tissues have revealed an overlapping of 41 genes affected during aging and AD. Among others, TGFβ1 is affected in pathways including interleukin signaling and the transcription of RNA polymerase II as well as immune signaling. Surprisingly, there is also an overlap between AD and longevity genes. Longevity genes are related to a longer lifespan and delayed aging. Here, again, 43 genes overlap that involve the TGFβ1 pathways [26].
In patients with AD, the levels of TGFβ1 in the CSF and plasma are significantly higher than in the controls [27]. Interestingly, in a mouse model of AD, the reduction in TGFβ1 led to a decrease in spine density, memory function, and overall synaptic plasticity [28]. Furthermore, the loss of TGFβ signaling in the microglia was shown to result in motor deficits and impaired myelination by disturbances in oligodendrocyte maturation [4]. The knockout of TGFβ in mice resulted in severe postnatal systemic inflammatory reactions, leading to premature death and impaired homeostasis [29][30][31]. A hallmark of AD is the decreased neurogenesis of neural stem cells in hippocampal formation [32][33]. In hippocampal microglia, TGFβ has been found to be a key player in their pro-neurogenic effects in chronic neurodegeneration processes [34]. Moreover, the overexpression of TGFβ1 was even able to recover hippocampal synaptic plasticity and memory function in an in vitro model of Aβ-induced toxicity [28][35]. The parabiosis of young wildtype and old transgenic AD-mice (18 month) resulted in a significant increase in TGFβ1 levels after 3 days, and the amyloid load decreased after 14 days [36]. Interestingly, this level increase was accompanied by the increase in the cell adhesion molecule fragment L1-70 [37]. L1-70 is a cleavage product of the full-length L1, which can enter the cytoplasm and even reach the nuclei [38]. L1 itself is involved in synaptic plasticity in the traumatized and regenerating CNS, thereby supporting neuronal activities [39]. Noteworthy, the expression of L1 and L1-70 has been observed to be low in old AD-mice, among which amyloid deposition was high. On the other hand, wildtype hippocampi have been shown to contain high levels of L1-70 and no amyloid plaques. Not surprisingly, L1 has been found to bind to Aβ species 40 and 42 itself, making it an interesting candidate for therapeutic treatments [40].
Moreover, TGFβ1 could be involved in this process, since it has been previously found to upregulate the expression of L1 in pancreatic duct cells [41]. A knockdown of TGFβ1 in neuroblastoma cells decreased the levels of L1-70 and the pro-inflammatory cytokine macrophage migration inhibitory factor (MIF) [36]. L1-70, in turn, is able to regulate MIF expression in the brain, thereby promoting microglia activation and amyloid clearance [42]. MIF strongly is related to CD74 expression in the microglia, since their interaction leads to the activation of numerous pathways involved in cell survival and proliferation [43]. MIF has been found to be expressed by neurons rather than glial cells upon interaction with Aβ oligomers, serving as a defense mechanism, and could be useful as a potential biomarker for AD rather than MCI [44]. The studies mentioned above indicate that TGFβ1 is responsible for the expression of L1 and, subsequently, MIF through CD74. After secretion, L1 is cleaved by a serine proteinase into the L1-70 fragment, which is necessary for the expression of MIF. MIF, in turn, acts during microglia activation and promotes amyloid clearance. Since in AD mice levels of TGFβ1, L1-70 and MIF are low, there is reduced microglia activation and a higher amyloid burden, making TGFβ1 a key modulator in this system. Moreover, TGFβ1 is able to downregulate CD74 in microglia. In TGFβR2-deficient microglia, high levels of CD74 are detectable [45].
In the brains of patients with AD, TGFβR2 expression is lower than in controls without AD [46]. A reduction in TGFβ receptor genes in microglia has also been observed in aged (12–22 months) C57Bl/6J mice, with the lowest expression of TGFβR1 being recorded in the hippocampus and striatum of 22-months-old mice, whereas TGFβR2 was lowest in the hippocampus and cerebellum [47]. In 9-month-old APP/PS1 mice, TGFβR2 levels were low compared to aged controls paired with an enhanced environment of neuroinflammation [48]. The TGFβR2−/− mice displayed more Aβ deposition in the hippocampus and a higher age-related neuronal degeneration rate [46]. Interestingly, in some AD cases, there is an ectopic localization of SMAD2 and SMAD3 with amyloid plaques and NFTs, thereby inhibiting its nuclear translocation and disrupting signal transduction [49][50]. Single-nucleotide polymorphisms (SNP) in the TGFβ1 gene are known to influence the expression level of the protein and thereby influence TGFβ signaling in general. In patients with AD, some functional SNPs have been found that are related to the occurrence of depression; in these cases, other SNPs were able to promote the progression from MCI to AD [51][52]. An overview of the different roles of TGFβ signaling in a healthy and AD brain is given in Figure 1.
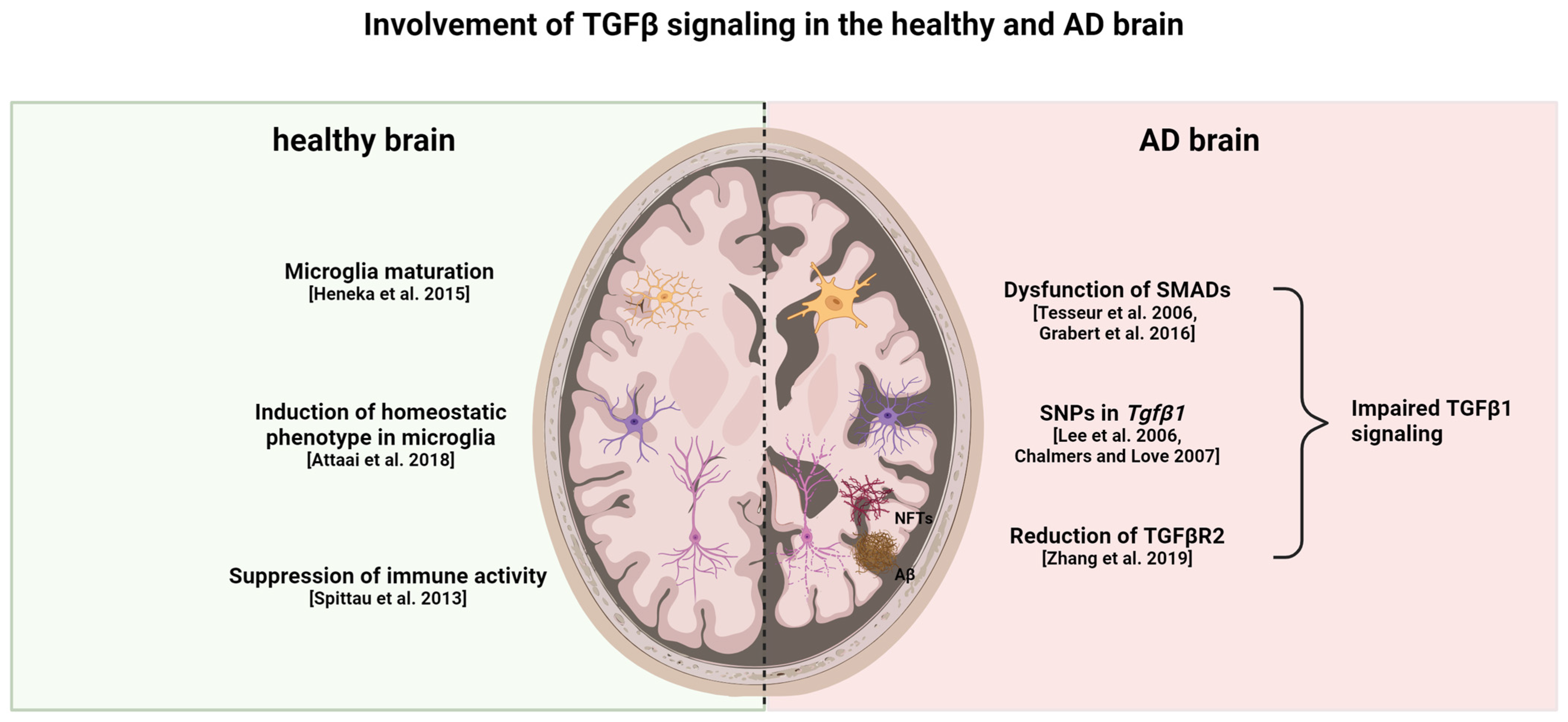
Figure 1. Healthy versus AD brain: differences in TGFβ signaling. TGFβ1 is important for microglia maturation and maintaining the homeostatic phenotype in healthy individuals. Moreover, it suppresses the activation of both astrocytes and microglia. Reduction in TGFβR2, dysfunction of SMAD proteins, and a specific genetic profile of TGFβ1 contribute to impaired TGFβ1 signaling in AD. This impaired signaling leads to enhanced glial cell activation in both microglia and astrocytes, thereby contributing to neuroinflammatory processes and neurodegeneration. In turn, the neurodegeneration and accumulation of Aβ and tau fibrils leads to increased gliosis and impairment of TGFβ signaling [44][46][47][49][50][53][54][55]. This figure was created with BioRender (https://biorender.com/, accessed on 20 February 2024).
Though there is an increase in TGFβ1 plasma and CSF levels, there is a decrease in TGFβR2 expression in patients with AD. Enhanced expression of TGFβ1 could be a mechanism to compensate for reduced TGFβ signaling due to the reduction in receptors. TGFβ signaling is able to decrease Aβ deposition and promote its clearance if not disrupted by chronic inflammatory events [56].
3. Apolipoprotein E–TREM 2 (–Transforming Growth Factor-Beta) Axis in Microglia
Overall, there is minor overlap between human and murine AD microglial gene signatures, except for APOE [57][58][59][60]. In APP-overexpressing mice, two main microglia gene clusters were identified. Cluster one lacked homeostatic microglial genes, and cluster 2 displayed the upregulation of inflammatory DAM genes. The authors of the study concluded that APOE and TGFβ are the major upstream regulators of MGnD. A specific SNP in Tgfβ1 was found to affect the expression level of APOE4, and this SNP is over-presented in patients with AD [61]. APOE suppresses the homeostatic signature of microglia that is regulated by TGFβ [62]. Not surprisingly, the expression of APOE by microglia themselves increases with the proximity of the plaques in APP-overexpressing mice [63][64]. Although APOE induction in 5xFAD mice was shown to be TREM2-independent, genetic targeting of Trem2 in APP-overexpressing mice suppressed APOE signaling and restored homeostatic microglia [62][65]. There is a strong correlation between TREM2 and APOE signaling. A lack of TREM2 was speculated to lock microglia in a homeostatic state, thereby blocking the defense mechanisms during AD progression [66]. APOE knockout studies revealed that the risk allele ε4 impairs the microglial response to neurodegenerative processes by directly repressing the transcription of MGnD genes [67]. Loss-of-function TREM2 variants in AD led to reduced APOE colocalization with amyloid deposits [68]. Overall, TREM2–APOE interaction has the potential to induce microglia activation towards MGnD (Figure 2) [62]. Understanding the intricate interplay of factors within this axis is crucial for developing effective therapeutic interventions. To delve deeper into this complex concept, it is essential to explore the nuances and interactions between the various components involved. Further investigations into the potential impact of this axis on therapeutic outcomes are necessary to elucidate its full significance. Additionally, identifying and evaluating therapeutic approaches that can modulate one or more factors within this axis will provide valuable insights into potential treatment strategies.
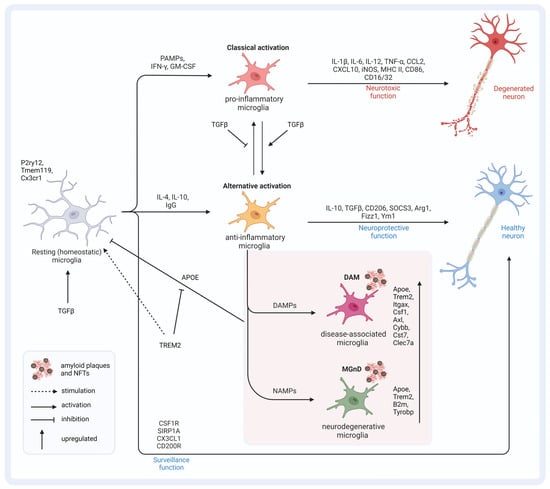
Figure 2. Microglia (re)activation states and the APOE–TREM2–TGFβ axis. TGFβ signaling is important for the induction of homeostatic microglia, which monitor and maintain a healthy environment for neurons. PAMPs lead to classically activated microglia of pro-inflammatory origins that exert neurotoxic function by releasing pro-inflammatory cytokines and factors. TGFβ signaling stimulates the transition to alternative activation states of anti-inflammatory origins, which exert a neuroprotective function, and inhibits the transition to pro-inflammatory cells. Depending on the signaling molecules, disease-associated microglia and microglia with a neurodegenerative phenotype can develop. Both conditions are characterized by an altered genetic profile compared to homeostatic microglia. The increased expression of APOE results in the inhibition of TGFβ-induced homeostatic microglia and favors the transition to DAMs and MGnDs. TREM2 inhibits APOE signaling and thereby favors the fixation of microglia in the homeostatic state. This figure was created with BioRender (https://biorender.com/, accessed on 20 February 2024).
It is important to note that the APOE isoforms differ in their impact on the amyloid burden. Lozupone and Panza’s review of the different isoforms of APOE sheds light on the intricate role of APOE in Aβ metabolism [69]. APOE3, the most prevalent isoform, is considered the neutral or “wild-type” variant. It is associated with an average lipid metabolism and is present in approximately 60–70% of the population. On the other hand, APOE4, the isoform linked to an increased risk for AD, exhibits less efficiency in lipid metabolism compared to APOE2 and APOE3. The impact of APOE4 on lipid metabolism may contribute to the buildup of cholesterol and other lipids in the brain, ultimately leading to the formation of amyloid plaques and neurodegeneration. This differential effect of APOE isoforms on health and disease susceptibility underscores the significance of genetic variations in lipid metabolism pathways. Unraveling the role of APOE isoforms in disease risk holds promise for identifying potential therapeutic targets for conditions such as AD. The comprehensive understanding of APOE isoforms and their nuanced effects on lipid metabolism and disease susceptibility is a crucial area of study in the pursuit of therapeutic interventions for neurodegenerative diseases.
Whereas the induced expression of APOE3 in APP-overexpressing mice led to the reduction in amyloid aggregation in the cortex and the hippocampus as well as soluble Aβ40 and Aβ42, APOE4 did not have an effect on the amyloid burden in this context at all [70]. Furthermore, microglial cells are able to produce short-length Aβ species themselves during the process of clearing unfolded Aβ molecules, which can give rise to even further Aβ42 aggregation [71]. Microglia-derived apoptosis-associated speck-like protein containing a CARD (ASC specks) are viewed as important triggers of Aβ aggregation and seeding. Injections of ASC specks into APP/PS1 mice resulted in an increased Aβ load compared to the control mice; likewise, injections of APP/PS brain homogenates into APP/PS1 mice led to an increase in Aβ-positive deposits, whereas the injection of the homogenates into APP/PS1 and ASC−/− mice did not result in any deposition. These results suggest that there is a strong correlation of ASC specks and Aβ deposition in vivo [72]. Microglia facing Aβ42 results in the release of ASC specks; this might be a mechanism of perpetuation and enhanced Aβ deposition. The deposition of Aβ species is thought to begin decades before the first clinical symptoms of AD arise [73][74]. Interestingly, APOE4 is known to play an essential role in this seeding process [64][75]. Moreover, in males carrying the APOE4 allele—the high-risk gene for AD—the authors of the above-mentioned study detected an upregulation of integrin beta-8 (ITGB8). ITGB8 is important in the activation of the latent TGFβ1 molecule [76]. However, in another study, the deletion of Itgfb8 was able restore the MGnD response and reduce plaque burden in AD mice, making the ITGB8-TGFβ signaling pathway a potential target for a therapeutic approach [67][77].
Targeting the TGFβ signaling pathway holds promising therapeutic implications in AD treatment due to its involvement in various aspects of the pathology, including neuroinflammation, synaptic dysfunction, and Aβ deposition. TGFβ signaling has been implicated in regulating the immune response in the brain, with the dysregulation of this pathway contributing to neuroinflammation, a key feature of AD pathology. Additionally, TGFβ signaling plays a role in synaptic plasticity and neuronal survival, processes which are disrupted in AD. Moreover, TGFβ signaling has been shown to modulate the production and clearance of Aβ, suggesting its involvement in the amyloid cascade. Challenges in targeting the TGFβ signaling pathway in AD treatment include the complex and context-dependent nature of TGFβ signaling, with both neuroprotective and neurotoxic effects reported in different stages of AD. Furthermore, the pleiotropic effects of TGFβ signaling in various cell types within the brain and peripheral tissues pose challenges in achieving the selective modulation of this pathway without causing off-target effects. Additionally, the BBB presents a challenge for delivering therapeutics targeting TGFβ signaling to the brain, necessitating the development of strategies to overcome this barrier. Potential strategies to modulate the TGFβ signaling pathway effectively in AD treatment include the development of selective small-molecule inhibitors or activators targeting specific components of the TGFβ signaling cascade. Moreover, approaches to enhance BBB permeability, such as the use of nanotechnology-based drug delivery systems or BBB-penetrating peptides, could facilitate the delivery of therapeutics targeting TGFβ signaling to the brain. Furthermore, combination therapies targeting multiple components of the AD pathology, including Aβ deposition, neuroinflammation, and synaptic dysfunction, may be necessary to achieve optimal therapeutic efficacy while minimizing off-target effects.
In conclusion, targeting the TGFβ signaling pathway represents a promising therapeutic approach for AD treatment, given its involvement in various aspects of AD pathology. However, addressing the challenges associated with modulating this pathway effectively, including its pleiotropic effects and BBB permeability, will be essential for the successful development of TGFβ-targeted therapeutics for AD. With the rapid advancements in imaging and sequencing technologies, researchers must stay updated with respect to the latest techniques to ensure accurate and comprehensive data collection. Additionally, it is crucial to extend research to human settings to bridge the gap between laboratory findings and real-world applications as well as consider gender-specific differences with regard to personalized medicine.
References
- Goldmann, T.; Wieghofer, P.; Jordão, M.J.C.; Prutek, F.; Hagemeyer, N.; Frenzel, K.; Amann, L.; Staszewski, O.; Kierdorf, K.; Krueger, M.; et al. Origin, Fate and Dynamics of Macrophages at Central Nervous System Interfaces. Nat. Immunol. 2016, 17, 797–805.
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Hölscher, C.; et al. Microglia Emerge from Erythromyeloid Precursors via Pu.1- and Irf8-Dependent Pathways. Nat. Neurosci. 2013, 16, 273–280.
- Aguzzi, A.; Barres, B.A.; Bennett, M.L. Microglia: Scapegoat, Saboteur, or Something Else? Science 2013, 339, 156–161.
- Arnold, T.D.; Lizama, C.O.; Cautivo, K.M.; Santander, N.; Lin, L.; Qiu, H.; Huang, E.J.; Liu, C.; Mukouyama, Y.-S.; Reichardt, L.F.; et al. Impaired AVβ8 and TGFβ Signaling Lead to Microglial Dysmaturation and Neuromotor Dysfunction. J. Exp. Med. 2019, 216, 900–915.
- Hristovska, I.; Pascual, O. Deciphering Resting Microglial Morphology and Process Motility from a Synaptic Prospect. Front. Integr. Neurosci. 2015, 9, 73.
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma In Vivo. Science 2005, 308, 1314–1318.
- Prinz, M.; Priller, J. Microglia and Brain Macrophages in the Molecular Age: From Origin to Neuropsychiatric Disease. Nat. Rev. Neurosci. 2014, 15, 300–312.
- Minogue, A.M. Role of Infiltrating Monocytes/Macrophages in Acute and Chronic Neuroinflammation: Effects on Cognition, Learning and Affective Behaviour. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 79, 15–18.
- Schultze, J.L. Transcriptional Programming of Human Macrophages: On the Way to Systems Immunology. J. Mol. Med. 2015, 93, 589–597.
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-Based Network Analysis Reveals a Spectrum Model of Human Macrophage Activation. Immunity 2014, 40, 274–288.
- Hart, A.D.; Wyttenbach, A.; Perry, V.H.; Teeling, J.L. Age Related Changes in Microglial Phenotype Vary between CNS Regions: Grey versus White Matter Differences. Brain Behav. Immun. 2012, 26, 754–765.
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the Distribution and Morphology of Microglia in the Normal Adult Mouse Brain. Neuroscience 1990, 39, 151–170.
- Batchelor, P.E.; Tan, S.; Wills, T.E.; Porritt, M.J.; Howells, D.W. Comparison of Inflammation in the Brain and Spinal Cord Following Mechanical Injury. J. Neurotrauma 2008, 25, 1217–1225.
- Murgas, P.; Godoy, B.; von Bernhardi, R. Aβ Potentiates Inflammatory Activation of Glial Cells Induced by Scavenger Receptor Ligands and Inflammatory Mediators in Culture. Neurotox. Res. 2012, 22, 69–78.
- Fassbender, K.; Walter, S.; Kühl, S.; Landmann, R.; Ishii, K.; Bertsch, T.; Stalder, A.K.; Muehlhauser, F.; Liu, Y.; Ulmer, A.J.; et al. The LPS Receptor (CD14) Links Innate Immunity with Alzheimer’s Disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 203–205.
- Liu, Y.; Walter, S.; Stagi, M.; Cherny, D.; Letiembre, M.; Schulz-Schaeffer, W.; Heine, H.; Penke, B.; Neumann, H.; Fassbender, K. LPS Receptor (CD14): A Receptor for Phagocytosis of Alzheimer’s Amyloid Peptide. Brain J. Neurol. 2005, 128, 1778–1789.
- Chellappa, R.C.; Rani, P. G82S RAGE Polymorphism Is Associated with Alzheimer’s Disease. Front. Biosci. Elite Ed. 2020, 12, 150–161.
- Monllor, P.; Giraldo, E.; Badia, M.-C.; de la Asuncion, J.G.; Alonso, M.-D.; Lloret, A.; Vina, J. Serum Levels of Clusterin, PKR, and RAGE Correlate with Amyloid Burden in Alzheimer’s Disease. J. Alzheimer’s Dis. JAD 2021, 80, 1067–1077.
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 Ligands Promote Sterile Inflammation through Assembly of a Toll-like Receptor 4 and 6 Heterodimer. Nat. Immunol. 2010, 11, 155–161.
- Edler, M.K.; Johnson, C.T.; Ahmed, H.S.; Richardson, J.R. Age, Sex, and Regional Differences in Scavenger Receptor CD36 in the Mouse Brain: Potential Relevance to Cerebral Amyloid Angiopathy and Alzheimer’s Disease. J. Comp. Neurol. 2021, 529, 2209–2226.
- Papageorgiou, I.E.; Lewen, A.; Galow, L.V.; Cesetti, T.; Scheffel, J.; Regen, T.; Hanisch, U.-K.; Kann, O. TLR4-Activated Microglia Require IFN-γ to Induce Severe Neuronal Dysfunction and Death In Situ. Proc. Natl. Acad. Sci. USA 2016, 113, 212–217.
- Tang, S.-C.; Lathia, J.D.; Selvaraj, P.K.; Jo, D.-G.; Mughal, M.R.; Cheng, A.; Siler, D.A.; Markesbery, W.R.; Arumugam, T.V.; Mattson, M.P. Toll-like Receptor-4 Mediates Neuronal Apoptosis Induced by Amyloid Beta-Peptide and the Membrane Lipid Peroxidation Product 4-Hydroxynonenal. Exp. Neurol. 2008, 213, 114–121.
- Liu, S.; Liu, Y.; Hao, W.; Wolf, L.; Kiliaan, A.J.; Penke, B.; Rübe, C.E.; Walter, J.; Heneka, M.T.; Hartmann, T.; et al. TLR2 Is a Primary Receptor for Alzheimer’s Amyloid β Peptide to Trigger Neuroinflammatory Activation. J. Immunol. 2012, 188, 1098–1107.
- Balducci, C.; Frasca, A.; Zotti, M.; La Vitola, P.; Mhillaj, E.; Grigoli, E.; Iacobellis, M.; Grandi, F.; Messa, M.; Colombo, L.; et al. Toll-like Receptor 4-Dependent Glial Cell Activation Mediates the Impairment in Memory Establishment Induced by β-Amyloid Oligomers in an Acute Mouse Model of Alzheimer’s Disease. Brain Behav. Immun. 2017, 60, 188–197.
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-Mediated Early Microglial Response Limits Diffusion and Toxicity of Amyloid Plaques. J. Exp. Med. 2016, 213, 667–675.
- Balmorez, T.; Sakazaki, A.; Murakami, S. Genetic Networks of Alzheimer’s Disease, Aging, and Longevity in Humans. Int. J. Mol. Sci. 2023, 24, 5178.
- Swardfager, W.; Lanctôt, K.; Rothenburg, L.; Wong, A.; Cappell, J.; Herrmann, N. A Meta-Analysis of Cytokines in Alzheimer’s Disease. Biol. Psychiatry 2010, 68, 930–941.
- Hu, Y.; Chen, W.; Wu, L.; Jiang, L.; Liang, N.; Tan, L.; Liang, M.; Tang, N. TGF-Β1 Restores Hippocampal Synaptic Plasticity and Memory in Alzheimer Model via the PI3K/Akt/Wnt/β-Catenin Signaling Pathway. J. Mol. Neurosci. 2019, 67, 142–149.
- Shull, M.M.; Ormsby, I.; Kier, A.B.; Pawlowski, S.; Diebold, R.J.; Yin, M.; Allen, R.; Sidman, C.; Proetzel, G.; Calvin, D.; et al. Targeted Disruption of the Mouse Transforming Growth Factor-Β1 Gene Results in Multifocal Inflammatory Disease. Nature 1992, 359, 693–699.
- Kulkarni, A.B.; Huh, C.G.; Becker, D.; Geiser, A.; Lyght, M.; Flanders, K.C.; Roberts, A.B.; Sporn, M.B.; Ward, J.M.; Karlsson, S. Transforming Growth Factor Beta 1 Null Mutation in Mice Causes Excessive Inflammatory Response and Early Death. Proc. Natl. Acad. Sci. USA 1993, 90, 770–774.
- Zöller, T.; Schneider, A.; Kleimeyer, C.; Masuda, T.; Potru, P.S.; Pfeifer, D.; Blank, T.; Prinz, M.; Spittau, B. Silencing of TGFβ Signalling in Microglia Results in Impaired Homeostasis. Nat. Commun. 2018, 9, 4011.
- Ekdahl, C.T.; Claasen, J.-H.; Bonde, S.; Kokaia, Z.; Lindvall, O. Inflammation Is Detrimental for Neurogenesis in Adult Brain. Proc. Natl. Acad. Sci. USA 2003, 100, 13632–13637.
- Moreno-Jiménez, E.P.; Flor-García, M.; Terreros-Roncal, J.; Rábano, A.; Cafini, F.; Pallas-Bazarra, N.; Ávila, J.; Llorens-Martín, M. Adult Hippocampal Neurogenesis Is Abundant in Neurologically Healthy Subjects and Drops Sharply in Patients with Alzheimer’s Disease. Nat. Med. 2019, 25, 554–560.
- De Lucia, C.; Rinchon, A.; Olmos-Alonso, A.; Riecken, K.; Fehse, B.; Boche, D.; Perry, V.H.; Gomez-Nicola, D. Microglia Regulate Hippocampal Neurogenesis during Chronic Neurodegeneration. Brain Behav. Immun. 2016, 55, 179–190.
- Caruso, G.; Fresta, C.G.; Musso, N.; Giambirtone, M.; Grasso, M.; Spampinato, S.F.; Merlo, S.; Drago, F.; Lazzarino, G.; Sortino, M.A.; et al. Carnosine Prevents Aβ-Induced Oxidative Stress and Inflammation in Microglial Cells: A Key Role of TGF-Β1. Cells 2019, 8, 64.
- Hu, J.; Lin, S.L.; Schachner, M. A Fragment of Cell Adhesion Molecule L1 Reduces Amyloid-β Plaques in a Mouse Model of Alzheimer’s Disease. Cell Death Dis. 2022, 13, 48.
- Sytnyk, V.; Leshchyns’ka, I.; Schachner, M. Neural Cell Adhesion Molecules of the Immunoglobulin Superfamily Regulate Synapse Formation, Maintenance, and Function. Trends Neurosci. 2017, 40, 295–308.
- Kraus, K.; Kleene, R.; Henis, M.; Braren, I.; Kataria, H.; Sharaf, A.; Loers, G.; Schachner, M.; Lutz, D. A Fragment of Adhesion Molecule L1 Binds to Nuclear Receptors to Regulate Synaptic Plasticity and Motor Coordination. Mol. Neurobiol. 2018, 55, 7164–7178.
- Ohyama, K.; Tan-Takeuchi, K.; Kutsche, M.; Schachner, M.; Uyemura, K.; Kawamura, K. Neural Cell Adhesion Molecule L1 Is Required for Fasciculation and Routing of Thalamocortical Fibres and Corticothalamic Fibres. Neurosci. Res. 2004, 48, 471–475.
- Djogo, N.; Jakovcevski, I.; Müller, C.; Lee, H.J.; Xu, J.-C.; Jakovcevski, M.; Kügler, S.; Loers, G.; Schachner, M. Adhesion Molecule L1 Binds to Amyloid Beta and Reduces Alzheimer’s Disease Pathology in Mice. Neurobiol. Dis. 2013, 56, 104–115.
- Geismann, C.; Morscheck, M.; Koch, D.; Bergmann, F.; Ungefroren, H.; Arlt, A.; Tsao, M.-S.; Bachem, M.G.; Altevogt, P.; Sipos, B.; et al. Up-Regulation of L1CAM in Pancreatic Duct Cells Is Transforming Growth Factor Beta1- and Slug-Dependent: Role in Malignant Transformation of Pancreatic Cancer. Cancer Res. 2009, 69, 4517–4526.
- Nasiri, E.; Sankowski, R.; Dietrich, H.; Oikonomidi, A.; Huerta, P.T.; Popp, J.; Al-Abed, Y.; Bacher, M. Key Role of MIF-Related Neuroinflammation in Neurodegeneration and Cognitive Impairment in Alzheimer’s Disease. Mol. Med. 2020, 26, 34.
- Potru, P.S.; Spittau, B. CD74: A Prospective Marker for Reactive Microglia? Neural Regen. Res. 2023, 18, 2673–2674.
- Zhang, S.; Zhao, J.; Zhang, Y.; Zhang, Y.; Cai, F.; Wang, L.; Song, W. Upregulation of MIF as a Defense Mechanism and a Biomarker of Alzheimer’s Disease. Alzheimer’s Res. Ther. 2019, 11, 54.
- Jahn, J.; Bollensdorf, A.; Kalischer, C.; Piecha, R.; Weiß-Müller, J.; Potru, P.S.; Ruß, T.; Spittau, B. Microglial CD74 Expression Is Regulated by TGFβ Signaling. Int. J. Mol. Sci. 2022, 23, 10247.
- Tesseur, I.; Zou, K.; Esposito, L.; Bard, F.; Berber, E.; Can, J.V.; Lin, A.H.; Crews, L.; Tremblay, P.; Mathews, P.; et al. Deficiency in Neuronal TGF-β Signaling Promotes Neurodegeneration and Alzheimer’s Pathology. J. Clin. Investig. 2006, 116, 3060–3069.
- Grabert, K.; Michoel, T.; Karavolos, M.H.; Clohisey, S.; Baillie, J.K.; Stevens, M.P.; Freeman, T.C.; Summers, K.M.; McColl, B.W. Microglial Brain Region-Dependent Diversity and Selective Regional Sensitivities to Aging. Nat. Neurosci. 2016, 19, 504–516.
- Song, L.; Gu, Y.; Jie, J.; Bai, X.; Yang, Y.; Liu, C.; Liu, Q. Dab2 Attenuates Brain Injury in APP/PS1 Mice via Targeting Transforming Growth Factor-Beta/SMAD Signaling. Neural Regen. Res. 2014, 9, 41–50.
- Lee, H.; Ueda, M.; Zhu, X.; Perry, G.; Smith, M.A. Ectopic Expression of Phospho-Smad2 in Alzheimer’s Disease: Uncoupling of the Transforming Growth Factor-Beta Pathway? J. Neurosci. Res. 2006, 84, 1856–1861.
- Chalmers, K.A.; Love, S. Neurofibrillary Tangles May Interfere with Smad 2/3 Signaling in Neurons. J. Neuropathol. Exp. Neurol. 2007, 66, 158–167.
- Caraci, F.; Bosco, P.; Signorelli, M.; Spada, R.S.; Cosentino, F.I.; Toscano, G.; Bonforte, C.; Muratore, S.; Prestianni, G.; Panerai, S.; et al. The CC Genotype of Transforming Growth Factor-Β1 Increases the Risk of Late-Onset Alzheimer’s Disease and Is Associated with AD-Related Depression. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2012, 22, 281–289.
- Arosio, B.; Bergamaschini, L.; Galimberti, L.; La Porta, C.; Zanetti, M.; Calabresi, C.; Scarpini, E.; Annoni, G.; Vergani, C. +10 T/C Polymorphisms in the Gene of Transforming Growth Factor-Β1 Are Associated with Neurodegeneration and Its Clinical Evolution. Mech. Ageing Dev. 2007, 128, 553–557.
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol. 2015, 14, 388–405.
- Attaai, A.; Neidert, N.; von Ehr, A.; Potru, P.S.; Zöller, T.; Spittau, B. Postnatal Maturation of Microglia Is Associated with Alternative Activation and Activated TGFβ Signaling. Glia 2018, 66, 1695–1708.
- Spittau, B.; Wullkopf, L.; Zhou, X.; Rilka, J.; Pfeifer, D.; Krieglstein, K. Endogenous Transforming Growth Factor-Beta Promotes Quiescence of Primary Microglia in Vitro. Glia 2013, 61, 287–300.
- Yang, C.; Xu, P. The Role of Transforming Growth Factor Β1 /Smad Pathway in Alzheimer’s Disease Inflammation Pathology. Mol. Biol. Rep. 2023, 50, 777–788.
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-Cell Transcriptomic Analysis of Alzheimer’s Disease. Nature 2019, 570, 332–337.
- Srinivasan, K.; Friedman, B.A.; Etxeberria, A.; Huntley, M.A.; van der Brug, M.P.; Foreman, O.; Paw, J.S.; Modrusan, Z.; Beach, T.G.; Serrano, G.E.; et al. Alzheimer’s Patient Microglia Exhibit Enhanced Aging and Unique Transcriptional Activation. Cell Rep. 2020, 31, 107843.
- Del-Aguila, J.L.; Li, Z.; Dube, U.; Mihindukulasuriya, K.A.; Budde, J.P.; Fernandez, M.V.; Ibanez, L.; Bradley, J.; Wang, F.; Bergmann, K.; et al. A Single-Nuclei RNA Sequencing Study of Mendelian and Sporadic AD in the Human Brain. Alzheimer’s Res. Ther. 2019, 11, 71.
- Lee, H.; Aylward, A.J.; Pearse, R.V.; Lish, A.M.; Hsieh, Y.-C.; Augur, Z.M.; Benoit, C.R.; Chou, V.; Knupp, A.; Pan, C.; et al. Cell-Type-Specific Regulation of APOE and CLU Levels in Human Neurons by the Alzheimer’s Disease Risk Gene SORL1. Cell Rep. 2023, 42, 112994.
- Dickson, M.R.; Perry, R.T.; Wiener, H.; Go, R.C.P. Association Studies of Transforming Growth Factor-Beta 1 and Alzheimer’s Disease. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. Off. Publ. Int. Soc. Psychiatr. Genet. 2005, 139, 38–41.
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9.
- Sala Frigerio, C.; Wolfs, L.; Fattorelli, N.; Thrupp, N.; Voytyuk, I.; Schmidt, I.; Mancuso, R.; Chen, W.-T.; Woodbury, M.E.; Srivastava, G.; et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Aβ Plaques. Cell Rep. 2019, 27, 1293–1306.e6.
- Ulrich, J.D.; Ulland, T.K.; Mahan, T.E.; Nyström, S.; Nilsson, K.P.; Song, W.M.; Zhou, Y.; Reinartz, M.; Choi, S.; Jiang, H.; et al. ApoE Facilitates the Microglial Response to Amyloid Plaque Pathology. J. Exp. Med. 2018, 215, 1047–1058.
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17.
- Mazaheri, F.; Snaidero, N.; Kleinberger, G.; Madore, C.; Daria, A.; Werner, G.; Krasemann, S.; Capell, A.; Trümbach, D.; Wurst, W.; et al. TREM2 Deficiency Impairs Chemotaxis and Microglial Responses to Neuronal Injury. EMBO Rep. 2017, 18, 1186–1198.
- Yin, Z.; Rosenzweig, N.; Kleemann, K.L.; Zhang, X.; Brandão, W.; Margeta, M.A.; Schroeder, C.; Sivanathan, K.N.; Silveira, S.; Gauthier, C.; et al. APOE4 Impairs the Microglial Response in Alzheimer’s Disease by Inducing TGFβ-Mediated Checkpoints. Nat. Immunol. 2023, 24, 1839–1853.
- Parhizkar, S.; Arzberger, T.; Brendel, M.; Kleinberger, G.; Deussing, M.; Focke, C.; Nuscher, B.; Xiong, M.; Ghasemigharagoz, A.; Katzmarski, N.; et al. Loss of TREM2 Function Increases Amyloid Seeding but Reduces Plaque-Associated ApoE. Nat. Neurosci. 2019, 22, 191–204.
- Lozupone, M.; Panza, F. Impact of Apolipoprotein E Isoforms on Sporadic Alzheimer’s Disease: Beyond the Role of Amyloid Beta. Neural Regen. Res. 2024, 19, 80.
- Liu, C.-C.; Wang, N.; Chen, Y.; Inoue, Y.; Shue, F.; Ren, Y.; Wang, M.; Qiao, W.; Ikezu, T.C.; Li, Z.; et al. Cell-Autonomous Effects of APOE4 in Restricting Microglial Response in Brain Homeostasis and Alzheimer’s Disease. Nat. Immunol. 2023, 24, 1854–1866.
- Mazzitelli, S.; Filipello, F.; Rasile, M.; Lauranzano, E.; Starvaggi-Cucuzza, C.; Tamborini, M.; Pozzi, D.; Barajon, I.; Giorgino, T.; Natalello, A.; et al. Amyloid-β 1-24 C-Terminal Truncated Fragment Promotes Amyloid-β 1-42 Aggregate Formation in the Healthy Brain. Acta Neuropathol. Commun. 2016, 4, 110.
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-Derived ASC Specks Cross-Seed Amyloid-β in Alzheimer’s Disease. Nature 2017, 552, 355–361.
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.S.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and Biomarker Changes in Dominantly Inherited Alzheimer’s Disease. N. Engl. J. Med. 2012, 367, 795–804.
- Jack, C.R.; Holtzman, D.M. Biomarker Modeling of Alzheimer’s Disease. Neuron 2013, 80, 1347–1358.
- Liu, C.-C.; Zhao, N.; Fu, Y.; Wang, N.; Linares, C.; Tsai, C.-W.; Bu, G. ApoE4 Accelerates Early Seeding of Amyloid Pathology. Neuron 2017, 96, 1024–1032.e3.
- Travis, M.A.; Reizis, B.; Melton, A.C.; Masteller, E.; Tang, Q.; Proctor, J.M.; Wang, Y.; Bernstein, X.; Huang, X.; Reichardt, L.F.; et al. Loss of Integrin Alpha(v)Beta8 on Dendritic Cells Causes Autoimmunity and Colitis in Mice. Nature 2007, 449, 361–365.
- Yin, Z.; Herron, S.; Silveira, S.; Kleemann, K.; Gauthier, C.; Mallah, D.; Cheng, Y.; Margeta, M.A.; Pitts, K.M.; Barry, J.-L.; et al. Identification of a Protective Microglial State Mediated by MiR-155 and Interferon-γ Signaling in a Mouse Model of Alzheimer’s Disease. Nat. Neurosci. 2023, 26, 1196–1207.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
428
Revisions:
2 times
(View History)
Update Date:
12 Mar 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No