 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Christina Piperi | -- | 3241 | 2024-03-08 10:41:22 | | | |
| 2 | Wendy Huang | Meta information modification | 3241 | 2024-03-08 12:50:04 | | |
Video Upload Options
Histone lysine methyltransferase SUV4-20H2, a member of the suppressor of variegation 4–20 homolog (SUV4-20) family, has a critical impact on the regulation of chromatin structure and gene expression. This methyltransferase establishes the trimethylation of histone H4 lysine 20 (H4K20me3), a repressive histone mark that affects several cellular processes. Deregulated SUV4-20H2 activity has been associated with altered chromatin dynamics, leading to the misregulation of key genes involved in cell cycle control, apoptosis and DNA repair. Emerging research evidence indicates that SUV4-20H2 acts as a potential epigenetic modifier, contributing to the development and progression of several malignancies, including breast, colon and lung cancer, as well as renal, hepatocellular and pancreatic cancer.
1. Introduction
1.1. DNA Methylation: A Key Epigenetic Mechanism in Cancer
1.2. The Role of Histone Modifications in Cancer
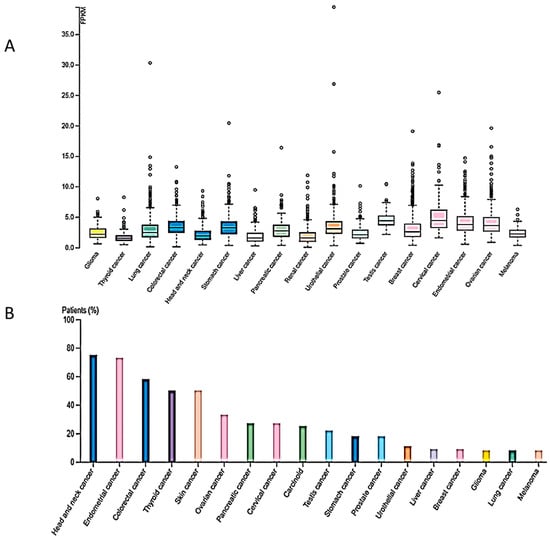
2. Cancer Types Exhibiting Reduced SUV4-20H2 Expression
2.1. Breast Cancer
2.2. Colon Cancer
2.3. Lung Cancer
3. Cancer Types Exhibiting Increased SUV4-20H2 Expression
3.1. Pancreatic Cancer
3.2. Renal Cell Carcinoma
| Cancer Type | SUV420H2 Expression | H4K20me3 Expression |
Main Effects | Reference |
|---|---|---|---|---|
| Breast | High levels in low-grade tumors Low levels in high-grade tumors |
Low | Repression of genes responsible for cell adhesion, tensin-3 blocked by miR-29a |
[36][37] |
| Colon | Low levels | Low | Enrichment of genes implicated in Wnt pathway and dissociation of LAD areas in mouse embryonic fibroblasts, attenuating gene repression | [28] |
| Lung | Low levels | Low | Loss of H4K20me3 correlates with worse survival in stage I adenocarcinoma patients | [29] |
| Pancreatic | High levels | High | Regulation of MET-associated transcriptional factors, FOXA1 and OVOL 1/2, regulation of epithelial or mesenchymal state | [30] |
| Renal cell carcinoma | High levels | High | Maintenance of H4K20me3 levels on DHRS2 promoter to maintain growth and avoid apoptosis | [31] |
| Hepatocellular | High levels in early stages, which drop with disease progression | Low | Reduction in epithelial marker E-cadherin and increase in mesenchymal markers SMA and MMP-9 | [33] |
3.3. Hepatocellular Carcinoma
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674.
- Shamsi, M.B.; Firoz, A.S.; Imam, S.N.; Alzaman, N.; Samman, M.A. Epigenetics of Human Diseases and Scope in Future Therapeutics. J. Taibah Univ. Med. Sci. 2017, 12, 205–211.
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in Cancer. Carcinogenesis 2010, 31, 27–36.
- Leick, M.B.; Shoff, C.J.; Wang, E.C.; Congress, J.L.; Gallicano, G.I. Loss of Imprinting of IGF2 and the Epigenetic Progenitor Model of Cancer. Am. J. Stem Cells 2012, 1, 59–74.
- Chen, J.F.; Yan, Q. The Roles of Epigenetics in Cancer Progression and Metastasis. Biochem. J. 2021, 478, 3373–3393.
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting Epigenetic Regulators for Cancer Therapy: Mechanisms and Advances in Clinical Trials. Signal Transduct. Target. Ther. 2019, 4, 62.
- Ahuja, N.; Sharma, A.R.; Baylin, S.B. Epigenetic Therapeutics: A New Weapon in the War Against Cancer. Annu. Rev. Med. 2016, 67, 73–89.
- Martisova, A.; Holcakova, J.; Izadi, N.; Sebuyoya, R.; Hrstka, R.; Bartosik, M. DNA Methylation in Solid Tumors: Functions and Methods of Detection. Int. J. Mol. Sci. 2021, 22, 4247.
- Nishiyama, A.; Nakanishi, M. Navigating the DNA Methylation Landscape of Cancer. Trends Genet. 2021, 37, 1012–1027.
- Xie, W.; Schultz, M.D.; Lister, R.; Hou, Z.; Rajagopal, N.; Ray, P.; Whitaker, J.W.; Tian, S.; Hawkins, R.D.; Leung, D.; et al. Epigenomic Analysis of Multilineage Differentiation of Human Embryonic Stem Cells. Cell 2013, 153, 1134–1148.
- Pappalardo, X.G.; Barra, V. Losing DNA Methylation at Repetitive Elements and Breaking Bad. Epigenetics Chromatin 2021, 14, 25.
- Messerschmidt, D.M.; Knowles, B.B.; Solter, D. DNA Methylation Dynamics during Epigenetic Reprogramming in the Germline and Preimplantation Embryos. Genes Dev. 2014, 28, 812–828.
- Cusack, M.; King, H.W.; Spingardi, P.; Kessler, B.M.; Klose, R.J.; Kriaucionis, S. Distinct Contributions of DNA Methylation and Histone Acetylation to the Genomic Occupancy of Transcription Factors. Genome Res. 2020, 30, 1393–1406.
- Nan, X.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional Repression by the Methyl-CpG-Binding Protein MeCP2 Involves a Histone Deacetylase Complex. Nature 1998, 393, 386–389.
- Wade, P.A.; Gegonne, A.; Jones, P.L.; Ballestar, E.; Aubry, F.; Wolffe, A.P. Mi-2 Complex Couples DNA Methylation to Chromatin Remodelling and Histone Deacetylation. Nat. Genet. 1999, 23, 62–66.
- Lee, H.-T.; Oh, S.; Ro, D.H.; Yoo, H.; Kwon, Y.-W. The Key Role of DNA Methylation and Histone Acetylation in Epigenetics of Atherosclerosis. J. Lipid Atheroscler. 2020, 9, 419–434.
- Kim, S. New and Emerging Factors in Tumorigenesis: An Overview. Cancer Manag. Res. 2015, 7, 225–239.
- Lakshminarasimhan, R.; Liang, G. The Role of DNA Methylation in Cancer. In DNA Methyltransferases—Role and Function; Jeltsch, A., Jurkowska, R.Z., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2016; Volume 945, pp. 151–172.
- Vitorakis, N.; Piperi, C. Insights into the Role of Histone Methylation in Brain Aging and Potential Therapeutic Interventions. Int. J. Mol. Sci. 2023, 24, 17339.
- Markouli, M.; Strepkos, D.; Piperi, C. Structure, Activity and Function of the SETDB1 Protein Methyltransferase. Life 2021, 11, 817.
- Markouli, M.; Strepkos, D.; Basdra, E.K.; Papavassiliou, A.G.; Piperi, C. Prominent Role of Histone Modifications in the Regulation of Tumor Metastasis. Int. J. Mol. Sci. 2021, 22, 2778.
- Angelopoulou, E.; Pyrgelis, E.-S.; Ahire, C.; Suman, P.; Mishra, A.; Piperi, C. Functional Implications of Protein Arginine Methyltransferases (PRMTs) in Neurodegenerative Diseases. Biology 2023, 12, 1257.
- Jarrold, J.; Davies, C.C. PRMTs and Arginine Methylation: Cancer’s Best-Kept Secret? Trends Mol. Med. 2019, 25, 993–1009.
- Liu, L.; Kimball, S.; Liu, H.; Holowatyj, A.; Yang, Z.-Q. Genetic Alterations of Histone Lysine Methyltransferases and Their Significance in Breast Cancer. Oncotarget 2015, 6, 2466–2482.
- Zhu, Z.; Jiang, L.; Ding, X. Advancing Breast Cancer Heterogeneity Analysis: Insights from Genomics, Transcriptomics and Proteomics at Bulk and Single-Cell Levels. Cancers 2023, 15, 4164.
- Yu, W.; Liu, N.; Song, X.; Chen, L.; Wang, M.; Xiao, G.; Li, T.; Wang, Z.; Zhang, Y. EZH2: An Accomplice of Gastric Cancer. Cancers 2023, 15, 425.
- Chen, Y.; Ren, B.; Yang, J.; Wang, H.; Yang, G.; Xu, R.; You, L.; Zhao, Y. The Role of Histone Methylation in the Development of Digestive Cancers: A Potential Direction for Cancer Management. Signal Transduct. Target. Ther. 2020, 5, 143.
- Tryndyak, V.P.; Kovalchuk, O.; Pogribny, I.P. Loss of DNA Methylation and Histone H4 Lysine 20 Trimethylation in Human Breast Cancer Cells Is Associated with Aberrant Expression of DNA Methyltransferase 1, Suv4-20h2 Histone Methyltransferase and Methyl-Binding Proteins. Cancer Biol. Ther. 2006, 5, 65–70.
- Isin, H.; Özgür, E.; Kelten Talu, C.; Can Trabulus, D.; Karaçetin, D.; Gezer, U. Impact of Histone Methyltransferase SUV420H2 in Breast Cancer. Biomed. Rep. 2020.
- Shinchi, Y.; Hieda, M.; Nishioka, Y.; Matsumoto, A.; Yokoyama, Y.; Kimura, H.; Matsuura, S.; Matsuura, N. SUV420H2 Suppresses Breast Cancer Cell Invasion through down Regulation of the SH2 Domain-Containing Focal Adhesion Protein Tensin-3. Exp. Cell Res. 2015, 334, 90–99.
- Wu, Y.; Shi, W.; Tang, T.; Wang, Y.; Yin, X.; Chen, Y.; Zhang, Y.; Xing, Y.; Shen, Y.; Xia, T.; et al. miR-29a Contributes to Breast Cancer Cells Epithelial–Mesenchymal Transition, Migration, and Invasion via down-Regulating Histone H4K20 Trimethylation through Directly Targeting SUV420H2. Cell Death Dis. 2019, 10, 176.
- Boonsanay, V.; Mosa, M.H.; Looso, M.; Weichenhan, D.; Ceteci, F.; Pudelko, L.; Lechel, A.; Michel, C.S.; Künne, C.; Farin, H.F.; et al. Loss of SUV420H2-Dependent Chromatin Compaction Drives Right-Sided Colon Cancer Progression. Gastroenterology 2023, 164, 214–227.
- Van Den Broeck, A.; Brambilla, E.; Moro-Sibilot, D.; Lantuejoul, S.; Brambilla, C.; Eymin, B.; Gazzeri, S. Loss of Histone H4K20 Trimethylation Occurs in Preneoplasia and Influences Prognosis of Non-Small Cell Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 7237–7245.
- Viotti, M.; Wilson, C.; McCleland, M.; Koeppen, H.; Haley, B.; Jhunjhunwala, S.; Klijn, C.; Modrusan, Z.; Arnott, D.; Classon, M.; et al. SUV420H2 Is an Epigenetic Regulator of Epithelial/Mesenchymal States in Pancreatic Cancer. J. Cell Biol. 2018, 217, 763–777.
- Ryu, T.Y.; Lee, J.; Kang, Y.; Son, M.-Y.; Kim, D.-S.; Lee, Y.S.; Kim, M.-Y.; Cho, H.-S. Epigenetic Regulation of DHRS2 by SUV420H2 Inhibits Cell Apoptosis in Renal Cell Carcinoma. Biochem. Biophys. Res. Commun. 2023, 663, 41–46.
- Rhodes, C.T.; Zunino, G.; Huang, S.-W.A.; Cardona, S.M.; Cardona, A.E.; Berger, M.S.; Lemmon, V.P.; Lin, C.-H.A. Region Specific Knock-out Reveals Distinct Roles of Chromatin Modifiers in Adult Neurogenic Niches. Cell Cycle 2018, 17, 377–389.
- Wickramasekara, R.; Stessman, H. Histone 4 Lysine 20 Methylation: A Case for Neurodevelopmental Disease. Biology 2019, 8, 11.
- Pogribny, I.P.; Ross, S.A.; Tryndyak, V.P.; Pogribna, M.; Poirier, L.A.; Karpinets, T.V. Histone H3 Lysine 9 and H4 Lysine 20 Trimethylation and the Expression of Suv4-20h2 and Suv-39h1 Histone Methyltransferases in Hepatocarcinogenesis Induced by Methyl Deficiency in Rats. Carcinogenesis 2006, 27, 1180–1186.
- Phoyen, S.; Sanpavat, A.; Ma-on, C.; Stein, U.; Hirankarn, N.; Tangkijvanich, P.; Jindatip, D.; Whongsiri, P.; Boonla, C. H4K20me3 Upregulated by Reactive Oxygen Species Is Associated with Tumor Progression and Poor Prognosis in Patients with Hepatocellular Carcinoma. Heliyon 2023, 9, e22589.

