Histone lysine methyltransferase SUV4-20H2, a member of the suppressor of variegation 4–20 homolog (SUV4-20) family, has a critical impact on the regulation of chromatin structure and gene expression. This methyltransferase establishes the trimethylation of histone H4 lysine 20 (H4K20me3), a repressive histone mark that affects several cellular processes. Deregulated SUV4-20H2 activity has been associated with altered chromatin dynamics, leading to the misregulation of key genes involved in cell cycle control, apoptosis and DNA repair. Emerging research evidence indicates that SUV4-20H2 acts as a potential epigenetic modifier, contributing to the development and progression of several malignancies, including breast, colon and lung cancer, as well as renal, hepatocellular and pancreatic cancer.
- histone modifications
- lysine methyltransferase
- SUV4-20H2
- DNA methylation
- epigenetic
- cancer
1. Introduction
1.1. DNA Methylation: A Key Epigenetic Mechanism in Cancer
Among the main epigenetic mechanisms, DNA methylation has been extensively studied and linked to cancer initiation and development, taking place at the 5′ ends of genes on cytosine residues of CpG dinucleotides in 60% of human genes [3,8][3][8]. DNA methylation is a central regulatory mechanism for gene expression during development, with CpG methylation levels varying according to the stage of the process, thus regulating genes necessary for the respective developmental stages [9,10][9][10]. High levels of DNA methylation are observed in repetitive genomic sequences, where heavy methylation attenuates genome instability. Examples of such genomic areas include transposable elements, noncoding DNA sequences and long interspersed nuclear elements [11]. In this background, DNA methylation serves as a stabilizing agent that protects chromatin structure and integrity. It is worth noting that DNA methylation can serve as a docking site for proteins while stabilizing chromatin structure. As an epigenetic mark present in gene promoters, DNA methylation serves as a repressor of transcription with a very important contribution not only in the correct regulation of development but also in the establishment of X-chromosome inactivation and repression of imprinted genes [12]. A reported mechanism that explains the repressive effect of DNA methylation, in some contexts and with regard to some transcription factors, involves the recruitment of proteins capable of recognizing CpG-methylated dinucleotides. These proteins also have the ability to simultaneously recruit histone deacetylases (HDACs), enzymes that remove acetylation marks from histones, thus creating a transcriptional repressive environment [13,14,15,16][13][14][15][16]. The establishment of DNA methylation takes place through three DNA methyltransferases (DNMTs), whereas removal of the mark is accomplished through the ten-eleven translocation family of dioxygenases (TET enzymes). In cancer, DNA methylation follows different patterns in distinct genomic regions. For example, in CG-rich promoters, DNA methylation can increase dramatically, thus repressing the expression of genes such as tumor suppressor or DNA repair- and cell cycle-related ones [8]. As a result, cell proliferation is left unchecked, either directly or indirectly. Examples include the breast cancer gene 1 (BRCA1) in ovarian and breast cancer, the Von Hippel–Lindau tumor suppressor (VHL) in clear renal cell carcinoma and the O-6-methylguanine-DNA methyltransferase (MGMT) repair gene in glioblastoma (GB) [17]. Hypomethylation, on the other hand, has been reported on imprinted genes such as IGF2/H19, which is responsible for the production of epidermal growth factor 2 (EGF2), in intragenic and intergenic regions where long or short interspersed elements can be found. Consequently, differential production of regulating factors and high motility of retrotransposons are observed, leading to increased proliferating signals and chromosome instability, respectively [18].1.2. The Role of Histone Modifications in Cancer
DNA methylation is not the only epigenetic mechanism with a prominent role in carcinogenesis. Histone modifications have also been detected in all cancer types, established post-translationally and exerting pivotal roles in gene expression regulation and chromatin structure. Histone modifications encompass a variety of chemical changes in N-terminal amino acids of histone proteins including methylation, acetylation, SUMOylation, ADP-ribosylation and ubiquitination [3]. Among them, histone methylation plays a prominent role in the activation as well as the suppression of gene transcription depending on the modified amino acid in the respective histone member. Methylation of histones is a well-studied mark, context-dependent and tightly associated with the aforementioned processes. It can take place on lysine or arginine residues of H3 and H4 in different states or degrees of methylation, with each level bearing distinct regulatory effects in normal physiology [19]. The establishment of histone methylation on lysine residues takes place through the enzymatic activity of six protein families, each one of them targeting different residues on histone molecules [20]. The same number of families exist also for the reverse reaction, without the protein family distinction representing substrate specificity. Lysine methylation can be grouped into activating signals such as H3K4, H3K36, H3K79 and repressive marks such as H3K9 and H4K20 [20]. Deregulation of enzymes involved in the addition or removal of methyl group(s) from lysine or arginine residues has been involved in cancer [21]. Arginine histone methylation is mediated through the respective group of enzymes named arginine methyltransferases (PRMTs), which can induce mono- or dimethylation of histones in a symmetrical and nonsymmetrical way [22]. The removal of arginine methylation marks takes place through deamination, yielding citrulline as an end product. In hematological and solid tumors, the levels of PRTMs appear to be significantly elevated and correlate with poor patient survival [23]. An intriguing fact about PRMTs is their ability to create both activating and repressive marks by cooperating with distinct proteins on different promoters. In the case of acute myeloid leukemia, H4R3me2a participates in epithelial–mesenchymal transition (EMT) by regulating zinc finger E-box binding homeobox 1 (ZEB1) promoter, while its demethylation under the influence of specific factors is implicated in leukemic transformation. Regarding histone lysine methylation, more data and genome-wide analyses are present that indicate a pivotal role in carcinogenesis. In breast cancer, a set of KMTs (histone lysine methyltransferases) was associated both on the mRNA and protein level with poor prognosis, bearing alterations in gene sequence as well as in expression levels and, thus, transforming the methylation levels of their respective histone lysine targets [24]. It is of note that in different types of breast cancer or in different stages of the disease, a distinct set of these enzymes is aberrantly expressed, demonstrating yet again the contextual nature of these marks [25]. In gastric cancers, high levels of enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2), a writer of H3K27me3, results in gene repression, whereas the epigenetic mark levels have been reduced when the core components of the polycomb complex are reduced, indicating the contextual nature of epigenetic marks in the heterogenous cancer background [26]. Additional examples of deregulated activity of histone methylation writers and erasers have been observed in digestive cancers [27]. Given the important role of H4K20me3 in vital cellular functions, it is evident that aberrant expression and activity of SUV4-20H2 may be associated with tumorigenesis. In fact, RNAseq expression analysis from the TCGA dataset revealed the presence of the KMT5C gene in several cancer types shown in Figure 1A. Moreover, SUV420H2 protein expression (HPA dataset) was higher in head and neck cancer, endometrial, colorectal, thyroid and skin cancer, as well as in several cases of ovarian, pancreatic, cervical, prostate and stomach cancers, exhibiting moderate to strong cytoplasmic staining along with nuclear positivity in some cases (Figure 1B).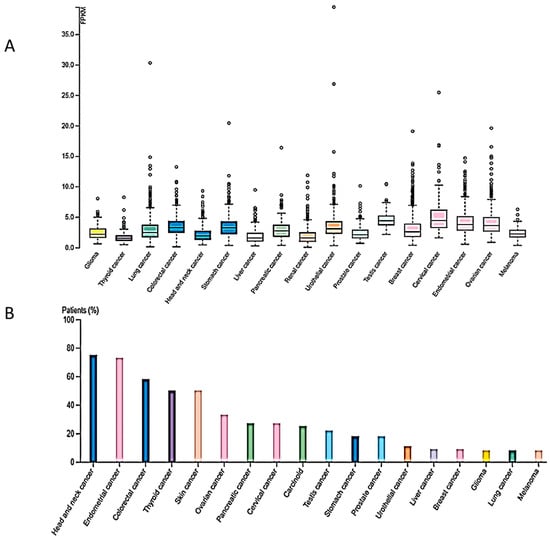
2. Cancer Types Exhibiting Reduced SUV4-20H2 Expression
Of importance, several cancer types including breast, colon and lung carcinoma exhibit loss of the heterochromatic mark H4K20me3 and reduced SUV4-20H2 expression with disease progression.2.1. Breast Cancer
More specifically, a study of breast cancer cell lines of different breast cancer subtypes showed that tumor progression was accompanied by significant epigenetic alterations such as loss of DNA methylation, loss of H4K20me3 and hyperacetylation of histone H4 [52][28]. In particular, MDA-MB-231 and MDA-MB-231(S30) cell lines, characterized by a more invasive and malignant phenotype, bear the aforementioned characteristics in a more exacerbated form as opposed to MCF-7 cells, which are indicative of a more subtle luminal subtype. In addition, lower H4K20me3 levels were accompanied by lower SUV4-20H2 levels as well as altered levels of DNA methyltransferase 1 (DNMT1), methyl-CpG binding protein 2 (MeCP2) and methyl-CpG binding domain protein 2 (MBD2), which are interacting proteins of the H4 trimethylation mark. These findings were recently confirmed in patient samples [53][29], where higher levels of the enzyme were detected in the neoplastic tissue as opposed to the adjacent healthy cells in the early stages of breast cancer. However, in more advanced stages of the disease, lower levels of SUV4-20H2 were observed compared to healthy tissue. A possible mechanism of SUV4-20H2 action involves transcriptional regulation and, more precisely, repression of genes associated with cell adhesion, such as tensin-3 [54][30]. It has been shown that SUV4-20H2 establishes its repressive mark upstream of the transcription start site of tensin-3, a focal adhesion protein associated with cancer cell migration, thus inhibiting its transcription and subsequently, the metastasis of breast cancer cells. Τhe tensin family proteins comprise four members, namely tensin 1, tensin 2, tensin 3 and c-ten. All the members of the family bear two important domains in their C-terminal part. The first is the Src homology domain (SH2), which is important for phosphorylation by the Src protein, and the second domain is the phosphotyrosine binding domain (PTB), which mediates interactions with β-integrin. These findings were confirmed through exogenous delivery of the methyltransferase in MDA-MB-231 cells as well as genetic manipulation of MDA-MB-231, resulting in overexpression of SUV4-20H2, while cell invasiveness was substantially diminished. Another mechanism that appears to regulate EMT in breast cancer is the induction of miR-29a by the basic fibroblast growth factor (bFGF) [55][31]. This protein is mainly produced by microvascular endothelial cells of tumor tissues and promotes cell invasiveness. In response to the production of bFGF, miR-29a is transcribed and binds to the 3′UTR of SUV4-20H2 mRNA, thus blocking its production. As a result, suppression of connective tissue growth factor (CTGF) and growth response protein-1 (EGR-1) were attenuated, leading to EMT progress of breast cancer cells. It is of note that miR-29a was detected to be elevated in breast cancer stem cells and involved in tumor formation, metastasis and drug resistance, indicating a possible therapeutic target.2.2. Colon Cancer
Studies addressing the role of SUV4-20H2 in colon cancer (CC) have shown that murine and patient-derived colorectal cancer organoids exhibit low levels of H4K20me3 in right-sided CC (RCC) patients and high chromatin accessibility [56][32]. However, it was reported that the methyltransferase levels did not differ between right and left CC patients, which was attributed to lower enzymatic activity. The higher chromatin accessibility was studied through MNase assay and the phenotype was rescued through methylstat, introduced to mice bearing tumors made from RCC patient-derived organoids. In addition, murine colon-derived organoids bearing mutations associated with RCC, namely Tgrbr2-deficient and K-ras G12D, exhibited decreased H4K20me3 and SUV4-20H2 levels, along with loosely packed chromatin. The compaction was ameliorated by applying methylstat, which resulted in larger tumors too. In a similar murine organoid of RCC, knockdown of SUV4-20H2 was shown to induce more aggressive orthotropic tumors, lower chromatin compaction and enrichment of genes targeted by the Wnt pathway such as leucine-rich repeat-containing G-protein coupled receptor 5 (Lgr5), SRY-box transcription factor 2 and 9 (Sox2, Sox9) and prominin-1 (CD133). It is worth highlighting that these genes are localized in LAD (lamina-associated domain) areas in mouse embryonic fibroblasts, leading to the hypothesis that the decrease in SUV4-20H2 levels allows for the dissociation of LAD areas from the nuclear lamina, thus attenuating gene repression. Chromatin immunoprecipitation experiments targeting the promoter of Lgr5 confirmed this assumption and the interplay of regulating gene expression through chromatin compaction and association with the nuclear membrane.2.3. Lung Cancer
A correlation between SUV4-20H2 levels and tumor development has been suggested in an immunohistochemical study of lung cancer patients, where H4K20me3 changes were observed in squamous cell carcinoma. H4K20me3 staining in early precursor lesions decreased significantly with disease progression and was associated with reduced SUV4-20H2 expression [57][33].3. Cancer Types Exhibiting Increased SUV4-20H2 Expression
3.1. Pancreatic Cancer
In pancreatic cancer, the role of SUV4-20H2 appears to be different and related to the regulation of genes responsible for mesenchymal phenotype, which is associated with higher invasiveness and drug resistance. While screening pancreatic ductal adenocarcinoma (PDAC) human samples, it was shown that the SUV4-20H2 reported reduced levels, contributing to the establishment of the epithelial state, as demonstrated through E-cadherin and epithelial cell adhesion molecule (EPCAM). On the contrary, higher levels of SUV4-20H2 establish the H4K20me3 repressive mark that blocks the expression of epithelial transcriptional marks and allows for the mesenchymal state to be developed, as reported through vimentin levels [58][34]. Furthermore, it was shown through bioinformatic analysis that knockdown of SUV4-20H2 results in the activation of genes involved in cell adhesion, cytoskeleton and extracellular matrix as well as markers related to epithelial state. Functional studies demonstrated that the knockdown of the methyltransferase is related to reduced migration and invasion potential. Moreover, higher sensitivity to drugs as well as lower levels of CD24 and CD44 were reported, indicating a reduction in cancer stem cells, which bear the ability to form tumors of pancreatic origin in xenografts [58][34]. It is worth pointing out that SUV4-20H2 exerts the aforementioned effects through transcriptional regulation of MET-associated transcriptional factors, namely forkhead box A1 (FOXA.1) and Ovo-like transcriptional repressor 1 and 2 (OVOL1/2) in pancreatic cancer, which in turn have the ability to activate different downstream signaling pathways. It is of interest that the epigenetic mark H4K20me3, established by SUV420H2, was differentially placed on gene loci associated with transcription factors implicated in mesenchymal and epithelial states. The levels of the mark dropped in the case of FOXA.1 and OVOL1/2, but no changes were reported for Cadherin 2 (CDH1) and EPCAM, suggesting the indirect regulatory effect of SUV4-20H2 on epithelial state-related genes. Furthermore, in pancreatic cell lines with a more epithelial-like phenotype, overexpression of SUV4-20H2 demonstrated the repressed levels of Cadherin 2 (CDH2) and EPCAM as well as increased levels of vimentin, implicating the enzyme in the regulation of the epithelial to mesenchymal transition. Finally, immunofluorescent staining of patient-derived tissues showed a correlation of SUV420H2 expression with the mesenchymal state and disease progression [58][34].3.2. Renal Cell Carcinoma
In renal cell carcinoma, SUV4-20H2 was shown to epigenetically regulate cell proliferation through targeting of dehydrogenase/reductase 2 (DHRS2) [59][35]. Using the A498 clear cell renal cancer cell line, representative of the majority of kidney cancers, it was demonstrated that silencing of the methyltransferase suppressed growth and induced cell apoptosis through attenuation of H4K20me3 levels on the promoter of DHRS2. It is of note that simultaneous silencing of KMT5C and DHRS2 rescued the phenotype, while the use of SUV4-20 family protein inhibitor A-196 resulted in cell apoptosis by increasing the levels of DHRS2. Furthermore, the research data highlight the higher levels of SUV4-20H2 in renal cell carcinoma patients (Table 1) and their association with poor prognosis [59][35].| Cancer Type | SUV420H2 Expression | H4K20me3 Expression |
Main Effects | Reference |
|---|---|---|---|---|
| Breast | High levels in low-grade tumors Low levels in high-grade tumors |
Low | Repression of genes responsible for cell adhesion, tensin-3 blocked by miR-29a |
[50,51][36][37] |
| Colon | Low levels | Low | Enrichment of genes implicated in Wnt pathway and dissociation of LAD areas in mouse embryonic fibroblasts, attenuating gene repression | [52][28] |
| Lung | Low levels | Low | Loss of H4K20me3 correlates with worse survival in stage I adenocarcinoma patients | [53][29] |
| Pancreatic | High levels | High | Regulation of MET-associated transcriptional factors, FOXA1 and OVOL 1/2, regulation of epithelial or mesenchymal state | [54][30] |
| Renal cell carcinoma | High levels | High | Maintenance of H4K20me3 levels on DHRS2 promoter to maintain growth and avoid apoptosis | [55][31] |
| Hepatocellular | High levels in early stages, which drop with disease progression | Low | Reduction in epithelial marker E-cadherin and increase in mesenchymal markers SMA and MMP-9 | [57][33] |
3.3. Hepatocellular Carcinoma
The implication of SUV4-20H2 in hepatocellular cancer was revealed in the F344 rat model of endogenous hepatocarcinogenesis [60][38] after treatment with the methyl-deficient diet (MDD), which showed a gradual loss of DNA methylation on repetitive elements, followed by the loss of H4K20me3 and the gradual decline of SUV4-20H2 levels with diseases progression. It was evident that hepatocellular carcinogenesis is associated with the gradual loss of histone H4 trimethylation due to lower enzyme activity (Table 1). Recently, it was shown that higher levels of H4K20me3 were associated with worse prognosis for hepatocellular carcinoma patients and suggested the reactive oxygen species (ROS) as one of the regulatory pathways for the induction of the epigenetic mark [61][39]. In HepG2 and Huh7 cell lines, the study demonstrated that during oxidative stress induced by the presence of H2O2, the epithelial marker E-cadherin is downregulated, whereas a-SMA and MMP-9 mesenchymal markers are increased. The repressive or activating pathways are initiated by ROS, but the underlying mechanism is not specified. In any case, cancer aggressiveness was substantially increased through the activation of EMT. These findings were further confirmed after treatment with the SUV4-20H2 inhibitor A-196, which resulted in decreased epigenetic mark levels and the EMT phenotype. Table 1 summarizes the studies indicating an implication of SUV4-20H2 in different cancer types.References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674.
- Shamsi, M.B.; Firoz, A.S.; Imam, S.N.; Alzaman, N.; Samman, M.A. Epigenetics of Human Diseases and Scope in Future Therapeutics. J. Taibah Univ. Med. Sci. 2017, 12, 205–211.
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in Cancer. Carcinogenesis 2010, 31, 27–36.
- Leick, M.B.; Shoff, C.J.; Wang, E.C.; Congress, J.L.; Gallicano, G.I. Loss of Imprinting of IGF2 and the Epigenetic Progenitor Model of Cancer. Am. J. Stem Cells 2012, 1, 59–74.
- Chen, J.F.; Yan, Q. The Roles of Epigenetics in Cancer Progression and Metastasis. Biochem. J. 2021, 478, 3373–3393.
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting Epigenetic Regulators for Cancer Therapy: Mechanisms and Advances in Clinical Trials. Signal Transduct. Target. Ther. 2019, 4, 62.
- Ahuja, N.; Sharma, A.R.; Baylin, S.B. Epigenetic Therapeutics: A New Weapon in the War Against Cancer. Annu. Rev. Med. 2016, 67, 73–89.
- Martisova, A.; Holcakova, J.; Izadi, N.; Sebuyoya, R.; Hrstka, R.; Bartosik, M. DNA Methylation in Solid Tumors: Functions and Methods of Detection. Int. J. Mol. Sci. 2021, 22, 4247.
- Nishiyama, A.; Nakanishi, M. Navigating the DNA Methylation Landscape of Cancer. Trends Genet. 2021, 37, 1012–1027.
- Xie, W.; Schultz, M.D.; Lister, R.; Hou, Z.; Rajagopal, N.; Ray, P.; Whitaker, J.W.; Tian, S.; Hawkins, R.D.; Leung, D.; et al. Epigenomic Analysis of Multilineage Differentiation of Human Embryonic Stem Cells. Cell 2013, 153, 1134–1148.
- Pappalardo, X.G.; Barra, V. Losing DNA Methylation at Repetitive Elements and Breaking Bad. Epigenetics Chromatin 2021, 14, 25.
- Messerschmidt, D.M.; Knowles, B.B.; Solter, D. DNA Methylation Dynamics during Epigenetic Reprogramming in the Germline and Preimplantation Embryos. Genes Dev. 2014, 28, 812–828.
- Cusack, M.; King, H.W.; Spingardi, P.; Kessler, B.M.; Klose, R.J.; Kriaucionis, S. Distinct Contributions of DNA Methylation and Histone Acetylation to the Genomic Occupancy of Transcription Factors. Genome Res. 2020, 30, 1393–1406.
- Nan, X.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional Repression by the Methyl-CpG-Binding Protein MeCP2 Involves a Histone Deacetylase Complex. Nature 1998, 393, 386–389.
- Wade, P.A.; Gegonne, A.; Jones, P.L.; Ballestar, E.; Aubry, F.; Wolffe, A.P. Mi-2 Complex Couples DNA Methylation to Chromatin Remodelling and Histone Deacetylation. Nat. Genet. 1999, 23, 62–66.
- Lee, H.-T.; Oh, S.; Ro, D.H.; Yoo, H.; Kwon, Y.-W. The Key Role of DNA Methylation and Histone Acetylation in Epigenetics of Atherosclerosis. J. Lipid Atheroscler. 2020, 9, 419–434.
- Kim, S. New and Emerging Factors in Tumorigenesis: An Overview. Cancer Manag. Res. 2015, 7, 225–239.
- Lakshminarasimhan, R.; Liang, G. The Role of DNA Methylation in Cancer. In DNA Methyltransferases—Role and Function; Jeltsch, A., Jurkowska, R.Z., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2016; Volume 945, pp. 151–172.
- Vitorakis, N.; Piperi, C. Insights into the Role of Histone Methylation in Brain Aging and Potential Therapeutic Interventions. Int. J. Mol. Sci. 2023, 24, 17339.
- Markouli, M.; Strepkos, D.; Piperi, C. Structure, Activity and Function of the SETDB1 Protein Methyltransferase. Life 2021, 11, 817.
- Markouli, M.; Strepkos, D.; Basdra, E.K.; Papavassiliou, A.G.; Piperi, C. Prominent Role of Histone Modifications in the Regulation of Tumor Metastasis. Int. J. Mol. Sci. 2021, 22, 2778.
- Angelopoulou, E.; Pyrgelis, E.-S.; Ahire, C.; Suman, P.; Mishra, A.; Piperi, C. Functional Implications of Protein Arginine Methyltransferases (PRMTs) in Neurodegenerative Diseases. Biology 2023, 12, 1257.
- Jarrold, J.; Davies, C.C. PRMTs and Arginine Methylation: Cancer’s Best-Kept Secret? Trends Mol. Med. 2019, 25, 993–1009.
- Liu, L.; Kimball, S.; Liu, H.; Holowatyj, A.; Yang, Z.-Q. Genetic Alterations of Histone Lysine Methyltransferases and Their Significance in Breast Cancer. Oncotarget 2015, 6, 2466–2482.
- Zhu, Z.; Jiang, L.; Ding, X. Advancing Breast Cancer Heterogeneity Analysis: Insights from Genomics, Transcriptomics and Proteomics at Bulk and Single-Cell Levels. Cancers 2023, 15, 4164.
- Yu, W.; Liu, N.; Song, X.; Chen, L.; Wang, M.; Xiao, G.; Li, T.; Wang, Z.; Zhang, Y. EZH2: An Accomplice of Gastric Cancer. Cancers 2023, 15, 425.
- Chen, Y.; Ren, B.; Yang, J.; Wang, H.; Yang, G.; Xu, R.; You, L.; Zhao, Y. The Role of Histone Methylation in the Development of Digestive Cancers: A Potential Direction for Cancer Management. Signal Transduct. Target. Ther. 2020, 5, 143.
- Tryndyak, V.P.; Kovalchuk, O.; Pogribny, I.P. Loss of DNA Methylation and Histone H4 Lysine 20 Trimethylation in Human Breast Cancer Cells Is Associated with Aberrant Expression of DNA Methyltransferase 1, Suv4-20h2 Histone Methyltransferase and Methyl-Binding Proteins. Cancer Biol. Ther. 2006, 5, 65–70.
- Isin, H.; Özgür, E.; Kelten Talu, C.; Can Trabulus, D.; Karaçetin, D.; Gezer, U. Impact of Histone Methyltransferase SUV420H2 in Breast Cancer. Biomed. Rep. 2020.
- Shinchi, Y.; Hieda, M.; Nishioka, Y.; Matsumoto, A.; Yokoyama, Y.; Kimura, H.; Matsuura, S.; Matsuura, N. SUV420H2 Suppresses Breast Cancer Cell Invasion through down Regulation of the SH2 Domain-Containing Focal Adhesion Protein Tensin-3. Exp. Cell Res. 2015, 334, 90–99.
- Wu, Y.; Shi, W.; Tang, T.; Wang, Y.; Yin, X.; Chen, Y.; Zhang, Y.; Xing, Y.; Shen, Y.; Xia, T.; et al. miR-29a Contributes to Breast Cancer Cells Epithelial–Mesenchymal Transition, Migration, and Invasion via down-Regulating Histone H4K20 Trimethylation through Directly Targeting SUV420H2. Cell Death Dis. 2019, 10, 176.
- Boonsanay, V.; Mosa, M.H.; Looso, M.; Weichenhan, D.; Ceteci, F.; Pudelko, L.; Lechel, A.; Michel, C.S.; Künne, C.; Farin, H.F.; et al. Loss of SUV420H2-Dependent Chromatin Compaction Drives Right-Sided Colon Cancer Progression. Gastroenterology 2023, 164, 214–227.
- Van Den Broeck, A.; Brambilla, E.; Moro-Sibilot, D.; Lantuejoul, S.; Brambilla, C.; Eymin, B.; Gazzeri, S. Loss of Histone H4K20 Trimethylation Occurs in Preneoplasia and Influences Prognosis of Non-Small Cell Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 7237–7245.
- Viotti, M.; Wilson, C.; McCleland, M.; Koeppen, H.; Haley, B.; Jhunjhunwala, S.; Klijn, C.; Modrusan, Z.; Arnott, D.; Classon, M.; et al. SUV420H2 Is an Epigenetic Regulator of Epithelial/Mesenchymal States in Pancreatic Cancer. J. Cell Biol. 2018, 217, 763–777.
- Ryu, T.Y.; Lee, J.; Kang, Y.; Son, M.-Y.; Kim, D.-S.; Lee, Y.S.; Kim, M.-Y.; Cho, H.-S. Epigenetic Regulation of DHRS2 by SUV420H2 Inhibits Cell Apoptosis in Renal Cell Carcinoma. Biochem. Biophys. Res. Commun. 2023, 663, 41–46.
- Rhodes, C.T.; Zunino, G.; Huang, S.-W.A.; Cardona, S.M.; Cardona, A.E.; Berger, M.S.; Lemmon, V.P.; Lin, C.-H.A. Region Specific Knock-out Reveals Distinct Roles of Chromatin Modifiers in Adult Neurogenic Niches. Cell Cycle 2018, 17, 377–389.
- Wickramasekara, R.; Stessman, H. Histone 4 Lysine 20 Methylation: A Case for Neurodevelopmental Disease. Biology 2019, 8, 11.
- Pogribny, I.P.; Ross, S.A.; Tryndyak, V.P.; Pogribna, M.; Poirier, L.A.; Karpinets, T.V. Histone H3 Lysine 9 and H4 Lysine 20 Trimethylation and the Expression of Suv4-20h2 and Suv-39h1 Histone Methyltransferases in Hepatocarcinogenesis Induced by Methyl Deficiency in Rats. Carcinogenesis 2006, 27, 1180–1186.
- Phoyen, S.; Sanpavat, A.; Ma-on, C.; Stein, U.; Hirankarn, N.; Tangkijvanich, P.; Jindatip, D.; Whongsiri, P.; Boonla, C. H4K20me3 Upregulated by Reactive Oxygen Species Is Associated with Tumor Progression and Poor Prognosis in Patients with Hepatocellular Carcinoma. Heliyon 2023, 9, e22589.