 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Pierluigi Scalia | -- | 3548 | 2024-03-07 21:04:24 | | | |
| 2 | Peter Tang | + 5 word(s) | 3553 | 2024-03-08 03:05:10 | | |
Video Upload Options
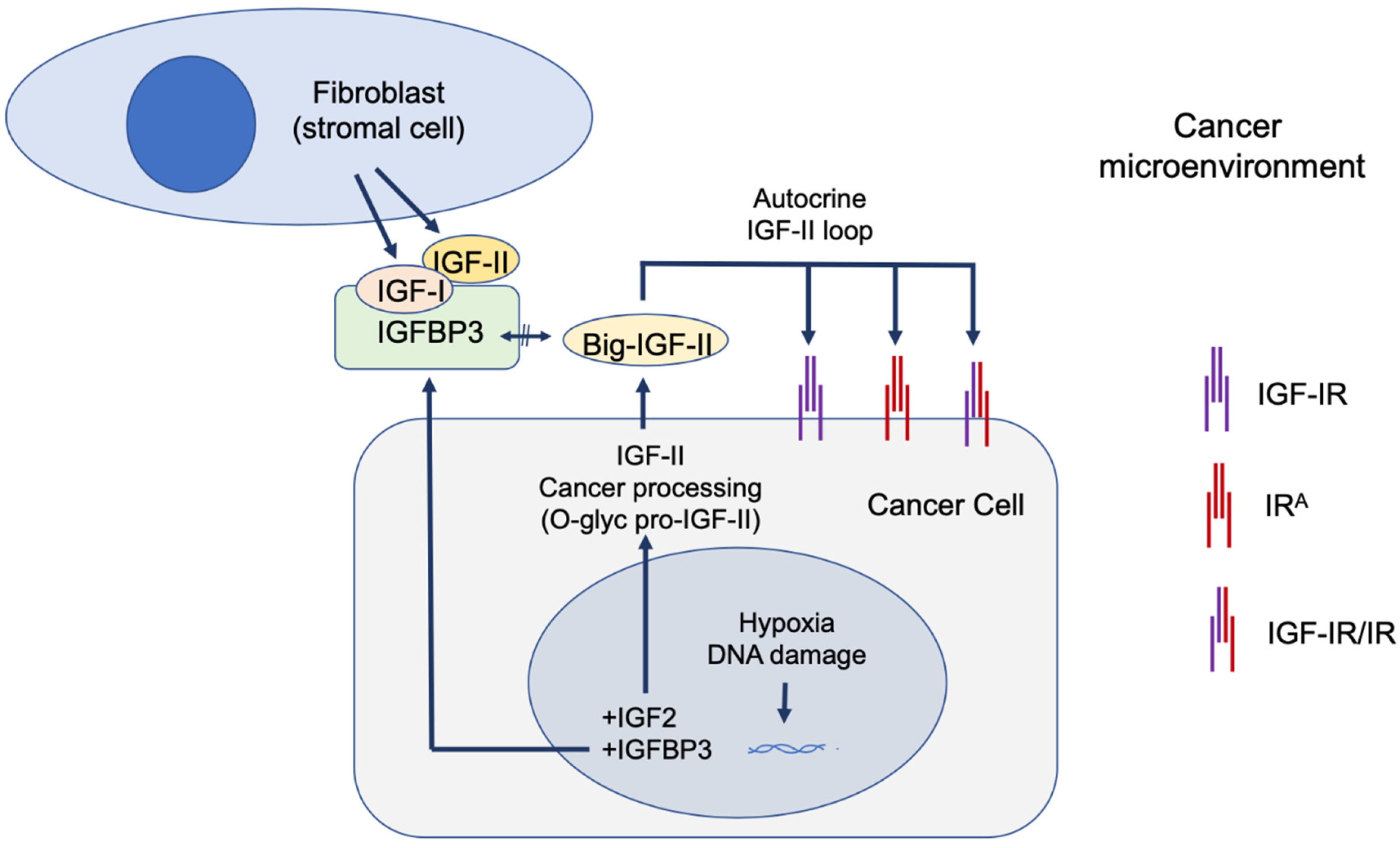
The paraneoplastic syndrome referred in the literature as non-islet-cell tumor hypoglycemia (NICTH) and extra-pancreatic tumor hypoglycemia (EPTH) was first reported almost a century ago, and the role of cancer-secreted IGF-II in causing this blood glucose-lowering condition has been widely established. The landscape emerging, based on molecular and cellular findings, supports a broader role for IGF-II in cancer biology beyond its involvement in the paraneoplastic syndrome. In particular, a few key findings are constantly observed during tumorigenesis, (a) a relative and absolute increase in fetal insulin receptor isoform (IRA) content, with (b) an increase in IGF-II high-molecular weight cancer-variants (big-IGF-II), and (c) a stage-progressive increase in the IGF-II autocrine signal in the cancer cell, mostly during the transition from benign to malignant growth. An increasing and still under-exploited combinatorial pattern of the IGF-II signal in cancer is shaping up in the literature with respect to its transducing receptorial system and effector intracellular network. Interestingly, while surgical and clinical reports have traditionally restricted IGF-II secretion to a small number of solid malignancies displaying paraneoplastic hypoglycemia, a retrospective literature analysis, along with publicly available expression data from patient-derived cancer cell lines conveyed in the present perspective, clearly suggests that IGF-II expression in cancer is a much more common event, especially in overt malignancy.
1. Introduction
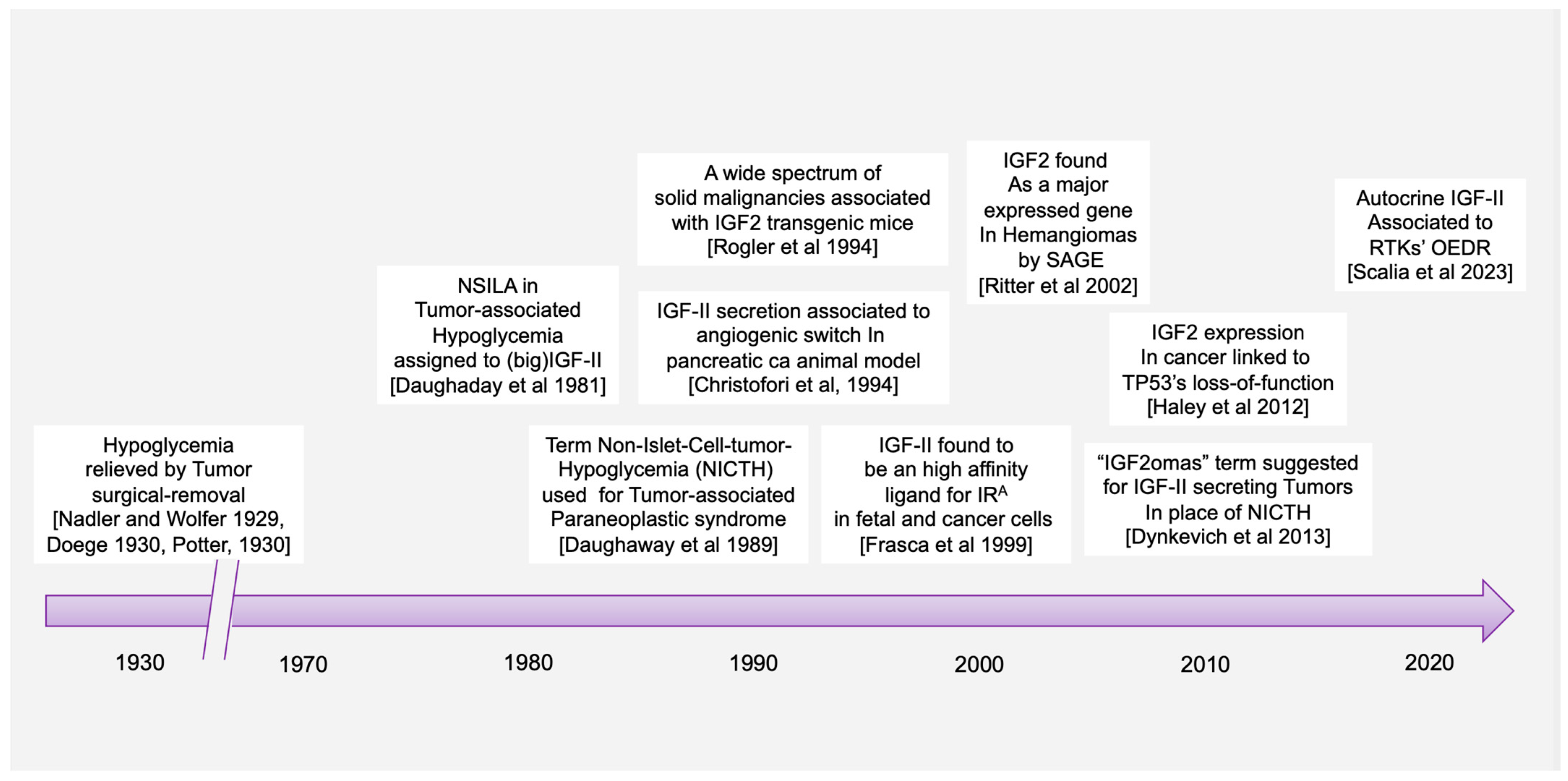
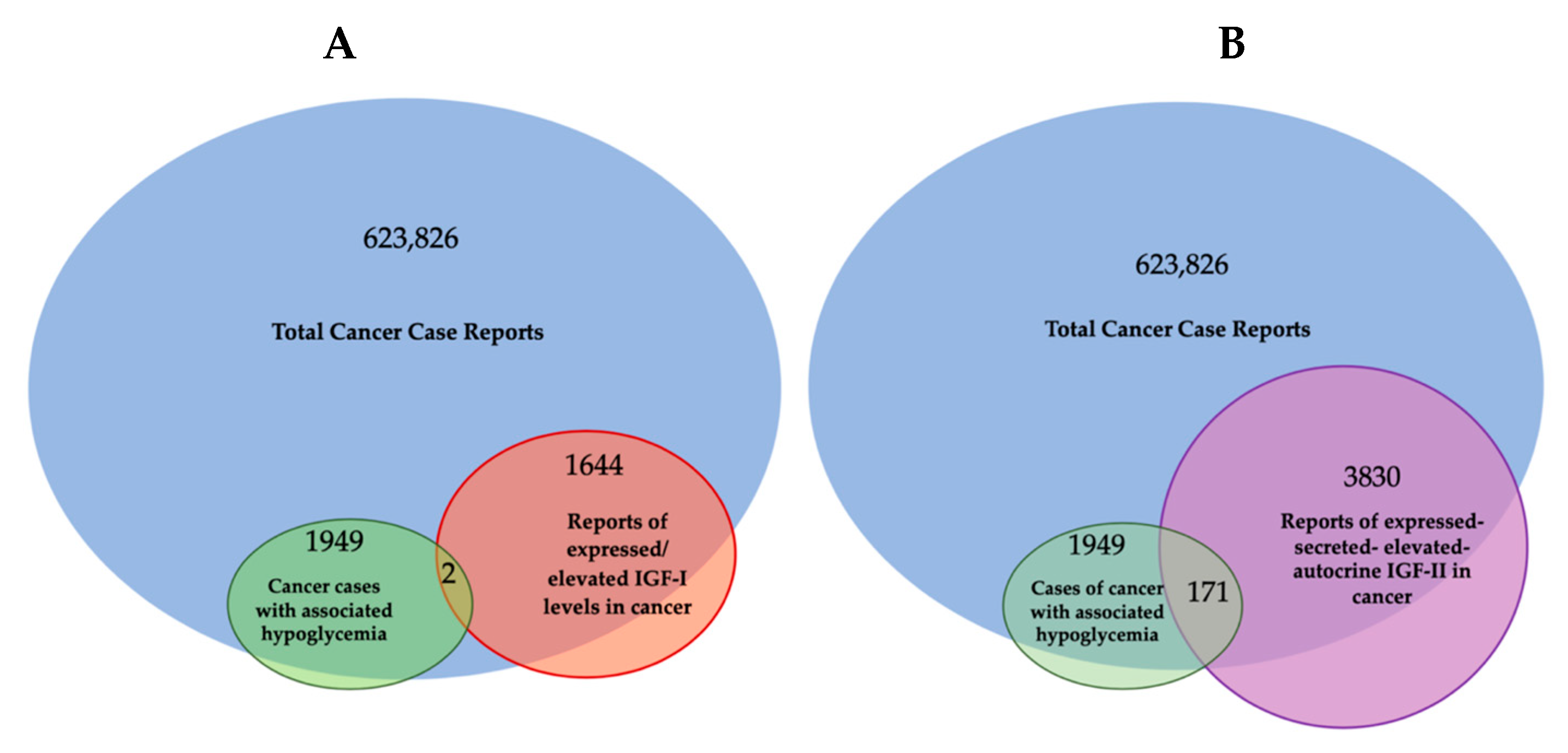
2. Cancer-Secreted IGF-II and Paraneoplastic Hypoglycemia: Is There Sufficient Evidence Supporting IGF-II as the Key IGF Ligand Involved in Solid Malignancy?
|
Cancer Associated Hypoglycemia |
Reports of Secreted Autocrine/Paracrine Growth Factor |
Reporting Elevated Plasma Growth Factor |
Reports of Elevated IGF Gene Transcripts Level in Underlying Tumor |
Cancer Case Report (1972) |
Hypoglycemia Case Reports |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
IGF-I |
IGF-II |
IGF-I |
IGF-II |
IGF-I |
IGF-II |
IGF-1 |
IGF-2 |
IGF-I |
IGF-II |
||
|
Cancer associated hypoglycemia |
Total cancer associated hypoglycemia cases = 1949 |
18 |
171 |
66 |
24 |
1 |
2 |
1690 |
1690 |
||
|
Protein expressing/Secreted IGF |
18 |
171 |
1644 * |
3830 |
301 |
201 |
312 |
136 |
136 |
1657 |
|
|
Reporting elevated plasma IGF |
66 |
24 |
322 |
201 |
893 |
892 |
172 |
16 |
22 |
64 |
|
|
Cancer (case report) |
1656 |
1656 |
980 |
1644 |
22 |
48 |
5 |
2 |
623,826 |
623,826 |
|
|
7616 |
|||||||||||
- (a)
-
It does not take in consideration the actual in vivo IGFs ligands and receptors co-expression context, which, taken together, supports a specific and independent role for cancer-secreted IGF-II and its autocrine loops;
- (b)
-
It does not succeed in explaining the failure of the individual pharmacological blockers of IGF-IR in clinical trials towards meeting the invoked therapeutic advantages suggested by the in vitro and epidemiologic studies;
- (c)
-
It has kept excluding alternative hypotheses and proper controls in experimental design which have been suggested by additional evidence available since the late nineties and proving the existence of an IGF-II- Insulin fetal receptor isoform (IRA) axis in mammalian fetal and cancer cells [16], as well as the expression and biological impact of IGF-IR/IR isoform-specific hybrids [25] in the studied cancer models.
3. IGF-II Over-Expression Is a Common Event in Cancer Cell-Lines
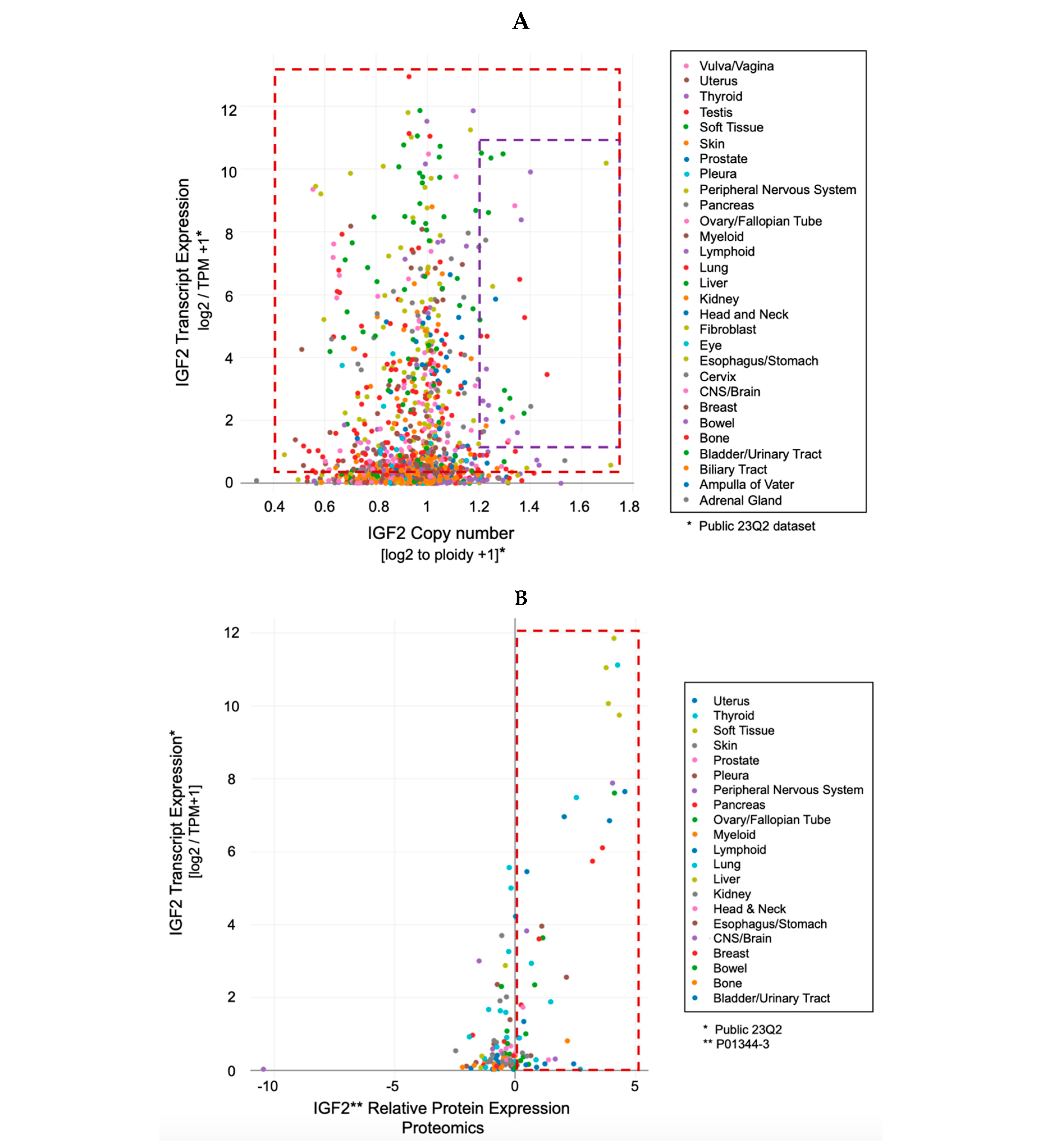
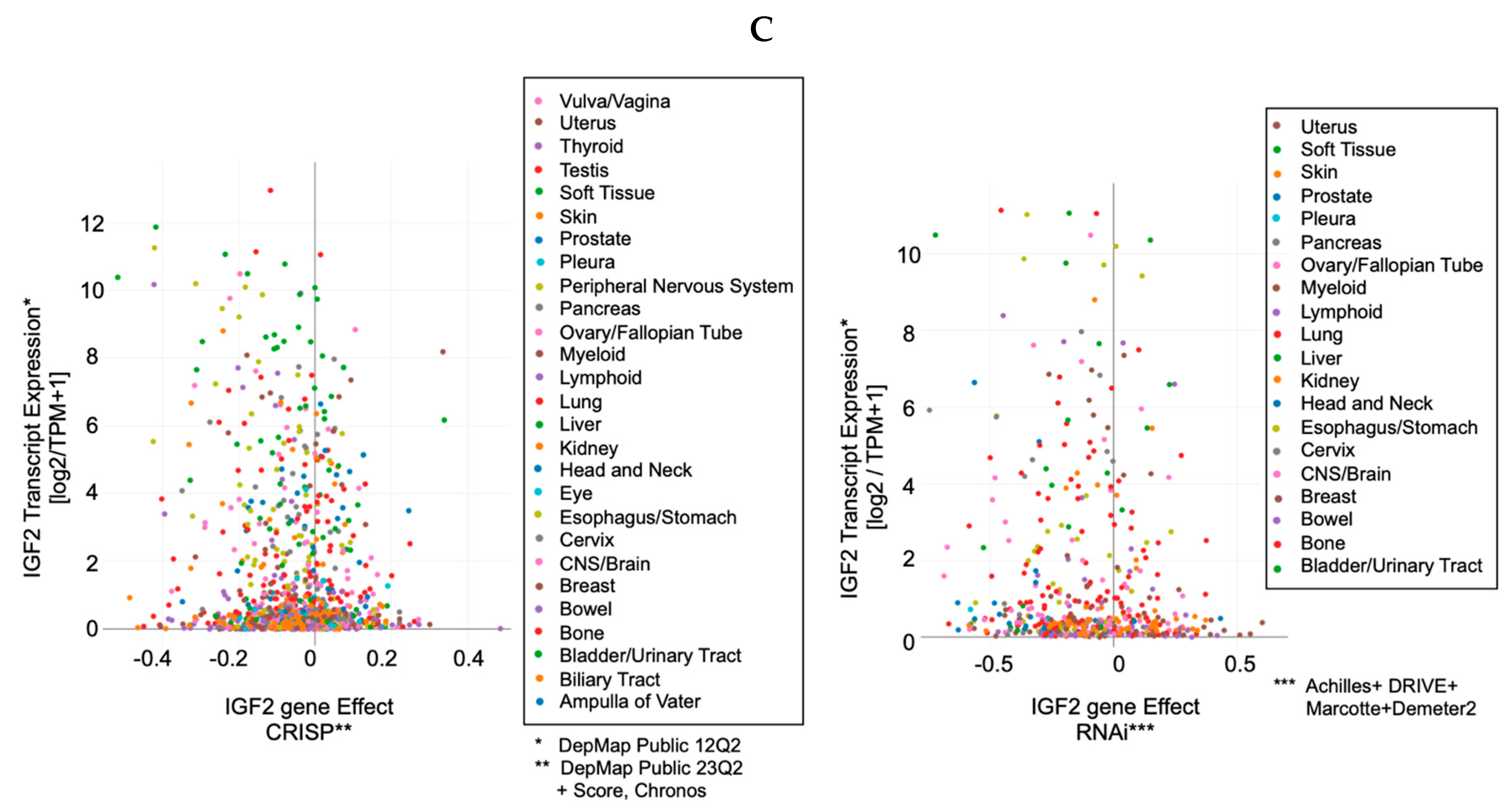
-
The IGF-II transcript expression in cancer cells exceeds the expression of normal cells and tissues by a range of 0.1- to 12-fold (Figure 3A–C);
-
The IGF-II transcript (mRNA) expression is not commonly associated with gene duplication events (Figure 3A);
-
The IGF-II protein expression in human-derived cancer cells exceeds normal cells/tissues by 0.1- to 5-fold (Figure 3B);
-
IGF-II gene editing and or transcript silencing negatively affects ~60–65% of cancer cells (Figure 3C).
4. The Role of IGF-II in Cancer Is Not Alternative to IGF-I
References
- Rinderknecht, E.; Humbel, R.E. Polypeptides with nonsuppressible insulin-like and cell-growth promoting activities in human serum: Isolation, chemical characterization, and some biological properties of forms I and II. Proc. Natl. Acad. Sci. USA 1976, 73, 2365–2369.
- Nadler, W.H.; Wolfer, J.A. Hepatogenic hypoglycemia associated with primary liver cell carcinoma. Arch. Intern. Med. 1929, 44, 700–710.
- Doege, K.W. Fibro-Sarcoma of the Mediastinum. Ann. Surg. 1930, 92, 955–960.
- Potter, R.P. Intrathoracic Tumors. Radiology 1930, 14, 60–61.
- Dynkevich, Y.; Rother, K.I.; Whitford, I.; Qureshi, S.; Galiveeti, S.; Szulc, A.L.; Danoff, A.; Breen, T.L.; Kaviani, N.; Shanik, M.H.; et al. Tumors, IGF-2, and hypoglycemia: Insights from the clinic, the laboratory, and the historical archive. Endocr. Rev. 2013, 34, 798–826.
- Daughaday, W.H.; Kapadia, M. Significance of abnormal serum binding of insulin-like growth factor II in the development of hypoglycemia in patients with non-islet-cell tumors. Proc. Natl. Acad. Sci. USA 1989, 86, 6778–6782.
- Daughaday, W.H.; Trivedi, B.; Baxter, R.C. Serum “big insulin-like growth factor II” from patients with tumor hypoglycemia lacks normal E-domain O-linked glycosylation, a possible determinant of normal propeptide processing. Proc. Natl. Acad. Sci. USA 1993, 90, 5823–5827.
- van Doorn, J. Insulin-like growth factor-II and bioactive proteins containing a part of the E-domain of pro-insulin-like growth factor-II. Biofactors 2020, 46, 563–578.
- Duguay, S.J.; Jin, Y.; Stein, J.; Duguay, A.N.; Gardner, P.; Steiner, D.F. Post-translational processing of the insulin-like growth factor-2 precursor. Analysis of O-glycosylation and endoproteolysis. J. Biol. Chem. 1998, 273, 18443–18451.
- Oka, Y.; Rozek, L.M.; Czech, M.P. Direct demonstration of rapid insulin-like growth factor II Receptor internalization and recycling in rat adipocytes. Insulin stimulates 125I-insulin-like growth factor II degradation by modulating the IGF-II receptor recycling process. J. Biol. Chem. 1985, 260, 9435–9442.
- Greenall, S.A.; Bentley, J.D.; Pearce, L.A.; Scoble, J.A.; Sparrow, L.G.; Bartone, N.A.; Xiao, X.; Baxter, R.C.; Cosgrove, L.J.; Adams, T.E. Biochemical characterization of individual human glycosylated pro-insulin-like growth factor (IGF)-II and big-IGF-II isoforms associated with cancer. J. Biol. Chem. 2013, 288, 59–68.
- Scalia, P.; Williams, S.J.; Fujita-Yamaguchi, Y.; Giordano, A. Cell cycle control by the insulin-like growth factor signal: At the crossroad between cell growth and mitotic regulation. Cell Cycle 2023, 22, 1–37.
- Daughaday, W.H.; Trivedi, B.; Kapadia, M. Measurement of insulin-like growth factor II by a specific radioreceptor assay in serum of normal individuals, patients with abnormal growth hormone secretion, and patients with tumor-associated hypoglycemia. J. Clin. Endocrinol. Metab. 1981, 53, 289–294.
- Rogler, C.; Yang, D.; Rossetti, L.; Donohoe, J.; Alt, E.; Chang, C.; Rosenfeld, R.; Neely, K.; Hintz, R. Altered body composition and increased frequency of diverse malignancies in insulin-like growth factor-II transgenic mice. J. Biol. Chem. 1994, 269, 13779–13784.
- Christofori, G.; Naik, P.; Hanahan, D. A second signal supplied by insulin-like growth factor II in oncogene-induced tumorigenesis. Nature 1994, 369, 414–418.
- Frasca, F.; Pandini, G.; Scalia, P.; Sciacca, L.; Mineo, R.; Costantino, A.; Goldfine, I.D.; Belfiore, A.; Vigneri, R. Insulin receptor isoform, A.; a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol. Cell. Biol. 1999, 19, 3278–3288.
- Ritter, M.R.; Dorrell, M.I.; Edmonds, J.; Friedlander, S.F.; Friedlander, M. Insulin-like growth factor 2 and potential regulators of hemangioma growth and involution identified by large-scale expression analysis. Proc. Natl. Acad. Sci. USA 2002, 99, 7455–7460.
- Haley, V.L.; Barnes, D.J.; Sandovici, I.; Constancia, M.; Graham, C.F.; Pezzella, F.; Bühnemann, C.; Carter, E.J.; Hassan, A.B. Igf2 pathway dependency of the Trp53 developmental and tumour phenotypes. EMBO Mol. Med. 2012, 4, 705–718.
- Scalia, P.; Williams, S.J. Over-expression by degradation rescue of RTKs via cancer-secreted autocrine growth factors: A Phospho-degron-driven actionable layer of post-translational regulation? Front. Oncol. 2023, 13, 1278402.
- Sell, C.; Dumenil, G.; Deveaud, C.; Miura, M.; Coppola, D.; DeAngelis, T.; Rubin, R.; Efstratiadis, A.; Baserga, R. Effect of a null mutation of the insulin-like growth factor I receptor gene on growth and transformation of mouse embryo fibroblasts. Mol. Cell. Biol. 1994, 14, 3604–3612.
- DeChiara, T.M.; Efstratiadis, A.; Robertson, E.J. A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature 1990, 345, 78–80.
- Baker, J.; Liu, J.P.; Robertson, E.J.; Efstratiadis, A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell 1993, 75, 73–82.
- Louvi, A.; Accili, D.; Efstratiadis, A. Growth-promoting interaction of IGF-II with the insulin receptor during mouse embryonic development. Dev. Biol. 1997, 189, 33–48.
- Ludwig, T.; Eggenschwiler, J.; Fisher, P.; D’Ercole, A.J.; Davenport, M.L.; Efstratiadis, A. Mouse mutants lacking the type 2 IGF receptor (IGF2R) are rescued from perinatal lethality in Igf2 and Igf1r null backgrounds. Dev. Biol. 1996, 177, 517–535.
- Pandini, G.; Frasca, F.; Mineo, R.; Sciacca, L.; Vigneri, R.; Belfiore, A. Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J. Biol. Chem. 2002, 277, 39684–39695.
- Rieger, L.; O’Shea, S.; Godsmark, G.; Stanicka, J.; Kelly, G.; O’Connor, R. IGF-1 receptor activity in the Golgi of migratory cancer cells depends on adhesion-dependent phosphorylation of Tyr(1250) and Tyr(1251). Sci. Signal. 2020, 13, 633.
- Crudden, C.; Girnita, L. The tale of a tail: The secret behind IGF-1R’s oncogenic power. Sci. Signal. 2020, 13, 633.
- Scalia, P.; Williams, S.J.; Fujita-Yamaguchi, Y. Human IGF2 Gene Epigenetic and Transcriptional Regulation: At the Core of Developmental Growth and Tumorigenic Behavior. Biomedicines 2023, 11, 1655.
- Soumerai, T.E.; Cote, G.M.; Goiffon, R.J.; Yerevanian, A.I.; Sy, A.L. Case 20-2023: A 52-Year-Old Man with a Solitary Fibrous Tumor and Hypoglycemia. N. Engl. J. Med. 2023, 388, 2467–2477.
- Feldser, D.; Agani, F.; Iyer, N.V.; Pak, B.; Ferreira, G.; Semenza, G.L. Reciprocal positive regulation of hypoxia-inducible factor 1alpha and insulin-like growth factor 2. Cancer Res. 1999, 59, 3915–3918.
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16.
- Zapf, J.; Walter, H.; Froesch, E.R. Radioimmunological determination of insulinlike growth factors I and II in normal subjects and in patients with growth disorders and extrapancreatic tumor hypoglycemia. J. Clin. Investig. 1981, 68, 1321–1330.
- Renehan, A.G.; Zwahlen, M.; Minder, C.; O’Dwyer, S.T.; Shalet, S.M.; Egger, M. Insulin-like growth factor (IGF)-I, IGF binding protein-3, and cancer risk: Systematic review and meta-regression analysis. Lancet 2004, 363, 1346–1353.
- Schernhammer, E.S.; Holly, J.M.; Pollak, M.N.; Hankinson, S.E. Circulating levels of insulin-like growth factors, their binding proteins, and breast cancer risk. Cancer Epidemiol. Biomark. Prev. 2005, 14, 699–704.
- Gao, Y.; Katki, H.; Graubard, B.; Pollak, M.; Martin, M.; Tao, Y.; Schoen, R.E.; Church, T.; Hayes, R.B.; Greene, M.H.; et al. Serum IGF1, IGF2 and IGFBP3 and risk of advanced colorectal adenoma. Int. J. Cancer 2012, 131, E105–E113.
- Cao, Y.; Lindström, S.; Schumacher, F.; Stevens, V.L.; Albanes, D.; Berndt, S.; Boeing, H.; Bueno-De-Mesquita, H.B.; Canzian, F.; Chamosa, S.; et al. Insulin-like growth factor pathway genetic polymorphisms, circulating IGF1 and IGFBP3, and prostate cancer survival. J. Natl. Cancer Inst. 2014, 106, dju085.
- Adamo, M.L.; Shao, Z.M.; Lanau, F.; Chen, J.C.; Clemmons, D.R.; Roberts, C.T., Jr.; LeRoith, D.; Fontana, J.A. Insulin-like growth factor-I (IGF-I) and retinoic acid modulation of IGF-binding proteins (IGFBPs): IGFBP-2, -3, and -4 gene expression and protein secretion in a breast cancer cell line. Endocrinology 1992, 131, 1858–1866.
- Milazzo, G.; Giorgino, F.; Damante, G.; Sung, C.; Stampfer, M.R.; Vigneri, R.; Goldfine, I.D.; Belfiore, A. Insulin receptor expression and function in human breast cancer cell lines. Cancer Res. 1992, 52, 3924–3930.
- Pandini, G.; Vigneri, R.; Costantino, A.; Frasca, F.; Ippolito, A.; Fujita-Yamaguchi, Y.; Siddle, K.; Goldfine, I.D.; Belfiore, A. Insulin and insulin-like growth factor-I (IGF-I) receptor overexpression in breast cancers leads to insulin/IGF-I hybrid receptor overexpression: Evidence for a second mechanism of IGF-I signaling. Clin. Cancer Res. 1999, 5, 1935–1944.
- Samani, A.A.; Yakar, S.; LeRoith, D.; Brodt, P. The role of the IGF system in cancer growth and metastasis: Overview and recent insights. Endocr. Rev. 2007, 28, 20–47.
- Gardner, D.G.; Shoback, D.M.; Greenspan, F.S. Greenspan’s Basic & Clinical Endocrinology, 10th ed.; McGraw-Hill Education LLC: New York, NY, USA, 2017.
- Werner, H.; Laron, Z. Insulin-like growth factors and aging: Lessons from Laron syndrome. Front. Endocrinol. 2023, 14, 1291812.
- Yamada, T.; De Souza, A.T.; Finkelstein, S.; Jirtle, R.L. Loss of the gene encoding mannose 6-phosphate/insulin-like growth factor II receptor is an early event in liver carcinogenesis. Proc. Natl. Acad. Sci. USA 1997, 94, 10351–10355.
- Chappell, S.A.; Walsh, T.; Walker, R.A.; Shaw, J.A. Loss of heterozygosity at the mannose 6-phosphate insulin-like growth factor 2 receptor gene correlates with poor differentiation in early breast carcinomas. Br. J. Cancer 1997, 76, 1558–1561.
- Hu, C.K.; McCall, S.; Madden, J.; Huang, H.; Clough, R.; Jirtle, R.L.; Anscher, M.S. Loss of heterozygosity of M6P/IGF2R gene is an early event in the development of prostate cancer. Prostate Cancer Prostatic Dis. 2006, 9, 62–67.
- Hankins, G.R.; DHankins, G.R.; De Souza, A.T.; Bentley, R.C.; Patel, M.R.; Marks, J.R.; Iglehart, J.D. M6P/IGF2 receptor: A candidate breast tumor suppressor gene. Oncogene 1996, 12, 2003–2009.
- Nissley, S.P.; Rechler, M.M. Somatomedin/insulin-like growth factor tissue receptors. Clin. Endocrinol. Metab. 1984, 13, 43–67.
- Ballard, F.J.; Ross, M.; Upton, F.M.; Francis, G.L. Specific binding of insulin-like growth factors 1 and 2 to the type 1 and type 2 receptors respectively. Biochem. J. 1988, 249, 721–726.
- Potalitsyn, P.; Mrázková, L.; Selicharová, I.; Tencerová, M.; Ferenčáková, M.; Chrudinová, M.; Turnovská, T.; Brzozowski, A.M.; Marek, A.; Kaminský, J.; et al. Non-glycosylated IGF2 prohormones are more mitogenic than native IGF2. Commun. Biol. 2023, 6, 863.
- Grimberg, A.; Coleman, C.M.; Burns, T.F.; Himelstein, B.P.; Koch, C.J.; Cohen, P.; El-Deiry, W.S. p53-Dependent and p53-independent induction of insulin-like growth factor binding protein-3 by deoxyribonucleic acid damage and hypoxia. J. Clin. Endocrinol. Metab. 2005, 90, 3568–3574.
- Gariboldi, M.B.; Ravizza, R.; Monti, E. The IGFR1 inhibitor NVP-AEW541 disrupts a pro-survival and pro-angiogenic IGF-STAT3-HIF1 pathway in human glioblastoma cells. Biochem. Pharmacol. 2010, 80, 455–462.
- Bond, J.J.; Meka, S.; Baxter, R.C. Binding characteristics of pro-insulin-like growth factor-II from cancer patients: Binary and ternary complex formation with IGF binding proteins-1 to -6. J. Endocrinol. 2000, 165, 253–260.
- Silha, J.V.; Sheppard, P.C.; Mishra, S.; Gui, Y.; Schwartz, J.; Dodd, J.G.; Murphy, L.J. Insulin-like growth factor (IGF) binding protein-3 attenuates prostate tumor growth by IGF-dependent and IGF-independent mechanisms. Endocrinology 2006, 147, 2112–2121.
- Takaoka, M.; Kim, S.-H.; Okawa, T.; Michaylira, C.Z.; Stairs, D.; Johnston, C.; Andl, C.D.; Rhoades, B.; Lee, J.; Klein-Szanto, A.; et al. IGFBP-3 regulates esophageal tumor growth through IGF-dependent and independent mechanisms. Cancer Biol. Ther. 2007, 6, 534–540.
- Takaoka, M.; Harada, H.; Andl, C.D.; Oyama, K.; Naomoto, Y.; Dempsey, K.L.; Klein-Szanto, A.J.; El-Deiry, W.S.; Grimberg, A.; Nakagawa, H. Epidermal growth factor receptor regulates aberrant expression of insulin-like growth factor-binding protein 3. Cancer Res. 2004, 64, 7711–7723.
- Ostrovsky, O.; Makarewich, C.A.; Snapp, E.L.; Argon, Y. An essential role for ATP binding and hydrolysis in the chaperone activity of GRP94 in cells. Proc. Natl. Acad. Sci. USA 2009, 106, 11600–11605.
- Ostrovsky, O.; Ahmed, N.T.; Argon, Y. The chaperone activity of GRP94 toward insulin-like growth factor II is necessary for the stress response to serum deprivation. Mol. Biol. Cell 2009, 20, 1855–1864.
- Argon, Y.; Bresson, S.E.; Marzec, M.T.; Grimberg, A. Glucose-Regulated Protein 94 (GRP94): A Novel Regulator of Insulin-Like Growth Factor Production. Cells 2020, 9, 1844.
- Reddy, R.K.; Dubeau, L.; Kleiner, H.; Parr, T.; Nichols, P.; Ko, B.; Dong, D.; Ko, H.; Mao, C.; DiGiovanni, J.; et al. Cancer-inducible transgene expression by the Grp94 promoter: Spontaneous activation in tumors of various origins and cancer-associated macrophages. Cancer Res. 2002, 62, 7207–7212.
- Dejeans, N.; Glorieux, C.; Guenin, S.; Beck, R.; Sid, B.; Rousseau, R.; Bisig, B.; Delvenne, P.; Calderon, P.B.; Verrax, J. Overexpression of GRP94 in breast cancer cells resistant to oxidative stress promotes high levels of cancer cell proliferation and migration: Implications for tumor recurrence. Free Radic. Biol. Med. 2012, 52, 993–1002.
- Bunn, R.C.; Fowlkes, J.L. Insulin-like growth factor binding protein proteolysis. Trends Endocrinol. Metab. 2003, 14, 176–181.
- Yee, D.; Paik, S.; Lebovic, G.S.; Marcus, R.R.; Favoni, R.E.; Cullen, K.J.; Lippman, M.E.; Rosen, N. Analysis of insulin-like growth factor I gene expression in malignancy: Evidence for a paracrine role in human breast cancer. Mol. Endocrinol. 1989, 3, 509–517.
- Cullen, K.J.; Allison, A.; Martire, I.; Ellis, M.; Singer, C. Insulin-like growth factor expression in breast cancer epithelium and stroma. Breast Cancer Res. Treat. 1992, 22, 21–29.
- Singer, C.; Rasmussen, A.; Smith, H.S.; Lippman, M.E.; Lynch, H.T.; Cullen, K.J. Malignant breast epithelium selects for insulin-like growth factor II expression in breast stroma: Evidence for paracrine function. Cancer Res. 1995, 55, 2448–2454.

