Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Claudio Pignata | -- | 2104 | 2024-03-07 10:06:56 | | | |
| 2 | Camila Xu | Meta information modification | 2104 | 2024-03-08 02:15:03 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Cillo, F.; Coppola, E.; Habetswallner, F.; Cecere, F.; Pignata, L.; Toriello, E.; De Rosa, A.; Grilli, L.; Ammendola, A.; Salerno, P.; et al. Genetic Features of 22q11.2 Deletion Syndrome. Encyclopedia. Available online: https://encyclopedia.pub/entry/55962 (accessed on 26 June 2026).
Cillo F, Coppola E, Habetswallner F, Cecere F, Pignata L, Toriello E, et al. Genetic Features of 22q11.2 Deletion Syndrome. Encyclopedia. Available at: https://encyclopedia.pub/entry/55962. Accessed June 26, 2026.
Cillo, Francesca, Emma Coppola, Federico Habetswallner, Francesco Cecere, Laura Pignata, Elisabetta Toriello, Antonio De Rosa, Laura Grilli, Antonio Ammendola, Paolo Salerno, et al. "Genetic Features of 22q11.2 Deletion Syndrome" Encyclopedia, https://encyclopedia.pub/entry/55962 (accessed June 26, 2026).
Cillo, F., Coppola, E., Habetswallner, F., Cecere, F., Pignata, L., Toriello, E., De Rosa, A., Grilli, L., Ammendola, A., Salerno, P., Romano, R., Cirillo, E., Merla, G., Riccio, A., Pignata, C., & Giardino, G. (2024, March 07). Genetic Features of 22q11.2 Deletion Syndrome. In Encyclopedia. https://encyclopedia.pub/entry/55962
Cillo, Francesca, et al. "Genetic Features of 22q11.2 Deletion Syndrome." Encyclopedia. Web. 07 March, 2024.
Copy Citation
The 22q11.2 region has a complex structure, characterized by low copy repeats (LCR22A, LCR22B, LCR22C, LCR22D) which share >96% of their sequence and are particularly prone to nonallelic homologous recombination during gametogenesis.
22q11.2 deletion syndrome
epigenetics
micro-RNAs
1. Introduction
Briefly, 22q11.2 deletion syndrome (22q11.2DS) is a complex and heterogeneous clinical syndrome. Under the 22q11.2DS definition are included several phenotypes such as the historically known DiGeorge syndrome (DGS), velocardiofacial syndrome (VCFS) and conotruncal anomaly face syndrome (CTAF) [1]. The primarily acknowledged presentation is the classic clinical triad including congenital heart defects (CHD) (75% of patients), T-cell compartment immunodeficiency due to hypoplastic/aplastic thymus (75% of patients) and hypocalcemia due to the developmental defect of parathyroid glands in 50% of cases, formerly referred to as DGS [1]. This term is now used for those individuals who show clinical phenotype of 22q11.2DS in the absence of an identified 22q11.2 deletion, in whom alternative pathogenetic alterations occur. Briefly, 22q11.2DS represents the most frequent microdeletion syndrome observed in the human genome, with an estimated incidence of 1 in 1.000 fetuses [2][3] or approximately 1 in 3.000–6.000 newborns [1]. However, a recent study conducted using DNA samples from dried blood spots for newborn screening reports an estimated minimum 22q11.2 DS prevalence of 1 in 2.148 live births [4]. Despite the significant incidence, no routine approach to prenatal screening for this condition has been established [5][6][7][8]. Newborn screening to measure the number of TREC copies successfully identifies 22q11.2 DS with T-cell lymphopenia, which can be helpful to prevent subsequent complications such as hypocalcemia [9][10]. In 90% of cases, the 22q11.2 deletion occurs de novo during gametogenesis as a consequence of nonallelic homologous recombination events. In 10% of patients, the syndrome is inherited in an autosomal-dominant fashion. The predominance of de novo cases may be partially explained by the impaired reproductive fitness of the patients carrying the deletion [11], especially males [12]. This hypothesis is also supported by the evidence that, in the familial forms, the disease is usually inherited from the mother [13][14]. Apart from the most recognizable aspects, more than 180 different phenotypic features have been described [15][16][17] in 22q11.2DS patients, and the syndrome is characterized by the extreme variability of the type and severity of the clinical manifestations, which can be also observed in members of the same family [1][15][18][19][20][21][22]. The phenotypic variability consists of a different combination of clinical manifestations, which compose a syndromic picture with various degrees of severity. Moreover, the same clinical abnormality can vary from mild to life-threatening in different subjects [23][24][25][26]. The syndrome can be associated with different size 22q11.2 region deletions. However, there is no correlation between the extension of the deletion and the severity of the syndrome [27]. Interestingly, microduplications of the 22q11.2 region result in a syndrome characterized by developmental delay, congenital heart defects, craniofacial dysmorphisms, behavioral alterations, visual and hearing impairment, and urogenital abnormalities, presenting with great clinical variability and absent genotype–phenotype predictability [28][29][30].
2. Genetic Features of 22q11.2DS
The 22q11.2 region has a complex structure, characterized by low copy repeats (LCR22A, LCR22B, LCR22C, LCR22D) which share >96% of their sequence and are particularly prone to nonallelic homologous recombination during gametogenesis [21]. In particular, LCR22A region is more susceptible to rearrangements, since it is characterized by hypervariability in the organization and in the copy number of duplicons which is human-specific and potentially variable in the population [31][32]. Depending on LCRs involved, the deletions causing 22q11.2DS may have different sizes and localizations. Almost 85% of patients share the so-called typical deletion of 3 Mb between LCR22A and LCR22D [27]. In patients with the typical deletion, the breakpoints within LCR22A and LCR22D are substantially clustered; they show small differences in genes not directly linked to clinical signs of the syndrome, thus not playing a major role in the variability of 22q11.2 DS [27]. Less frequently, the syndrome is caused by atypical, proximal or distal deletions [1].
In 90% of cases with typical 3 Mb or 1.5 Mb deletions the meiotic error occurs de novo. On the contrary, smaller size proximal or distal deletions are more frequently inherited [33]. These deletions are less penetrant and may be unrecognized since the patients are usually less symptomatic and the deletion cannot be identified using FISH (Fluorescent In Situ Hybridization). CMA (chromosomal microarray) is the most useful genomic testing method that allows to determine the copy number of sequences and to detect the recurrent deletion in a proband. The ability to size the deletion depends on the type of microarray used and the density of probes in the 22q11.2. Cardiovascular manifestation is found in approximately two-thirds of children with 22q11.2DS, so it represents one of the major diagnostic clues for 22q11.2DS [34][35].
FISH with a probe that targets the proximal fragment of the region (LCR 22A–22B) can also be used for the diagnosis.
The 3 Mb typically deleted region includes 90 genes: 46 protein-coding genes, 7 microRNAs, 10 non-coding RNAs and 27 pseudogenes [36] (Figure 1 and Table 1).
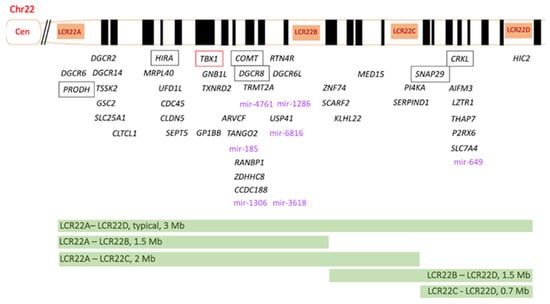
Figure 1. Schematic representation of the 22q11.2 region, including the four low-copy repeats (LCRs) LCR22A-LCR22D. The 46 protein-coding genes are indicated in black. TBX1 (T-box 1) is highlighted in red, since it is considered the main genetic driver of 22q11.2 DS. The potential pathogenetic role of PRODH, HIRA, COMT, DGCR8, SNAP29 and CRKL genes (in the box) is discussed in the text. The 7 micro-RNAs are indicated in violet. The size and the localization of the different deletions are shown at the bottom of the figure. Mir, microRNA.
Table 1. 46 protein-coding genes located in 22q11.2 region with the associated phenotype, genomic coordinates, and inheritance (omim.org).
| Associated Phenotype | Genomic Coordinates | Inheritance | |
|---|---|---|---|
| DGCR6 | - | 22:18,906,319-18,912,087 | - |
| PRODH | Hyperprolinemia type 1 | 22:18,912,780-18,936,552 | AR |
| DGCR2 | - | 22:19,036,285-19,122,453 | - |
| DGCR14 | - | 22:19,130,278-19,144,725 | - |
| TSSK2 | - | 22:19,131,307-19,132,621 | - |
| GSC2 | - | 22:19,146,992-19,150,291 | - |
| SLC25A1 | Combined D-2, L-2 hydroxyglutaric aciduria; Presynaptic Congenital Myasthenic Syndrome 23 |
22:19,175,580-19,178,735 | AR AR |
| CLTCL1 | - | 22:19,179,472-19,291,718 | - |
| HIRA | - | 22:19,330,697-19,431,732 | - |
| MRPL40 | - | 22:19,432,544-19,436,074 | - |
| UFD1L | - | 22:19,449,910-19,479,192 | - |
| CDC45 | Meier-Gorlin Syndrome | 22:19,479,293-19,520,611 | AR |
| CLDN5 | - | 22:19,523,023-19,525,336 | - |
| SEPT5 | - | 22:19,714,502-19,723,318 | - |
| TBX1 | - | 22:19,756,702-19,783,592 | - |
| GNB1L | - | 22:19,783,222-19,854,873 | - |
| TXNRD2 | Glucocorticoid deficiency? | 22:19,875,521-19,941,817 | - |
| GP1BB | Bernard-Soulier Syndrome, type B; Giant platelet disorder |
22:19,723,538-19,724,770 | AR AR |
| COMT | schizophrenia, susceptibility | 22:19,941,771-19,969,97 | AD |
| ARVCF | - | 22:19,966,726-20,016,822 | - |
| TANGO2 | Metabolic encephalomyopathic crises, recurrent, with rhabdomyolisis, cardiac arrhythmias and neurodegeneration | 22:20,016,999-20,067,163 | AR |
| DGCR8 | - | 22:20,080,240-20,111,871 | - |
| TRMT2A | - | 22:20,111,871-20,117,253 | - |
| RANBP1 | - | 22:20,116,103-20,127,354 | - |
| ZDHHC8 | - | 22:20,131,803-20,148,006 | - |
| CCDC188 | - | 22:20,148,113-20,151,828 | - |
| RTN4R | schizophrenia, susceptibility | 22:20,241,414-20,268,317 | AD |
| DGCR6L | - | 22:20,314,237-20,320,059 | - |
| USP41 | - | 22:20,350,578-20,390,758 | - |
| ZNF74 | - | 22:20,394,150-20,408,454 | - |
| SCARF2 | Van den Ende-Gupta Syndrome | 22:20,424,583-20,437,824 | AR |
| KLHL22 | - | 22:20,441,518-20,497,304 | - |
| MED15 | - | 22:20,507,581-20,587,620 | - |
| PI4KA | Gastrointestinal defects and immunodeficiency syndrome 2; perisylvian polymicrogyria with cerebellar hypoplasia and arthrogryposis; spastic paraplegia 84 |
22:20,707,690-20,858,811 | AR |
| SERPIND1 | Thrombophilia 10 due to heparin cofactor II deficiency | 22:20,774,112-20,787,719 | AD |
| SNAP29 | CEDNIK Syndrome | 22:20,859,006-20,891,213 | AR |
| CRKL | - | 22:20,917,406-20,953,746 | - |
| AIFM3 | - | 22:20,965,171-20,981,357 | - |
| LZTR1 | Noonan Syndrome 10; Noonan Syndrome 2; Schwannomatosis 2, susceptibility |
22:20,982,296-20,999,031 | AD AR AD |
| THAP7 | - | 22:20,999,103-21,002,117 | - |
| P2RX6 | - | 22:21,009,699-21,028,013 | - |
| SLC7A4 | - | 22:21,028,717-21,032,560 | - |
| HIC2 | - | 22:21,417,370-21,451,462 | - |
Among the most studied protein-coding genes, TBX1 (T-box transcription factor 1), located at the proximal side of the 22q11.2 region, has been shown to play a crucial role in the pathogenesis of 22q11.2DS [37]. TBX1 is implicated in DNA transcriptional regulation, acting on chromatin accessibility through the interaction with histone modifiers and chromatin remodeling complexes, with a direct effect on H3K4me1 levels [38]. TBX1 regulates monomethylation of histone 3 lysine 4 (H3K4me1) through interaction with and recruitment of histone methyltransferases and demethylases. It has been proposed as a priming factor that plays a role in keeping targeted chromatin accessible to other regulatory factors, which may be activators or repressors [38] and it is involved in the regulation of developmental processes [39]. Heterozygous Tbx1 mouse mutants (Tbx1+/−) show low penetrance of cardiovascular abnormalities with normal thymus gland, while Tbx1−/− knockout is embryonic lethal and mice show abnormal development of pharyngeal arches and pouches [40]. TBX1 is required for the characteristic segmentation of the pharyngeal apparatus in arches and pouches [41]. A strict relationship between TBX1 dosage and retinoic acid signaling pathway during embryonic development has been described [42][43]. The vitamin A active metabolite is a key morphogen involved in pharyngeal apparatus segmentation [44], as demonstrated by teratogenesis evidence associated with its exposure during pregnancy. Likewise, vitamin B12 has been identified as a positive regulator of TBX1 gene expression. Studies conducted using mouse models have shown that vitamin B12 can partially rescue the haploinsufficiency phenotype [45]. Furthermore, TBX1 finely regulates the interaction between VEGFR2 (vascular endothelial growth factor receptor 2) and VEGFR3 (vascular endothelial growth factor receptor 3) during brain microvascular organization and is implicated in cerebral cortex development [46].
Another protein-coding gene involved in 22q11.2 pathogenesis is CRKL (V-crk avian sarcoma virus CT10 oncogene homologue-like). Due to its central role in kidneys and urinary tract development, CRKL is considered the genetic driver of CAKUT occurring in 22q11.2DS patients [47]. In patients with “partial DGS”, characterized by a normal or slightly reduced number of T-lymphocytes, CRKL deficiency is involved in the mechanisms leading to impaired T-cell proliferation, something that has been shown even in the absence of lymphopenia [48]. Indeed, proliferative response in 22q11.2DS patients is relatively unaffected when assays are normalized for T-cells, and likewise, standard mitogen proliferation tests are usually impaired due to extremely low T-cell counts [49]. Furthermore, CRKL is required for natural killer cells’ physiological activity, since its haploinsufficiency is associated with the functional deficiency of this lymphocyte subpopulation [50].
HIRA (histone cell cycle regulator) regulates gene expression, modulating the incorporation of the H3.3 histone into the chromatinic structure [51].
Evidence suggests that genes deleted in the 22q11.2 region participate in complex networks of interactions influencing, with their altered dosage, a plethora of different signaling pathways. Since 22q11.2 hemizygosity alone does not explain the genetic basis of the phenotypic variability observed in patients, due to the evidence that patients sharing the same deletion present with different clinical phenotypes, additional mechanisms have been proposed. These include epigenetic mechanisms, which are better explained in the following sections. In particular, epigenetic regulation is extremely variable as a consequence of TBX1 hemizygosity, which creates a random epigenetic marking that varies from cell to cell [45][52]. In some, the deletion may unmask recessive mutations in genes located in the intact 22q11.2 region, leading to atypical and more severe presentations of 22q11.2DS [21]. Furthermore, recent evidence demonstrates that 1% of patients with 22q11.2DS may be affected by a second genetic condition in the context of a dual diagnosis [53][54][55].
Recent studies suggest that copy number variants (CNVs) of genes located outside the 22q11.2 region may partially explain the variability and complexity of different phenotypes observed in patients sharing the same deletion, increasing the risk of developing certain pathological manifestations. In 22q11.2DS patients, CNVs of the genes GPR98 (G-protein-coupled receptor 98) [56], KANSL1 (KAT8 regulatory NSL complex subunit 1 gene) [57] and SC2A3 (solute carrier family 2 facilitated glucose transporter member 3) [58] have been described as risk factors for congenital heart anomalies.
Some conditions presenting with a phenotype overlapping 22q11.2DS, but without 22q11.2 region anomalies, have been described. The so-called phenocopies of 22q11.2DS are a useful model with which to investigate the potential role of other regions of the genome in the pathogenesis of the main clinical aspects observed in 22q11.2DS patients. Table 2 summarizes the most common clinical manifestation described in 22q11.2DS patients, compared with those observed in patients with other cytogenetic alterations sharing the DiGeorge-like phenotype. In mice, HoxA3 (class 1 homeobox gene A3) knockout (HoxA3−/−) reproduces the typical clinical defects of the DiGeorge phenotype [59]. Similarly, mutations in specific Vegf isoforms are responsible for the same congenital abnormalities caused by Tbx1 knockout [60]. Furthermore, Cirillo et al. [61] identified the duplication of 15q11.2 region and the deletion of the 22q13.3 and 14q32.1 chromosomal regions in patients with the DiGeorge phenotype not presenting 22q11.2 deletion. The region on chromosome 15 is involved in Prader–Willi/Angelman syndromes, while deleted genes on chromosomes 22 and 14 participate in immune system functions.
Table 2. Schematic representation of the most common clinical manifestation described in 22q11.2DS patients, compared with those observed in patients with other cytogenetic alterations sharing the DiGeorge-like phenotype.
| Clinical Manifestations | 10p13-14 DGS2 Locus |
3p10.3 | 4q34.1-35.2 | Del2p11.2 | Microdup 22q11.2 |
Del22q13.33 Phelan-McDermid Syndrome |
|---|---|---|---|---|---|---|
| Congenital Heart Disease (CHD) | 82% | Yes | 15% | No | Yes | 3–25% |
| Hypocalcemia (hypoparathyroidism) | 22% | Yes | Na | Yes | Yes | Na |
| Immune Deficiency | 17% | Yes | Na | Yes | Yes | Na |
| Craniofacial dysmorphisms | 50% | Yes | 95–99% | Yes | Yes | >75% |
| Renal anomalies | 5% | Yes | Na | No | Yes | 38% |
| Skeletal defects | 30–80% | Na | 88% | Yes | Yes | >75% |
| Learning problems/ Developmental delay |
80–99% | Yes | 65% | Yes | Yes | >75% |
| Psychiatric disorders | Na | Na | Na | Na | Yes | >75% |
| Gastrointestinal abnormalities | Na | Na | Na | No | Yes | >25% |
| Genes mapping in the region | Ni | FOXI3 | See Table 3 | SHANK3 |
References
- McDonald-McGinn, D.M.; Sullivan, K.E.; Marino, B.; Philip, N.; Swillen, A.; Vorstman, J.A.; Zackai, E.H.; Emanuel, B.S.; Vermeesch, J.R.; Morrow, B.E.; et al. 22q11.2 deletion syndrome. Nat. Rev. Dis. Prim. 2015, 1, 15071.
- Grati, F.R.; Molina Gomes, D.; Ferreira, J.C.; Dupont, C.; Alesi, V.; Gouas, L.; Horelli-Kuitunen, N.; Choy, K.W.; García-Herrero, S.; de la Vega, A.G.; et al. Prevalence of recurrent pathogenic microdeletions and microduplications in over 9500 pregnancies. Prenat. Diagn. 2015, 35, 801–809.
- Gross, S.J.; Stosic, M.; McDonald-McGinn, D.M.; Bassett, A.S.; Norvez, A.; Dhamankar, R.; Kobara, K.; Kirkizlar, E.; Zimmermann, B.; Wayham, N.; et al. Clinical experience with single-nucleotide polymorphism-based non-invasive prenatal screening for 22q11.2 deletion syndrome. Ultrasound Obstet. Gynecol. 2016, 47, 177–183.
- Blagojevic, C.; Heung, T.; Theriault, M.; Tomita-Mitchell, A.; Chakraborty, P.; Kernohan, K.; Bulman, D.E.; Bassett, A.S. Estimate of the contemporary live-birth prevalence of recurrent 22q11.2 deletions: A cross-sectional analysis from population-based newborn screening. CMAJ Open 2021, 9, E802–E809.
- Bevilacqua, E.; Jani, J.C.; Chaoui, R.; Suk, E.A.; Palma-Dias, R.; Ko, T.M.; Warsof, S.; Stokowski, R.; Jones, K.J.; Grati, F.R.; et al. Performance of a targeted cell-free DNA prenatal test for 22q11.2 deletion in a large clinical cohort. Ultrasound Obstet. Gynecol. 2021, 58, 597–602.
- Dar, P.; Norton, M.E. Performance of noninvasive prenatal screening for 22q11.2 deletion syndrome in the SMART study. Am. J. Obstet. Gynecol. 2022, 227, 124–125.
- Kagan, K.O.; Hoopmann, M.; Pfaff, T.; Prodan, N.; Wagner, P.; Schmid, M.; Dufke, A.; Mau-Holzmann, U.; Brucker, S.; Marcato, L.; et al. First Trimester Screening for Common Trisomies and Microdeletion 22q11.2 Syndrome Using Cell-Free DNA: A Prospective Clinical Study. Fetal Diagn. Ther. 2020, 47, 841–852.
- Blagowidow, N.; Nowakowska, B.; Schindewolf, E.; Grati, F.R.; Putotto, C.; Breckpot, J.; Swillen, A.; Crowley, T.B.; Loo, J.C.Y.; Lairson, L.A.; et al. Prenatal Screening and Diagnostic Considerations for 22q11.2 Microdeletions. Genes 2023, 14, 160.
- Kwan, A.; Abraham, R.S.; Currier, R.; Brower, A.; Andruszewski, K.; Abbott, J.K.; Baker, M.; Ballow, M.; Bartoshesky, L.E.; Bonilla, F.A.; et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA 2014, 312, 729–738.
- Barry, J.C.; Crowley, T.B.; Jyonouchi, S.; Heimall, J.; Zackai, E.H.; Sullivan, K.E.; McDonald-McGinn, D.M. Identification of 22q11.2 Deletion Syndrome via Newborn Screening for Severe Combined Immunodeficiency. J. Clin. Immunol. 2017, 37, 476–485.
- Palmer, L.D.; McManus, Z.; Heung, T.; McAlpine, G.; Blagojevic, C.; Corral, M.; Bassett, A.S. Reproductive Outcomes in Adults with 22q11.2 Deletion Syndrome. Genes 2022, 13, 2126.
- Kapadia, R.K.; Bassett, A.S. Recognizing a common genetic syndrome: 22q11.2 deletion syndrome. CMAJ 2008, 178, 391–393.
- Delio, M.; Guo, T.; McDonald-McGinn, D.M.; Zackai, E.; Herman, S.; Kaminetzky, M.; Higgins, A.M.; Coleman, K.; Chow, C.; Jalbrzikowski, M.; et al. Enhanced maternal origin of the 22q11.2 deletion in velocardiofacial and DiGeorge syndromes. Am. J. Hum. Genet. 2013, 92, 439–447.
- Costain, G.; Chow, E.W.; Silversides, C.K.; Bassett, A.S. Sex differences in reproductive fitness contribute to preferential maternal transmission of 22q11.2 deletions. J. Med. Genet. 2011, 48, 819–824.
- Cancrini, C.; Puliafito, P.; Digilio, M.C.; Soresina, A.; Martino, S.; Rondelli, R.; Consolini, R.; Ruga, E.M.; Cardinale, F.; Finocchi, A.; et al. Clinical features and follow-up in patients with 22q11.2 deletion syndrome. J. Pediatr. 2014, 164, 1475–1480.e2.
- Robin, N.H.; Shprintzen, R.J. Defining the clinical spectrum of deletion 22q11.2. J. Pediatr. 2005, 147, 90–96.
- Shprintzen, R.J.; Higgins, A.M.; Antshel, K.; Fremont, W.; Roizen, N.; Kates, W. Velo-cardio-facial syndrome. Curr. Opin. Pediatr. 2005, 17, 725–730.
- McDonald-McGinn, D.M.; Tonnesen, M.K.; Laufer-Cahana, A.; Finucane, B.; Driscoll, D.A.; Emanuel, B.S.; Zackai, E.H. Phenotype of the 22q11.2 deletion in individuals identified through an affected relative: Cast a wide FISHing net! Genet. Med. 2001, 3, 23–29.
- Cirillo, E.; Giardino, G.; Gallo, V.; Puliafito, P.; Azzari, C.; Bacchetta, R.; Cardinale, F.; Cicalese, M.P.; Consolini, R.; Martino, S.; et al. Intergenerational and intrafamilial phenotypic variability in 22q11.2 deletion syndrome subjects. BMC Med. Genet. 2014, 15, 1.
- Vergaelen, E.; Swillen, A.; Van Esch, H.; Claes, S.; Van Goethem, G.; Devriendt, K. 3 generation pedigree with paternal transmission of the 22q11.2 deletion syndrome: Intrafamilial phenotypic variability. Eur. J. Med. Genet. 2015, 58, 244–248.
- Morrow, B.E.; McDonald-McGinn, D.M.; Emanuel, B.S.; Vermeesch, J.R.; Scambler, P.J. Molecular genetics of 22q11.2 deletion syndrome. Am. J. Med. Genet. Part A 2018, 176, 2070–2081.
- Szczawińska-Popłonyk, A.; Schwartzmann, E.; Chmara, Z.; Głukowska, A.; Krysa, T.; Majchrzycki, M.; Olejnicki, M.; Ostrowska, P.; Babik, J. Chromosome 22q11.2 Deletion Syndrome: A Comprehensive Review of Molecular Genetics in the Context of Multidisciplinary Clinical Approach. Int. J. Mol. Sci. 2023, 24, 8317.
- McDonald-McGinn, D.M.; Sullivan, K.E. Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Medicine 2011, 90, 1–18.
- Repetto, G.M.; Guzmán, M.L.; Delgado, I.; Loyola, H.; Palomares, M.; Lay-Son, G.; Vial, C.; Benavides, F.; Espinoza, K.; Alvarez, P. Case fatality rate and associated factors in patients with 22q11 microdeletion syndrome: A retrospective cohort study. BMJ Open 2014, 4, e005041.
- Sullivan, K.E. Chromosome 22q11.2 deletion syndrome and DiGeorge syndrome. Immunol. Rev. 2019, 287, 186–201.
- Karbarz, M. Consequences of 22q11.2 Microdeletion on the Genome, Individual and Population Levels. Genes 2020, 11, 977.
- Bertini, V.; Azzarà, A.; Legitimo, A.; Milone, R.; Battini, R.; Consolini, R.; Valetto, A. Deletion Extents Are Not the Cause of Clinical Variability in 22q11.2 Deletion Syndrome: Does the Interaction between DGCR8 and miRNA-CNVs Play a Major Role? Front. Genet. 2017, 8, 47.
- Bartik, L.E.; Hughes, S.S.; Tracy, M.; Feldt, M.M.; Zhang, L.; Arganbright, J.; Kaye, A. 22q11.2 duplications: Expanding the clinical presentation. Am. J. Med. Genet. Part A 2022, 188, 779–787.
- Yu, A.; Turbiville, D.; Xu, F.; Ray, J.W.; Britt, A.D.; Lupo, P.J.; Jain, S.K.; Shattuck, K.E.; Robinson, S.S.; Dong, J. Genotypic and phenotypic variability of 22q11.2 microduplications: An institutional experience. Am. J. Med. Genet. Part A 2019, 179, 2178–2189.
- Meneses, Z.; Durant, J.; Ale, H. The Unique Experience of a New Multidisciplinary Program for 22q Deletion and Duplication Syndromes in a Community Hospital in Florida: A Reaffirmation That Multidisciplinary Care Is Essential for Best Outcomes in These Patients. Genes 2022, 13, 1949.
- Vervoort, L.; Dierckxsens, N.; Pereboom, Z.; Capozzi, O.; Rocchi, M.; Shaikh, T.H.; Vermeesch, J.R. 22q11.2 Low Copy Repeats Expanded in the Human Lineage. Front. Genet. 2021, 12, 706641.
- Demaerel, W.; Mostovoy, Y.; Yilmaz, F.; Vervoort, L.; Pastor, S.; Hestand, M.S.; Swillen, A.; Vergaelen, E.; Geiger, E.A.; Coughlin, C.R.; et al. The 22q11 low copy repeats are characterized by unprecedented size and structural variability. Genome Res. 2019, 29, 1389–1401.
- Adeyinka, A.; Stockero, K.J.; Flynn, H.C.; Lorentz, C.P.; Ketterling, R.P.; Jalal, S.M. Familial 22q11.2 deletions in DiGeorge/velocardiofacial syndrome are predominantly smaller than the commonly observed 3Mb. Genet. Med. 2004, 6, 517–520.
- McDonald-McGinn, D.M.; Hain, H.S.; Emanuel, B.S.; Zackai, E.H. 22q11.2 Deletion Syndrome. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993.
- Óskarsdóttir, S.; Boot, E.; Crowley, T.B.; Loo, J.C.Y.; Arganbright, J.M.; Armando, M.; Baylis, A.L.; Breetvelt, E.J.; Castelein, R.M.; Chadehumbe, M.; et al. Updated clinical practice recommendations for managing children with 22q11.2 deletion syndrome. Genet. Med. 2023, 25, 100338.
- Guna, A.; Butcher, N.J.; Bassett, A.S. Comparative mapping of the 22q11.2 deletion region and the potential of simple model organisms. J. Neurodev. Disord. 2015, 7, 18.
- Jerome, L.A.; Papaioannou, V.E. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat. Genet. 2001, 27, 286–291.
- Fulcoli, F.G.; Franzese, M.; Liu, X.; Zhang, Z.; Angelini, C.; Baldini, A. Rebalancing gene haploinsufficiency in vivo by targeting chromatin. Nat. Commun. 2016, 7, 11688.
- Yagi, H.; Furutani, Y.; Hamada, H.; Sasaki, T.; Asakawa, S.; Minoshima, S.; Ichida, F.; Joo, K.; Kimura, M.; Imamura, S.; et al. Role of TBX1 in human del22q11.2 syndrome. Lancet 2003, 362, 1366–1373.
- Funato, N. Craniofacial Phenotypes and Genetics of DiGeorge Syndrome. J. Dev. Biol. 2022, 10, 18.
- Baldini, A.; Fulcoli, F.G.; Illingworth, E. Tbx1: Transcriptional and Developmental Functions. Curr. Top. Dev. Biol. 2017, 122, 223–243.
- Roberts, C.; Ivins, S.M.; James, C.T.; Scambler, P.J. Retinoic acid down-regulates Tbx1 expression in vivo and in vitro. Dev. Dyn. 2005, 232, 928–938.
- Ryckebüsch, L.; Bertrand, N.; Mesbah, K.; Bajolle, F.; Niederreither, K.; Kelly, R.G.; Zaffran, S. Decreased levels of embryonic retinoic acid synthesis accelerate recovery from arterial growth delay in a mouse model of DiGeorge syndrome. Circ. Res. 2010, 106, 686–694.
- Mark, M.; Ghyselinck, N.B.; Chambon, P. Retinoic acid signalling in the development of branchial arches. Curr. Opin. Genet. Dev. 2004, 14, 591–598.
- Lania, G.; Bresciani, A.; Bisbocci, M.; Francone, A.; Colonna, V.; Altamura, S.; Baldini, A. Vitamin B12 ameliorates the phenotype of a mouse model of DiGeorge syndrome. Hum. Mol. Genet. 2016, 25, 4369–4375.
- Cioffi, S.; Martucciello, S.; Fulcoli, F.G.; Bilio, M.; Ferrentino, R.; Nusco, E.; Illingworth, E. Tbx1 regulates brain vascularization. Hum. Mol. Genet. 2014, 23, 78–89.
- Homans, J.F.; Tromp, I.N.; Colo, D.; Schlösser, T.P.C.; Kruyt, M.C.; Deeney, V.F.X.; Crowley, T.B.; McDonald-McGinn, D.M.; Castelein, R.M. Orthopaedic manifestations within the 22q11.2 Deletion syndrome: A systematic review. Am. J. Med. Genet. Part A 2018, 176, 2104–2120.
- Giacomelli, M.; Kumar, R.; Soresina, A.; Tamassia, N.; Lorenzini, T.; Moratto, D.; Gasperini, S.; Cassatella, M.; Plebani, A.; Lougaris, V.; et al. Reduction of CRKL expression in patients with partial DiGeorge syndrome is associated with impairment of T-cell functions. J. Allergy Clin. Immunol. 2016, 138, 229–240.e3.
- Mustillo, P.J.; Sullivan, K.E.; Chinn, I.K.; Notarangelo, L.D.; Haddad, E.; Davies, E.G.; de la Morena, M.T.; Hartog, N.; Yu, J.E.; Hernandez-Trujillo, V.P.; et al. Clinical Practice Guidelines for the Immunological Management of Chromosome 22q11.2 Deletion Syndrome and Other Defects in Thymic Development. J. Clin. Immunol. 2023, 43, 247–270.
- Zheng, P.; Noroski, L.M.; Hanson, I.C.; Chen, Y.; Lee, M.E.; Huang, Y.; Zhu, M.X.; Banerjee, P.P.; Makedonas, G.; Orange, J.S.; et al. Molecular mechanisms of functional natural killer deficiency in patients with partial DiGeorge syndrome. J. Allergy Clin. Immunol. 2015, 135, 1293–1302.
- Ray-Gallet, D.; Quivy, J.P.; Scamps, C.; Martini, E.M.; Lipinski, M.; Almouzni, G. HIRA is critical for a nucleosome assembly pathway independent of DNA synthesis. Mol. Cell 2002, 9, 1091–1100.
- Romano, R.; Cillo, F.; Moracas, C.; Pignata, L.; Nannola, C.; Toriello, E.; De Rosa, A.; Cirillo, E.; Coppola, E.; Giardino, G.; et al. Epigenetic Alterations in Inborn Errors of Immunity. J. Clin. Med. 2022, 11, 1261.
- Cohen, J.L.; Crowley, T.B.; McGinn, D.E.; McDougall, C.; Unolt, M.; Lambert, M.P.; Emanuel, B.S.; Zackai, E.H.; McDonald-McGinn, D.M. 22q and two: 22q11.2 deletion syndrome and coexisting conditions. Am. J. Med. Genet. Part A 2018, 176, 2203–2214.
- Cecere, F.; Pignata, L.; Hay Mele, B.; Saadat, A.; D’Angelo, E.; Palumbo, O.; Palumbo, P.; Carella, M.; Scarano, G.; Rossi, G.B.; et al. Co-Occurrence of Beckwith-Wiedemann Syndrome and Early-Onset Colorectal Cancer. Cancers 2023, 15, 1944.
- Carli, D.; Operti, M.; Russo, S.; Cocchi, G.; Milani, D.; Leoni, C.; Prada, E.; Melis, D.; Falco, M.; Spina, J.; et al. Clinical and molecular characterization of patients affected by Beckwith-Wiedemann spectrum conceived through assisted reproduction techniques. Clin. Genet. 2022, 102, 314–323.
- Guo, T.; Repetto, G.M.; McDonald-McGinn, D.M.; Chung, J.H.; Nomaru, H.; Campbell, C.L.; Blonska, A.; Bassett, A.S.; Chow, E.W.C.; Mlynarski, E.E.; et al. Genome-Wide Association Study to Find Modifiers for Tetralogy of Fallot in the 22q11.2 Deletion Syndrome Identifies Variants in the GPR98 Locus on 5q14.3. Circ. Cardiovasc. Genet. 2017, 10, e001690.
- León, L.E.; Benavides, F.; Espinoza, K.; Vial, C.; Alvarez, P.; Palomares, M.; Lay-Son, G.; Miranda, M.; Repetto, G.M. Partial microduplication in the histone acetyltransferase complex member KANSL1 is associated with congenital heart defects in 22q11.2 microdeletion syndrome patients. Sci. Rep. 2017, 7, 1795.
- Mlynarski, E.E.; Sheridan, M.B.; Xie, M.; Guo, T.; Racedo, S.E.; McDonald-McGinn, D.M.; Gai, X.; Chow, E.W.; Vorstman, J.; Swillen, A.; et al. Copy-Number Variation of the Glucose Transporter Gene SLC2A3 and Congenital Heart Defects in the 22q11.2 Deletion Syndrome. Am. J. Hum. Genet. 2015, 96, 753–764.
- Chisaka, O.; Capecchi, M.R. Regionally restricted developmental defects resulting from targeted disruption of the mouse homeobox gene HOX-1.5. Nature 1991, 350, 473–479.
- Stalmans, I.; Lambrechts, D.; De Smet, F.; Jansen, S.; Wang, J.; Maity, S.; Kneer, P.; von der Ohe, M.; Swillen, A.; Maes, C.; et al. VEGF: A modifier of the del22q11 (DiGeorge) syndrome? Nat. Med. 2003, 9, 173–182.
- Cirillo, E.; Prencipe, M.R.; Giardino, G.; Romano, R.; Scalia, G.; Genesio, R.; Nitsch, L.; Pignata, C. Clinical Phenotype, Immunological Abnormalities, and Genomic Findings in Patients with DiGeorge Spectrum Phenotype without 22q11.2 Deletion. J. Allergy Clin. Immunol. Pract. 2020, 8, 3112–3120.
- Du, Q.; de la Morena, M.T.; van Oers, N.S.C. The Genetics and Epigenetics of 22q11.2 Deletion Syndrome. Front. Genet. 2020, 10, 1365.
- Phelan, K.; Rogers, R.C.; Boccuto, L. Phelan-McDermid Syndrome. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993.
- Butensky, A.; de Rinaldis, C.P.; Patel, S.; Edman, S.; Bailey, A.; McGinn, D.E.; Zackai, E.; Crowley, T.B.; McDonald-McGinn, D.M.; Min, J.; et al. Cardiac evaluation of patients with 22q11.2 duplication syndrome. Am. J. Med. Genet. Part A 2021, 185, 753–758.
More
Information
Subjects:
Medicine, General & Internal
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
564
Revisions:
2 times
(View History)
Update Date:
08 Mar 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No