Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Indra Ramasamy | -- | 2433 | 2024-03-07 09:46:40 | | | |
| 2 | Rita Xu | Meta information modification | 2433 | 2024-05-27 10:11:03 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ramasamy, I. Appetite Regulation and Bariatric Surgery. Encyclopedia. Available online: https://encyclopedia.pub/entry/55959 (accessed on 25 July 2026).
Ramasamy I. Appetite Regulation and Bariatric Surgery. Encyclopedia. Available at: https://encyclopedia.pub/entry/55959. Accessed July 25, 2026.
Ramasamy, Indra. "Appetite Regulation and Bariatric Surgery" Encyclopedia, https://encyclopedia.pub/entry/55959 (accessed July 25, 2026).
Ramasamy, I. (2024, March 07). Appetite Regulation and Bariatric Surgery. In Encyclopedia. https://encyclopedia.pub/entry/55959
Ramasamy, Indra. "Appetite Regulation and Bariatric Surgery." Encyclopedia. Web. 07 March, 2024.
Copy Citation
Obesity remains a common metabolic disorder and a threat to health as it is associated with numerous complications. Lifestyle modifications and caloric restriction can achieve limited weight loss. Bariatric surgery is an effective way of achieving substantial weight loss as well as glycemic control secondary to weight-related type 2 diabetes mellitus.
hunger centre
satiety centre
leptin
incretin
1. Introduction
The gut endocrine system and the gut-central nervous system axis function to digest and absorb food and, consequently, to regulate appetite. The gut-pancreatic endocrine system is one of the largest endocrine systems in the body. Most of the gut hormones secreted (incretins, cholecystokinin, and pancreatic polypeptide), except ghrelin, act to increase satiety and decrease food intake [1]. The adipose tissue is widely distributed in the body; has distinct functions both in energy homeostasis and as an endocrine organ. Leptin, an adipokine secreted by the adipose tissue, has a satiety effect on the central nervous system [2]. Many gut hormones act on the hypothalamus and brain stem areas of appetite control. Within the arcuate nucleus of the hypothalamus are the proopiomelanocortin (POMC)-secreting neurons, which suppress appetite, and a second population of neurons, which increase food intake and co-express neuropeptide Y (NPY) and agouti-related protein (AgRP) [3].
Obesity has become a major contributor to the global burden of chronic disease. Obesity is associated with several disease states, which include, among others, type 2 diabetes (T2DM), hypertension, dyslipidemia, cardiovascular disease, non-alcoholic fatty liver disease, and obstructive sleep apnea [4]. This has led to the development of several obesity treatment methods, including bariatric surgery.
Appropriate response to changes in the homeostatic control of energy intake is obligatory for a stable energetic state and weight maintenance. Understanding the mechanisms controlling appetite and weight transformation may facilitate the understanding of appetite changes as well as weight loss and hormonal changes following bariatric surgery. Bariatric surgery is associated with side effects such as dumping syndrome, postprandial hypoglycaemia, partial T2DM remission or T2DM relapse, and weight regain. Knowledge of the mechanisms of appetite homeostasis may facilitate a greater understanding of changes following bariatric surgery, the development of therapeutic approaches for avoiding the side effects of bariatric surgery, and the advancement of less invasive weight loss strategies [5].
2. Neurons and the Control of Appetite
Several neuronal populations, which are distributed throughout the brain, affect the capacity for food intake. Specific areas of the hypothalamus are believed to control feeding behaviour. Two distinct neuronal populations are thought to be significant in regulating energy balance. These are found in the arcuate nucleus of the hypothalamus (ARH), the anorexigenic (appetite suppressing) proopiomelanocortin (POMC) neurons, and the orexigenic (appetite-increasing) neuropeptide Y (NPY)/agouti-related peptide (AgRP) co-expressing neurons. The neurons are positioned to receive signals from both peripheral organs due to the rich blood supply to the arcuate nucleus as well as input from various parts of the central nervous system [6]. POMC-expressing neurons are also located in the nucleus tractus solitaries (NTS) and have different behavioral energy homeostasis functions in the ARH and NTS [7]. POMC neurons have been investigated extensively because of their central role in energy balance. POMC neurones in the ARH extend fibres to other ARH neurones and other multiple brain regions, including the paraventricular nucleus of the hypothalamus, lateral hypothalamus, commissural nucleus of the solitary tract, bed nucleus of the stria terminalis, nucleus accumbens, septal nucleus, ventral tegmental area, central amygdala, periaqueductal grey, and dorsal raphe nucleus [8]. The ARH contains distinct populations of POMC neurons that are either GABAergic or glutamatergic neurotransmitters and are activated by serotonin 5-HT2Cs, insulin, and leptin receptors (among others) [8]. The evidence for leptin acting on a different population of POMC neurons than insulin is conflicting [9]. Other studies suggest a heterogeneous population for POMC and that POMC neurons form several molecularly distinct clusters [10]. POMC and AgRP neurons change neuronal activity in response to glucose fluctuations [11]. Specific responses may be segregated into distinct populations of POMC neurons (Figure 1a–c). POMC neurons suppress appetite, and following processing of POMC by proprotein convertase (coded by PCSK1), release α-melanocyte stimulating hormone (α-MSH). The hormone α-MSH is an agonist of the melanocortin-4-receptors (MC4R), found in the paraventricular nucleus. The MC4R receptor has anorectic activity [6][12]. AgRP inhibits the MC4R agonist activity of melanocortin peptides (Figure 1a–c). In addition, a functional role in energy homeostasis for central and peripheral MC3R has been suggested [8]. In this system, hormones of the fed state (insulin and leptin) released by the pancreas and adipocytes (respectively) bind to their receptors on the POMC neurons and support the processing of proopiomelanocortin to α-MSH, promoting a signal to decrease food intake. Leptin also binds to AgRP /NPY neurons to inhibit their orexigenic activity. It is likely that leptin binding to neuronal cells other than the POMC neurons contributes to energy homeostasis [13] and that AgRP neurons engage other circuits that coordinate feeding. Axon projections of AgRP neurons extend to neurons in the bed nucleus of the stria terminalis, lateral hypothalamus, and parabrachial nuclei that control insulin sensitivity in brown adipose tissue. The axon projections of the AgRP neurons may trigger the motivational and autonomic circuits that contribute to feeding behavior. It is likely that multiple pathways function in the brain, one for homeostatic control of energy balance and feeding and another for non-homeostatic, reward-driven, hedonic feeding [13]. One suggestion is that the hedonic circuitry, once activated, can override the homeostatic energy balance circuitry and the chronic inhibition of AgRP [14]. The effects of lifestyle on epigenetic modifications of genes in the hypothalamus and its role in the regulation of energy homeostasis require further investigation [15].
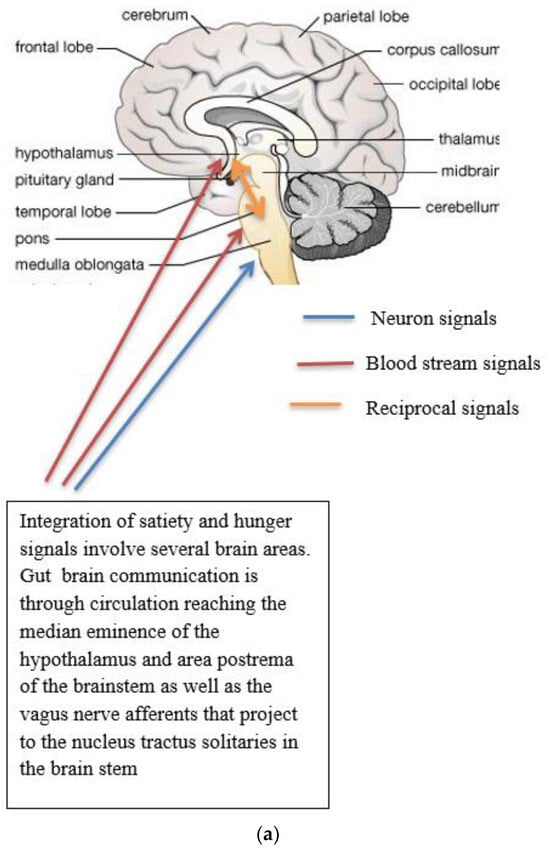
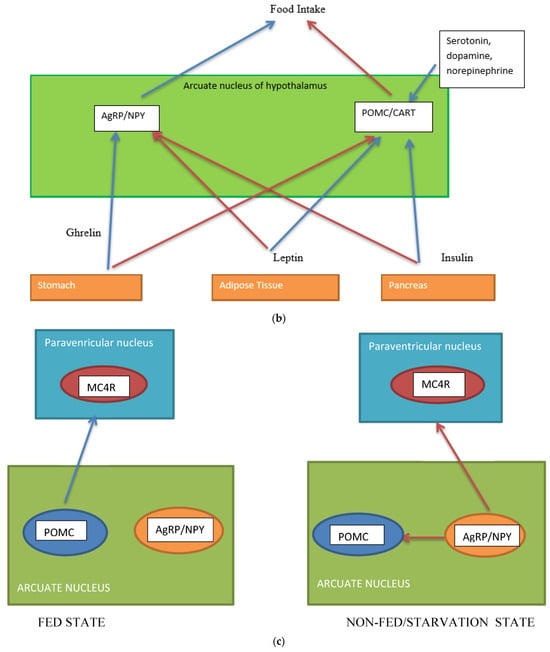
Figure 1. (a) Gut-brain communication underlying appetite control; (b) neuron function in appetite control, AgRP/NPY (neuropeptide Y/agouti-related peptide neurons), POMC (proopiomelanocortin),  activation,
activation,  inhibition; (c) neuron interaction in appetite control, activation , inhibition . Populations of POMC neurons express different receptors. e.g., leptin and insulin. In the fed state, POMC neurons act to decrease food intake and increase energy expenditure. In the fasted state, AgRP/NPY neurons inhibit the activity of MC4R neurons and POMC neurons.
inhibition; (c) neuron interaction in appetite control, activation , inhibition . Populations of POMC neurons express different receptors. e.g., leptin and insulin. In the fed state, POMC neurons act to decrease food intake and increase energy expenditure. In the fasted state, AgRP/NPY neurons inhibit the activity of MC4R neurons and POMC neurons.
activation, inhibition; (c) neuron interaction in appetite control, activation , inhibition . Populations of POMC neurons express different receptors. e.g., leptin and insulin. In the fed state, POMC neurons act to decrease food intake and increase energy expenditure. In the fasted state, AgRP/NPY neurons inhibit the activity of MC4R neurons and POMC neurons.Complex neurological and endocrine signals are relayed between the gut and the brain to regulate hunger and satiety. The gut neuroendocrine system and the gut-brain axis function to optimise digestion and absorption and regulate appetite. Numerous reciprocal connections exist between the brain stem (particularly the nucleus of the tractus solitaries) and the hypothalamus [16]. The brain stem receives vagus nerve afferents from the gastrointestinal tract and endocrine signals from the blood due to its closeness to the blood-brain barrier. It is positioned to act as a site of integration between endocrine and neuronal signals (Figure 1a). Afferent neurons of the vagus nerve targets for gut hormones [17].
3. The Endocrine System of the Gut and the Control of Appetite
Gut hormones are secreted by the enteroendocrine cells scattered throughout the epithelial cells of the gut. Gut hormones can act as both hormones and neurotransmitters. The incretin effect is described as the increase in insulin secretion by pancreatic β cells by exposure to intestinal absorption of glucose compared to isoglycemic levels by intravenous infusion. Gut derived hormones are the proglucagon-derived peptides, glucagon-like peptide 1 (GLP-1), GLP2, and gastric inhibitory peptide, also known as glucose-dependent insulinotropic polypeptide (GIP), as well as cholecystokinin (CCK) and peptide YY (PYY), released by the gastrointestinal tract into the general circulation, which mediate effects of feeding [18]. Both GIP and GLP-1, the real incretins, exert their effects by binding to their specific receptors, the GIP receptor (GIPR) and the GLP1 receptor (GLP1R), in pancreatic β cells and boosting the secretion of glucose-dependent insulin release [19][20]. Evidence points out that some enteroendocrine cells, which constitute approximately 1% of intestinal epithelial cells, are plurihormonal. In mice, GIP is secreted from K cells found predominantly in the duodenum, and GLP-1 is secreted from L cells located in the lower small intestine and colon. The presence of food (carbohydrates, protein, and fats) stimulates incretin hormone secretion. Similar to other gut peptides, GLP-1 is a neurotransmitter. Evidence for the involvement of GLP-1 in signalling in the CNS is the wide distribution of the GLP-1 receptor [21]. The GLP1R receptor is widely distributed in the body, for example, in the cardiovascular system, gastrointestinal tract, adipose tissue, brain, and bone [22]. Similarly, some studies report the GLP2R receptor to be widely expressed [23]. This highlights the diversity of incretin functions. A wide range of pharmacological drugs and pre-clinical studies have been used to clarify GLP1R/GIPR physiological function [24].
3.1. Physiology of Incretin Secretion
Release of incretins (GLP-1 and GLP2 are cosecreted) requires the presence of nutrients, carbohydrates, proteins, and fat in the intestinal lumen and their absorption through the enterocytes [25]. To understand the process of altered incretin secretion following bariatric surgery, an understanding of the detailed molecular mechanism underlying its secretion is valuable and is included.
3.2. Secretion and Metabolism of Incretins
In the pancreas, the proglucagon molecule is processed by proconvertase 2 (PC2) to glucagon (Figure 2) and by proconvertase 1/3 (PC1/3) in the gut into GLP-1 (amino acids 1–37) and GLP2 (amino acids 1–33) [26]. Glucagon is secreted by the pancreatic α cell, stimulates glycogenolysis and gluconeogenesis in the liver, and opposes the hypoglycemic action of insulin. Bioactive GLP-1 (7–37) is generated from GLP-1 (1–37). Cleavage by PC1/3 leaves two basic residues at the C-terminus of GLP-1, which are removed by carboxypeptidase prior to carboxyamidation. Several immunoreactive forms of GLP-1 (including GLP-1 7–36 amide and GLP-1 7–37, which are thought to be equipotent), are released in vivo [27]. GLP-1 is inactivated by dipeptidyl peptidase IV (DPP-IV), which cleaves the N-terminal end. GLP-1 (7–36 amide) is thought to be the majority of circulating active GLP-1 in human plasma [28]. Degradation and inactivation of human GLP-2 (amino acids 1–33) with the enzyme DPP-IV resulted in the liberation of GLP-2 (amino acids 3–33) [29].
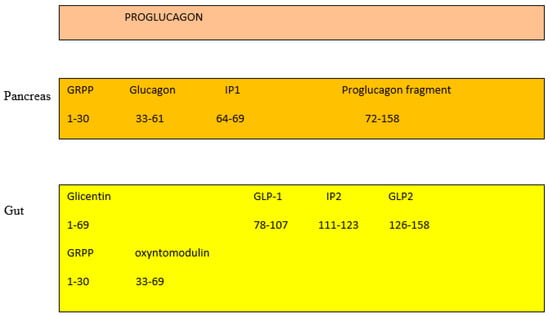
Figure 2. Gut and pancreas processing of the proglucagon molecule.
GIP is derived by proteolytic processing of a 153-residue precursor, preproGIP [30]. GIP is a 42 amino acid, and as it contains an alanine in position 2, it is physiologically degraded and inactivated by DPP-IV to its bioinactive form (amino acid 3–42) within minutes of secretion [31]. Both inactive GIP and GLP-1 are cleared by the kidney [32]. In healthy humans, the half-lives of GLP-1 is 2–3 min, GIP 5 min [33], and GLP-2 7 min [34]. Which of the two, GLP-1 or GIP, is the most prominent is still a matter of debate. Depending on the method protocol, it has been suggested that either GIP or GLP-1 contributes equally or that either GIP or GLP-1 contribute the majority of the incretin effect in humans [35]. One suggestion is that the contribution to the incretin effect is GIP: approximately 44%, GLP-1: approximately 22%, Glucose: approximately 33%, and neural transmission: negligible [36].
3.3. Control of Incretin Secretion by Nutrients
It has been shown that the presence of macronutrients (carbohydrates, proteins, and fats) in the gut regulates incretin hormone secretion.
3.3.1. Carbohydrates
Glucose is a recognised stimulant of incretins and acts through the sodium glucose cotransporter 1 (SGLT1) with the influx of sodium ions, which depolarises the plasma membrane, opening the voltage-sensitive calcium channels, which is followed by exocytosis of GLP-1-containing secretory vesicles [37]. Facilitative glucose transport through the GLUT2 transporter appears to be of less importance in incretin secretion [38].
3.3.2. Proteins
The pathways underlying the mechanism of protein stimulation of gut hormones are still not clearly understood. At the molecular level, meat hydrolysate has been shown to recruit the G-protein coupled calcium sensing receptor (CaSR) to cause GLP-1 release from the rodent intestine [39], though it is likely that other sensory pathways are involved, e.g., an increase in the anorectic hormone PYY [39][40].
3.3.3. Fats
Fats also potentially stimulate CCK, PYY, and GLP-1 release. The release of CCK, PYY, and GLP-1 in plasma is attenuated by inhibitors of lipase. CCK, GLP-1, and PYY release were found to be dependent on fatty acid chain length, with only fatty acids greater than C10 being effective in stimulating hormone secretion [41][42]. Several G-protein-coupled receptors (GPCR) have been associated with the sensing of fatty acids and the release of gut hormones. GPR40 is a receptor for medium- and long-chain fatty acids. The distribution of GPR40 suggests it may act to regulate pancreatic islet and neurological cell function [43]. Close to the GPR40 locus are the GPR41 and GPR43 genes encoding receptors activated by short-chain fatty acids [44]. GPCR receptors linked to GIP secretion are GPR120 and GPR40, expressed in K cells in the upper small intestine [45][46]. GPR40, GPR41, GPR43, GPR119, and GP120 were identified as sensing receptors for fatty acids with a role in GLP-1 secretion [47][48][49][50][51].
3.4. Other Variables That Regulate Gut Hormones
- (i)
- (ii)
-
Bile acids: Using a pharmacological approach, the bile acid-responsive receptor, G protein-coupled bile acid receptor 1 (GPBAR1/TGR5), has been shown to promote GLP-1 and PYY secretion from intestinal L-cells [54][55]. While bile acids are recognised to signal through specific nuclear acid receptors [56], GPBAR1 is a cell surface receptor. GPBAR1 is a GPCR that is activated by bile acids, resulting in stimulation of Gαs proteins and downstream cAMP signalling pathways. Recent findings support the hypothesis that additional pathways such as elevation of intracellular Ca2+ concentrations [57] and closure of ATP-sensitive potassium (KATP) channels may play a role in GLP-1 release [58]. Available data suggest that bile acids were more effective if they were applied to the basolateral GPBAR1 (vascular side) on L-cells and that luminal-applied bile acids are effective after absorption across the intestinal epithelial layer [59].
- (iii)
-
Proinflammatory cytokines TNFα, IL6, and Regulated on Activation, Normal T-cell Expressed and Secreted (RANTES) are increased in obesity in both rodents and humans. Acute treatment with TNFα increases GLP-1 release, and chronic elevation of TNFα decreases GLP-1 release. It has been suggested that TNFα alters cell signalling through the TNF receptor-NFκβ pathway [60]. IL6 regulates glucose homeostasis by stimulating L cells and pancreatic α cells to secrete GLP-1 and, as a result, increase insulin secretion. One suggestion is that acute effects of IL6 are caused by increased GLP-1 exocytosis in L cells and chronic effects by increased glucose responsiveness [61][62]. The chemokine RANTES reduced glucose and stimulated GLP-1 secretion in humans. One proposal is that RANTES acts through the CCR1 receptor to reduce cAMP levels and PKA activity [63]. The response of the L-cell is likely an integrated response to different cytokines using different signalling pathways.
- (iv)
-
Other suspected stimulants of GLP-1 secretion are progesterone (studies on cell lines suggest stimulation through the extracellular signal-related kinase 1/2 (ERK1/2) [64], insulin (studies on cell lines suggest a role for phosphatidylinositol 3 kinase-Akt and MAPK kinase (MEK)-ERK1/2 pathways [65], and glucocorticoid (reduced GLP-1 secretion in rodents) [66].
The contribution of each of these additional effects to incretin secretion is unclear, and further studies are required to understand the mechanism underlying these effects.
References
- Gribble, F.M.; Reimann, F. Function and mechanisms of enteroendocrine cells and gut hormones in metabolism. Nat. Rev. Endocrinol. 2019, 15, 226–237.
- Park, H.-K.; Ahima, R.S. Physiology of leptin: Energy homeostasis, neuroendocrine function and metabolism. Metabolism 2015, 64, 24–34.
- Vohra, M.S.; Benchoula, K.; Serpell, C.J.; Hwa, W.E. AgRP/NPY and POMC neurons in the arcuate nucleus and their potential role in treatment of obesity. Eur. J. Pharmacol. 2022, 915, 174611.
- Ward, Z.J.; Bleich, S.N.; Cradock, A.L.; Barrett, J.L.; Giles, C.M.; Flax, C.; Long, M.W.; Gortmaker, S.L. Projected U.S. state-level prevalence of adult obesity and severe obesity. N. Engl. J. Med. 2019, 381, 2440–2450.
- Ruban, A.; Stoenchev, K.; Ashrafian, H.; Teare, J. Current treatments for obesity. Clin. Med. 2019, 19, 205–212.
- Sohn, J.-W. Network of hypothalamic neurons that control appetite. BMB Rep. 2015, 48, 229–233.
- Zhan, C.; Zhou, J.; Feng, Q.; Zhang, J.-E.; Lin, S.; Bao, J.; Wu, P.; Luo, M. Acute and Long-Term Suppression of Feeding Behavior by POMC Neurons in the Brainstem and Hypothalamus, Respectively. J. Neurosci. 2013, 33, 3624–3632.
- Mountjoy, K.G. Pro-Opiomelanocortin (POMC) Neurones, POMC-Derived Peptides, Melanocortin Receptors and Obesity: How Understanding of this System has Changed Over the Last Decade. J. Neuroendocr. 2015, 27, 406–418.
- Qiu, J.; Zhang, C.; Borgquist, A.; Nestor, C.C.; Smith, A.W.; Bosch, M.A.; Ku, S.; Wagner, E.J.; Rønnekleiv, O.K.; Kelly, M.J. Insulin Excites Anorexigenic Proopiomelanocortin Neurons via Activation of Canonical Transient Receptor Potential Channels. Cell Metab. 2014, 19, 682–693.
- Quarta, C.; Claret, M.; Zeltser, L.M.; Williams, K.W.; Yeo, G.S.H.; Tschöp, M.H.; Diano, S.; Brüning, J.C.; Cota, D. POMC neuronal heterogeneity in energy balance and beyond: An integrated view. Nat. Metab. 2021, 3, 299–308.
- Yoon, N.A.; Diano, S. Hypothalamic glucose-sensing mechanisms. Diabetologia 2021, 64, 985–993.
- Sohn, Y.B. Genetic obesity: An update with emerging therapeutic approaches. Ann. Pediatr. Endocrinol. Metab. 2022, 27, 169–175.
- Baldini, G.; Phelan, K.D. The melanocortin pathway and control of appetite-progress and therapeutic implications. J. Endocrinol. 2019, 241, R1–R33.
- Denis, R.G.; Joly-Amado, A.; Webber, E.; Langlet, F.; Schaeffer, M.; Padilla, S.L.; Cansell, C.; Dehouck, B.; Castel, J.; Delbès, A.-S.; et al. Palatability Can Drive Feeding Independent of AgRP Neurons. Cell Metab. 2015, 22, 646–657, Erratum in Cell Metab. 2017, 25, 975.
- Benite-Ribeiro, S.A.; Putt, D.A.; Soares-Filho, M.C.; Santos, J.M. The link between hypothalamic epigenetic modifications and long-term feeding control. Appetite 2016, 107, 445–453.
- Schwartz, M.W.; Woods, S.C.; Porte, D., Jr.; Seeley, R.J.; Baskin, D.G. Central nervous system control of food intake. Nature 2000, 404, 661–671.
- Khandekar, N.; Berning, B.A.; Sainsbury, A.; Lin, S. The role of pancreatic polypeptide in the regulation of energy homeostasis. Mol. Cell. Endocrinol. 2015, 418 Pt 1, 33–41.
- Pais, R.; Gribble, F.M.; Reimann, F. Stimulation of incretin secreting cells. Ther. Adv. Endocrinol. Metab. 2016, 7, 24–42.
- El, K.; Campbell, J.E. The role of GIP in α-cells and glucagon secretion. Peptides 2020, 125, 170213.
- McLean, M.A.; Wong, C.K.; Campbell, J.E.; Hodson, D.J.; Trapp, S.; Drucker, D.J. Revisiting the Complexity of GLP-1 Action from Sites of Synthesis to Receptor Activation. Endocr. Rev. 2021, 42, 101–132.
- Chaudhri, O.; Small, C.; Bloom, S. Gastrointestinal hormones regulating appetite. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 1187–1209.
- Nauck, M.A.; Quast, D.R.; Wefers, J.; Pfeiffer, A.F.H. The evolving story of incretins (GIP and GLP-1) in metabolic and cardiovascular disease: A pathophysiological update. Diabetes Obes. Metab. 2021, 23 (Suppl. S3), 5–29.
- El-Jamal, N.; Erdual, E.; Neunlist, M.; Koriche, D.; Dubuquoy, C.; Maggiotto, F.; Chevalier, J.; Berrebi, D.; Dubuquoy, L.; Boulanger, E.; et al. Glugacon-like peptide-2: Broad receptor expression, limited therapeutic effect on intestinal inflammation and novel role in liver regeneration. Am. J. Physiol. Liver Physiol. 2014, 307, G274–G285.
- Zhao, X.; Wang, M.; Wen, Z.; Lu, Z.; Cui, L.; Fu, C.; Xue, H.; Liu, Y.; Zhang, Y. GLP-1 Receptor Agonists: Beyond Their Pancreatic Effects. Front. Endocrinol. 2021, 12, 721135.
- Seino, Y.; Fukushima, M.; Yabe, D. GIP and GLP-1, the two incretin hormones: Similarities and differences. J. Diabetes Investig. 2010, 1, 8–23.
- Rouillé, Y.; Martin, S.; Steiner, D.F. Differential Processing of Proglucagon by the Subtilisin-like Prohormone Convertases PC2 and PC3 to Generate either Glucagon or Glucagon-like Peptide. J. Biol. Chem. 1995, 270, 26488–26496.
- Friis-Hansen, L.; Lacourse, K.; Samuelson, L.; Holst, J. Attenuated processing of proglucagon and glucagon-like peptide-1 in carboxypeptidase E-deficient mice. J. Endocrinol. 2001, 169, 595–602.
- Ørskov, C.; Rabenhøj, L.; Wettergren, A.; Kofod, H.; Holst, J.J. Tissue and Plasma Concentrations of Amidated and Glycine-Extended Glucagon-Like Peptide I in Humans. Diabetes 1994, 43, 535–539.
- Brubaker, P.L.; Crivici, A.; Izzo, A.; Ehrlich, P.; Tsai, C.-H.; Drucker, D.J. Circulating and Tissue Forms of the Intestinal Growth Factor, Glucagon-Like Peptide-2*. Endocrinology 1997, 138, 4837–4843.
- Takeda, J.; Seino, Y.; Tanaka, K.; Fukumoto, H.; Kayano, T.; Takahashi, H.; Mitani, T.; Kurono, M.; Suzuki, T.; Tobe, T. Sequence of an intestinal cDNA encoding human gastric inhibitory polypeptide precursor. Proc. Natl. Acad. Sci. USA 1987, 84, 7005–7008.
- Kieffer, T.J.; McIntosh, C.H.; Pederson, R.A. Degradation of glucose-dependent insulinotropic polypeptide and truncated glucagon-like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV. Endocrinology 1995, 136, 3585–3596.
- Mentlein, R. Mechanisms underlying the rapid degradation and elimination of the incretin hormones GLP-1 and GIP. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 443–452.
- Meier, J.J.; Nauck, M.A.; Kranz, D.; Holst, J.J.; Deacon, C.F.; Gaeckler, D.; Schmidt, W.E.; Gallwitz, B. Secretion, Degradation, and Elimination of Glucagon-Like Peptide 1 and Gastric Inhibitory Polypeptide in Patients with Chronic Renal Insufficiency and Healthy Control Subjects. Diabetes 2004, 53, 654–662.
- Hartmann, B.; Harr, M.B.; Jeppesen, P.B.; Wojdemann, M.; Deacon, C.F.; Mortensen, P.B.; Holst, J.J. In Vivo and in Vitro Degradation of Glucagon-Like Peptide-2 in Humans1. J. Clin. Endocrinol. Metab. 2000, 85, 2884–2888.
- Gasbjerg, L.S.; Helsted, M.M.; Hartmann, B.; Jensen, M.H.; Gabe, M.B.N.; Sparre-Ulrich, A.H.; Veedfald, S.; Stensen, S.; Lanng, A.R.; Bergmann, N.C.; et al. Separate and Combined Glucometabolic Effects of Endogenous Glucose-Dependent Insulinotropic Polypeptide and Glucagon-like Peptide 1 in Healthy Individuals. Diabetes 2019, 68, 906–917.
- Nauck, M.A.; Meier, J.J. GIP and GLP-1: Stepsiblings Rather Than Monozygotic Twins Within the Incretin Family. Diabetes 2019, 68, 897–900.
- Gribble, F.M.; Williams, L.; Simpson, A.K.; Reimann, F. A Novel Glucose-Sensing Mechanism Contributing to Glucagon-Like Peptide-1 Secretion from the GLUTag Cell Line. Diabetes 2003, 52, 1147–1154.
- Röder, P.V.; Geillinger, K.E.; Zietek, T.S.; Thorens, B.; Koepsell, H.; Daniel, H. The Role of SGLT1 and GLUT2 in Intestinal Glucose Transport and Sensing. PLoS ONE 2014, 9, e89977.
- Pais, R.; Gribble, F.M.; Reimann, F. Signalling pathways involved in the detection of peptones by murine small intestinal enteroendocrine L-cells. Peptides 2016, 77, 9–15.
- Batterham, R.L.; Heffron, H.; Kapoor, S.; Chivers, J.E.; Chandarana, K.; Herzog, H.; Le Roux, C.W.; Thomas, E.L.; Bell, J.D.; Withers, D.J. Critical role for peptide YY in protein-mediated satiation and body-weight regulation. Cell Metab. 2006, 4, 223–233.
- Feltrin, K.L.; Little, T.J.; Meyer, J.H.; Horowitz, M.; Smout, A.J.P.M.; Wishart, J.; Pilichiewicz, A.N.; Rades, T.; Chapman, I.M.; Feinle-Bisset, C. Effects of intraduodenal fatty acids on appetite, antropyloroduodenal motility, and plasma CCK and GLP-1 in humans vary with their chain length. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R524–R533.
- Feltrin, K.L.; Little, T.J.; Meyer, J.H.; Horowitz, M.; Rades, T.; Wishart, J.; Feinle-Bisset, C. Effects of lauric acid on upper gut motility, plasma cholecystokinin and peptide YY, and energy intake are load, but not concentration, dependent in humans. J. Physiol. 2007, 581 Pt 2, 767–777.
- Briscoe, C.P.; Tadayyon, M.; Andrews, J.L.; Benson, W.G.; Chambers, J.K.; Eilert, M.M.; Ellis, C.; Elshourbagy, N.A.; Goetz, A.S.; Minnick, D.T.; et al. The Orphan G Protein-coupled Receptor GPR40 Is Activated by Medium and Long Chain Fatty Acids. J. Biol. Chem. 2003, 278, 11303–11311.
- Ridner, G.; Bartoov-Shifman, R.; Zalogin, T.; Avnit-Sagi, T.; Bahar, K.; Sharivkin, R.; Kantorovich, L.; Weiss, S.; Walker, M.D. Regulation of the GPR40 locus: Towards a molecular understanding. Biochem. Soc. Trans. 2008, 36 Pt 3, 360–362.
- Iwasaki, K.; Harada, N.; Sasaki, K.; Yamane, S.; Iida, K.; Suzuki, K.; Hamasaki, A.; Nasteska, D.; Shibue, K.; Joo, E.; et al. Free Fatty Acid Receptor GPR120 Is Highly Expressed in Enteroendocrine K Cells of the Upper Small Intestine and Has a Critical Role in GIP Secretion After Fat Ingestion. Endocrinology 2015, 156, 837–846.
- Sankoda, A.; Harada, N.; Kato, T.; Ikeguchi, E.; Iwasaki, K.; Yamane, S.; Murata, Y.; Hirasawa, A.; Inagaki, N. Free fatty acid receptors, G protein-coupled receptor 120 and G protein-coupled receptor 40, are essential for oil-induced gastric inhibitory polypeptide secretion. J. Diabetes Investig. 2019, 10, 1430–1437.
- Overton, H.A.; Babbs, A.J.; Doel, S.M.; Fyfe, M.C.; Gardner, L.S.; Griffin, G.; Jackson, H.C.; Procter, M.J.; Rasamison, C.M.; Tang-Christensen, M.; et al. Deorphanization of a G protein-coupled receptor for oleoylethanolamide and its use in the discovery of small-molecule hypophagic agents. Cell Metab. 2006, 3, 167–175.
- Edfalk, S.; Steneberg, P.; Edlund, H. Gpr40 Is Expressed in Enteroendocrine Cells and Mediates Free Fatty Acid Stimulation of Incretin Secretion. Diabetes 2008, 57, 2280–2287.
- Hirasawa, A.; Tsumaya, K.; Awaji, T.; Katsuma, S.; Adachi, T.; Yamada, M.; Sugimoto, Y.; Miyazaki, S.; Tsujimoto, G. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat. Med. 2005, 11, 90–94.
- Chu, Z.-L.; Carroll, C.; Alfonso, J.; Gutierrez, V.; He, H.; Lucman, A.; Pedraza, M.; Mondala, H.; Gao, H.; Bagnol, D.; et al. A Role for Intestinal Endocrine Cell-Expressed G Protein-Coupled Receptor 119 in Glycemic Control by Enhancing Glucagon-Like Peptide-1 and Glucose-Dependent Insulinotropic Peptide Release. Endocrinology 2008, 149, 2038–2047.
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; Cameron, J.; Grosse, J.; Reimann, F.; Gribble, F.M. Short-Chain Fatty Acids Stimulate Glucagon-Like Peptide-1 Secretion via the G-Protein-Coupled Receptor FFAR2. Diabetes 2012, 61, 364–371.
- Tremaroli, V.; Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249.
- Wichmann, A.; Allahyar, A.; Greiner, T.U.; Plovier, H.; Lundén, G.; Larsson, T.; Drucker, D.J.; Delzenne, N.M.; Cani, P.D.; Bäckhed, F. Microbial Modulation of Energy Availability in the Colon Regulates Intestinal Transit. Cell Host Microbe 2013, 14, 582–590.
- Katsuma, S.; Hirasawa, A.; Tsujimoto, G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem. Biophys. Res. Commun. 2005, 329, 386–390.
- Ullmer, C.; Sanchez, R.A.; Sprecher, U.; Raab, S.; Mattei, P.; Dehmlow, H.; Sewing, S.; Iglesias, A.; Beauchamp, J.; Conde-Knape, K. Systemic bile acid sensing by G protein-coupled bile acid receptor 1 (GPBAR1) promotes PYY and GLP-1 release. Br. J. Pharmacol. 2013, 169, 671–684.
- Hylemon, P.B.; Zhou, H.; Pandak, W.M.; Ren, S.; Gil, G.; Dent, P. Bile acids as regulatory molecules. J. Lipid Res. 2009, 50, 1509–1520.
- Parker, H.; Wallis, K.; le Roux, C.; Wong, K.; Reimann, F.; Gribble, F. Molecular mechanisms underlying bile acid-stimulated glucagon-like peptide-1 secretion. Br. J. Pharmacol. 2012, 165, 414–423.
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.; et al. TGR5-Mediated Bile Acid Sensing Controls Glucose Homeostasis. Cell Metab. 2009, 10, 167–177.
- Brighton, C.A.; Rievaj, J.; Kuhre, R.E.; Glass, L.L.; Schoonjans, K.; Holst, J.J.; Gribble, F.M.; Reimann, F. Bile Acids Trigger GLP-1 Release Predominantly by Accessing Basolaterally Located G Protein–Coupled Bile Acid Receptors. Endocrinology 2015, 156, 3961–3970.
- Gagnon, J.; Sauvé, M.; Zhao, W.; Stacey, H.M.; Wiber, S.C.; Bolz, S.-S.; Brubaker, P.L. Chronic Exposure to TNFα Impairs Secretion of Glucagon-Like Peptide-1. Endocrinology 2015, 156, 3950–3960.
- Allen, T.L.; Whitham, M.; Febbraio, M.A. IL-6 Muscles in on the Gut and Pancreas to Enhance Insulin Secretion. Cell Metab. 2012, 15, 8–9.
- Ellingsgaard, H.; Hauselmann, I.; Schuler, B.; Habib, A.M.; Baggio, L.L.; Meier, D.T.; Eppler, E.; Bouzakri, K.; Wueest, S.; Muller, Y.D.; et al. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat. Med. 2011, 17, 1481–1489.
- Pais, R.; Zietek, T.; Hauner, H.; Daniel, H.; Skurk, T. RANTES (CCL5) reduces glucose-dependent secretion of glucagon-like peptides 1 and 2 and impairs glucose-induced insulin secretion in mice. Am. J. Physiol. Liver Physiol. 2014, 307, G330–G337.
- Flock, G.B.; Cao, X.; Maziarz, M.; Drucker, D.J. Activation of Enteroendocrine Membrane Progesterone Receptors Promotes Incretin Secretion and Improves Glucose Tolerance in Mice. Diabetes 2012, 62, 283–290.
- Lim, G.E.; Huang, G.J.; Flora, N.; LeRoith, D.; Rhodes, C.J.; Brubaker, P.L. Insulin Regulates Glucagon-Like Peptide-1 Secretion from the Enteroendocrine L Cell. Endocrinology 2009, 150, 580–591.
- Kappe, C.; Fransson, L.; Wolbert, P.; Ortsäter, H. Glucocorticoids suppress GLP-1 secretion: Possible contribution to their diabetogenic effects. Clin. Sci. 2015, 129, 405–414.
More
Information
Subjects:
Gastroenterology & Hepatology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
504
Revisions:
2 times
(View History)
Update Date:
27 May 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No