Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Eric D. Hamlett | -- | 2566 | 2024-03-06 19:13:28 | | | |
| 2 | Peter Tang | + 5 word(s) | 2571 | 2024-03-07 02:50:47 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Granholm, A.; Hamlett, E.D. Tau Pathology in Alzheimer’s Disease and Down Syndrome. Encyclopedia. Available online: https://encyclopedia.pub/entry/55935 (accessed on 10 August 2026).
Granholm A, Hamlett ED. Tau Pathology in Alzheimer’s Disease and Down Syndrome. Encyclopedia. Available at: https://encyclopedia.pub/entry/55935. Accessed August 10, 2026.
Granholm, Ann-Charlotte, Eric D. Hamlett. "Tau Pathology in Alzheimer’s Disease and Down Syndrome" Encyclopedia, https://encyclopedia.pub/entry/55935 (accessed August 10, 2026).
Granholm, A., & Hamlett, E.D. (2024, March 06). Tau Pathology in Alzheimer’s Disease and Down Syndrome. In Encyclopedia. https://encyclopedia.pub/entry/55935
Granholm, Ann-Charlotte and Eric D. Hamlett. "Tau Pathology in Alzheimer’s Disease and Down Syndrome." Encyclopedia. Web. 06 March, 2024.
Copy Citation
Individuals with Down syndrome (DS) exhibit an almost complete penetrance of Alzheimer’s disease (AD) pathology but are underrepresented in clinical trials for AD. The Tau protein is associated with microtubule function in the neuron and is crucial for normal axonal transport. In several different neurodegenerative disorders, Tau misfolding leads to hyper-phosphorylation of Tau (p-Tau), which may seed pathology to bystander cells and spread.
Down syndrome (DS)
Alzheimer’s disease (AD)
neurofibrillary tangles (NFTs)
seeding competent p-Tau
neuropathology
1. Introduction
Individuals with Down syndrome (DS) exhibit Alzheimer’s disease (AD)-related pathology in the brain early in life, leading to the development of dementia in their 40s or 50s, with few exceptions [1][2][3]. The AD pathology that develops in the DS brain includes widespread amyloid plaques, neuroinflammation, cell death, and neurofibrillary tangles (NFTs, [1][4][5][6]). Amyloid pathology is naturally occurring in the DS brain because the amyloid precursor protein gene (APP) is located on Chromosome 21 (Chr. 21), leading to increased APP and amyloid production early in life, both in humans with DS and DS mouse models [3][7][8][9][10]. Neuroinflammation is also an inherent trait of the DS brain due to several different genes encoded on Chr. 21, including four of six interferon receptors [11][12] and the superoxide dismutase 1 gene (SOD-1) [13], among other factors. Therefore, cells obtained from the DS brain exhibit early signs of inflammatory activation as well as oxidative stress [3][4][6][14][15].
The spreading of misfolded Tau is associated with AD and DS-AD pathology, including hyperphosphorylated Tau (p-Tau), which spreads from region to region in the brain, finally leading to a Braak stage of V-VI in the final stages of DS-AD pathology [16][17]. While AD is considered a sporadic disease of aging or as occurring earlier in life, due to the inheritance of rare genetic mutations, DS-AD presents early age-dependent kinetics of amyloidogenesis and Tau seeding, perhaps as early as the teenage years [18]. It is difficult to compare DS-AD with familial or late onset AD, since they have different biological mechanisms. This points to an absolute need for more research and clinical trials, including on those with DS, since we still do not know the mechanisms involved in the propagation of AD pathology in the Trisomy 21 brain.
2. Microtubule-Associated Protein Tau (MAPT) Structure and Function
The Tau protein is a highly conserved protein in mammals that is known to have six isoforms, which is caused by alternate splicing of exons 2, 3, and 10 of the microtubule-associated protein tau (MAPT) gene [19][20][21][22]. Tau is a microtubule-associated protein that contributes to the stability of microtubules in neurites and is crucial for the maintenance of normal microtubule structure and function, as well as axonal transport, in the neuron [23]. The Tau protein interacts with microtubules via either three or four microtubule binding repeats (3R or 4R, respectively), and the balance between 3R and 4R Tau proteins is altered during pathological states. When Tau undergoes modifications, this can lead to destabilization of neuronal microtubules and, consequently, to neuronal dysfunction and death [24]. Proteinaceous filaments of p-Tau are known to spread in a prion-like fashion in the human brain, leading to the detrimental formation of neurofibrillary tangles (NFTs) that contribute to AD pathology, correlate significantly with cognitive deficits in Alzheimer’s disease (AD), and contribute to distinguishing differences between AD and other tauopathies [25][26]. A distinct set of modifications occur in different neurodegenerative conditions, of which AD is the most common [23]. A precise balance between four-repeat (4R) and three-repeat (3R) isoforms of Tau are found in normal conditions, while dysregulation of the 3R:4R ratio is associated with different forms of tauopathy [20][27][28][29][30].
Several other Tau isoforms have a noteworthy impact on aggregation in AD. García-Escudero et al. recently discovered a new, human-specific truncated form of Tau generated by intron 12 retention [31]. This intron 12 retention generates a truncated Tau protein, followed by 18 extra amino acids, dramatically reducing this isoform’s ability to aggregate. While this Tau isoform exhibits similar biochemical properties and microtubule binding affinity, the variation seems to stabilize Tau, and is therefore considered to have a beneficial role. Interestingly, Cuadros et al. discovered that this form of Tau, called “W-Tau”, is reduced in AD brain at later stages of the disease. Another Tau isoform is the so-called “big-Tau”, a long-known high-molecular-weight isoform that contains an additional large exon termed 4a, which is sometimes without exon 6. Big-Tau expression and selective distribution is associated with neuronal development and regeneration [32][33], particularly in dorsal root ganglia. Chung et al. proposed that big-Tau expression is a feature of neurons that tend to be protected as AD progresses. In future studies, it would be prudent to study these isoforms in various regions of the AD-affected brain, considering that the occipital region often shows resilience to accumulation of AD pathology compared to the temporal and frontal regions, even within different portions of cortex.
3. Tauopathy Seeding and Its Spread in the DS Brain
Dr. Stanely Prusiner and his research group have minted AD as a “double-prion” disease, in acknowledgement of the prion-like activities conducted by both aggregating amyloid and Tau isoforms [34]. Interestingly, Condello and Prusiner examined prion activities in frozen brain tissue from individuals with DS of different ages by selectively precipitating Aβ and Tau from DS brain homogenates and using cellular bioassays to measure the number of prions. They discovered that while brain tissue from individuals with early-onset Alzheimer’s disease (EOAD) or LOAD exhibited reduced prion activities with age, this is not the case for DS brain tissue, where the levels of Aβ and Tau seeds increased with age [18]. These findings suggest a partially different seeding mechanism for Tau aggregates in DS, versus other forms of AD.
As mentioned above, truncated, fibrous, and oligomeric Tau are seeding competent and can seed and spread, both in vivo and in vitro, when given the opportunity to propagate in a prion-like manner [29][35]. Recent studies have implicated fibrillar, truncated and oligomeric Tau in the seeding, aggregation, and propagation of Tau pathology in the brain [20][29][30][36][37][38]. Interestingly, oligomeric and fibrillar Tau appear to be equivalent in potency, in terms of seeding competency, and are both known to be taken up by local neurons after intracranial injection [39]. However, oligomeric Tau appears to drive a more potent glial response in the brain and a more rapid propagation of misfolded Tau to other brain regions [39] (see Figure 1 below).
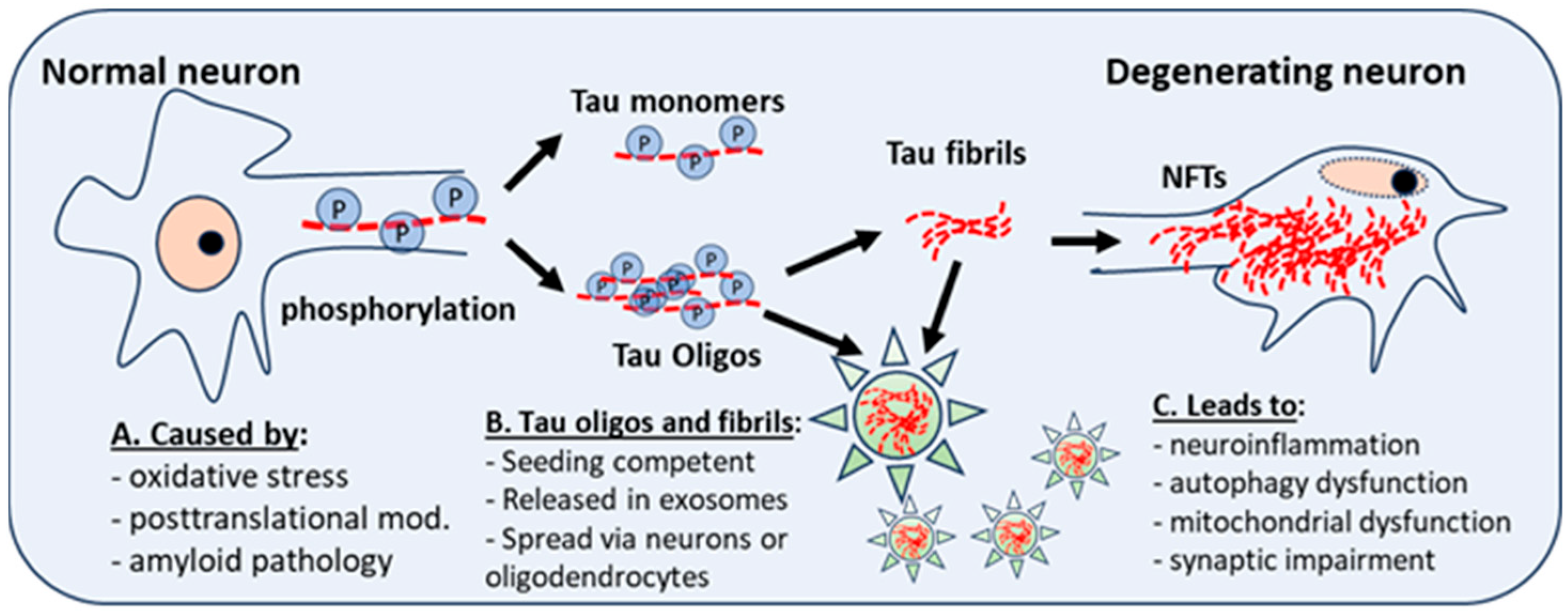
Figure 1. Posttranslational modifications and neuroinflammation or oxidative stress give rise to abnormal phosphorylation of Tau (P). This affects the stability of microtubules, and the development of Tau monomers and oligomers, which have aggregation potential and can form Tau fibrils, which aggregate into neurofibrillary tangles (NFTs), a hallmark of AD and DS-AD. Intra-neuronal NFT accumulation leads to neuroinflammation, autophagy dysfunction, and mitochondrial dysfunction. Tau fibrils and oligomers, seeding competent and secreted in exosomes (star), spread to other neurons and astrocytes, microglia, or oligodendrocytes. Oligos = oligomers.
4. Tau Biomarkers in Biofluids
Individuals with DS develop dementia and AD pathology at a variable age, but earlier than in LOAD and perhaps earlier than in EOAD. Given that as much as 90% of individuals with DS develop AD-like dementia by the fifth decade of life [40], one might hypothesize that Tau seeding and spread within the DS brain may occur decades earlier. Recent improvements in measuring P-Tau by immunoassays, seeding assays and Positron Emission tomography (PET) have enabled this hypothesis to be tested and led to several breakthroughs, while producing new questions about when Tau gains seeding “prion-like” capacities.
4.1. Ultrasensitive Immunoassays
With the recent development of new sensitive assays such as the single molecule array (Simoa [41]), investigators are now capable of measuring even minute amounts of biomarkers in plasma in different neurological conditions. The researchers performed a research focused on exosome biomarkers in individuals with DS, showing increased amyloid exosomal levels already in early childhood, which increased through adulthood [42]. However, the researchers also unexpectedly found an increase in p-Tau (S396 and T181) when comparing all DS and all control participants at an early age, and noted an increase of p-Tau with age (T181) [43]. These findings suggested that misfolding and phosphorylation of Tau may be an early event in DS and could be used to predict the onset of dementia, and also improve treatment efficacy for this population. Measuring biomarkers in exosomes proved to be a sensitive and possibly less variable assay of dementia-related biomarkers than when the same biomarkers were applied in plasma, which was due to the relatively low levels of AD-related proteins observed in plasma.
In engaging with older adults with DS, Alzheimer Biomarkers Consortium—Down Syndrome (ABC–DS) demonstrated early changes in persons with DS by using different p-Tau isoforms in plasma [44]. The Petersen et al. group [44] studied more than 300 participants with DS at different ages, showing that plasma levels of total Tau and neurofilament light (NfL) were highly predictive of both AD pathology and clinical status in those with DS at different ages. In another recent biomarker study for the European DS clinical network Horizon 21, Carmona and collaborators examined neurofilament light (NfL) [45], and found that NfL plasma levels had excellent diagnostic performance and a highly similar temporal distribution of change, compared to that seen in autosomal dominant AD [45]. Phospho-Tau biomarkers in plasma have also been examined by using ultrasensitive methods [46][47]. And Plasma p-Tau (T217), glial fibrillary acidic protein (GFAP), amyloid beta peptides 42 and 40 (Aβ42/Aβ40), NfL, and total Tau (t-Tau) were assayed. The study, which included 300 participants with DS and 37 non-DS siblings, found that higher p-Tau (T217) levels and no other biomarkers were associated with worse performance in the DS mental status examination and cued recall test, suggesting that plasma p-Tau (T217) is an accurate blood-based biomarker of both Tau and Aβ pathological brain changes in DS that could be used to include individuals with DS in AD clinical trials, especially when combined with age as a covariate [46]. Since plasma levels of AD-related pathology are easier to accomplish in the clinic than in NDE assessments, this newly developed measure may lend itself better to large clinical studies than to more cumbersome exosomal biomarker studies [43][48].
4.2. Tau Seeding and Aggregation Assays
Promising techniques have been developed for the quantitation of Tau seeding activity in human biofluid. Holme et al. (2014) developed a cell-based assay, where a Tau-containing biosample is incubated with a cell line overexpressing tau linked to a fluorescent protein [49]. Upon aggregation, energy transfers between fluorescent proteins (FRET) allowed for the detection of seeding capacity signals using flow cytometry. Using this FRET method, robust Tau seeding activity could be observed one month before histopathological stains showed NFTs, suggesting that tau seeding is an early signature of tauopathy. Cell-free assays have been developed by real-time quaking-induced conversion (RT-QuIC) to measure prion seeding [50] and, more recently, Tau seeding [51]. Tau seed-containing material from biofluids or brain samples is incubated with recombinant tau substrate and thioflavin T in optimized conditions. When RT-QuIC is used, seeding-competent material induces the aggregation of the substrate, which generates a fluorescent signal.
Jin et al. (2022) recently developed a Tau seeding activity assay using truncated HA-tagged Tau151-391 peptide and cellular transformation [52]. Cells transiently express the HA-tagged Tau151-391 peptide, which is readily captured and aggregated by oligomeric Tau derived from postmortem AD brain samples. The captured Tau is then quantified using traditional immunoblot methods. They employed these assays on AD and DS brain samples in two different studies [53] and discovered that only brain extracts from AD or DS brain samples contained hyperphosphorylated Tau seeded Tau aggregation in cultured cells. Interestingly, Tau extracts of DS corpus callosum showed low tau seeding activity, and no detectable tau seeding activity was observed in DS cerebellar cortex. Again, Tau seeding ability was found to be highly correlated with phosphorylation, but the group did not study the effects of other Tau PTMs in this assay.
4.3. Tau Binding Studies Using Positron Emission Tomography (PET) Ligands
Tau PET imaging is emerging as an important clinical tool for early diagnosis of AD and other tauopathies, as p-Tau appears to correlate more closely than amyloid with symptomatic dementia progression. In the last ten years, multiple Tau PET ligands have been developed. Tau-specific ligands for use in PET include first-generation ligands and, consequently, second-generation ligands (see Leuzy et al. [54]). Several of these Tau PET ligands have been tested in patients with DS and AD including, for example, the [18F]-AV-1451 Tau PET ligand [55]. Rafii et al. demonstrated that amyloid-negative participants who were DS imaged were all Tau-negative, and that both amyloid and Tau burden correlated with age [55]. They also found that Tau binding in the brain correlated significantly with cognitive decline, suggesting that this clinical measure can be used to predict the onset or progression of dementia in DS-AD. Recently, Dr. Bradley Christian and his research group have performed longitudinal PET analyses in a DS cohort with the aim of better defining a timeline for the progression of amyloid and Tau burden through the conventional Braak stages [56][57][58]. By using PET scans, they revealed early and rapid Tau elevation following the onset of amyloid-positive PET imaging. While Tau elevation is highly variable in AD cohorts, individuals with DS displayed uniform increases in Tau within 2.5 years of the onset of amyloid binding in the brain. Although very early in life, the spatial progression of neurofibrillary Tau tangles and paired helical filament Tau in DS closely follows the hierarchical staging pattern outlined by Braak and Braak for LOAD studies [59]. This pattern has been well described by Davidson et al., who observe that Tau pathology usually commences after 35-years-of-age, in the entorhinal cortex, hippocampal formation, and in subcortical brainstem regions such as the locus coeruleus and dorsal raphe nucleus. Later, Tau pathology spreads throughout the neocortex, sparing the occipital lobes until the mid-50 years of age [18]. Although this pattern is generally recognized in individuals with DS, there are reports showing that dysregulation of Tau phosphorylation can occur quite early in life in those with DS [60], which is something that cannot be detected by Tau PET imaging, which serves to demonstrate the need for detailed postmortem tissue studies.
To further quantify the binding of Tau PET ligands in specific brain regions, the researchers performed an autoradiographic binding research of Tau PET ligand binding in postmortem, fixed and paraffin-embedded brain tissue sections from individuals that received a neuropathological diagnosis of either LOAD, EOAD, or DS-AD, and compared them against age-matched controls [61]. Interestingly, the researchers found that the binding of both the first generation (THK5117) and the second generation (MK6240) Tau PET ligand exhibited a significant increase in frontal cortex (middle frontal gyrus) in DS–AD postmortem cases, compared to both EOAD and LOAD cases. In addition, autoradiographic binding of both of these Tau ligands correlated significantly with AT8 p-Tau immunostaining of adjacent sections, strongly suggesting that on-target binding was more prevalent than off-target binding, at least in these fixed and paraffin-embedded sections [61]. Other studies have suggested that in fresh frozen materials and in vivo, there is off-target binding of Tau ligands to, for example, monoamine oxidase B (MAO-B) [62], which did not appear to be the case in the research using fixed materials. Investigators have identified a binding site for all the Tau tracers on MAO-B [62], which could make clinical studies difficult to undertake unless a better understanding of off-target binding is first achieved. However, it was reported by Murugan et al [62] that a second-generation Tau PET ligand, MK6240, has a lower affinity for MAO-B than the first-generation tracers. Nevertheless, in the binding research, the researchers found a significant correlation between p-Tau immunostaining (AT8 antibodies), with both the first and second-generation Tau ligands used in this research [61], suggesting less off-target binding in fixed tissues than what was previously reported in vivo.
References
- Alldred, M.J.; Martini, A.C.; Patterson, D.; Hendrix, J.; Granholm, A.C. Aging with Down Syndrome—Where Are We Now and Where Are We Going? J. Clin. Med. 2021, 10, 4687.
- Delabar, J.M.; Allinquant, B.; Bianchi, D.; Blumenthal, T.; Dekker, A.; Edgin, J.; O’Bryan, J.; Dierssen, M.; Potier, M.C.; Wiseman, F.; et al. Changing Paradigms in Down Syndrome: The First International Conference of the Trisomy 21 Research Society. Mol. Syndromol. 2016, 7, 251–261.
- Head, E.; Lott, I.T.; Wilcock, D.M.; Lemere, C.A. Aging in Down Syndrome and the Development of Alzheimer’s Disease Neuropathology. Curr. Alzheimer Res. 2016, 13, 18–29.
- Flores-Aguilar, L.; Iulita, M.F.; Kovecses, O.; Torres, M.D.; Levi, S.M.; Zhang, Y.; Askenazi, M.; Wisniewski, T.; Busciglio, J.; Cuello, A.C. Evolution of neuroinflammation across the lifespan of individuals with Down Syndrome. Brain 2020, 143, 3653–3671.
- Fortea, J.; Zaman, S.H.; Hartley, S.; Rafii, M.S.; Head, E.; Carmona-Iragui, M. Alzheimer’s disease associated with Down Syndrome: A genetic form of dementia. Lancet Neurol. 2021, 20, 930–942.
- Hartley, D.; Blumenthal, T.; Carrillo, M.; DiPaolo, G.; Esralew, L.; Gardiner, K.; Granholm, A.C.; Iqbal, K.; Krams, M.; Lemere, C.; et al. Down Syndrome and Alzheimer’s disease: Common pathways, common goals. Alzheimers Dement. 2015, 11, 700–709.
- Bartley, M.G.; Marquardt, K.; Kirchhof, D.; Wilkins, H.M.; Patterson, D.; Linseman, D.A. Overexpression of amyloid-beta protein precursor induces mitochondrial oxidative stress and activates the intrinsic apoptotic cascade. J. Alzheimers Dis. 2012, 28, 855–868.
- Helman, A.M.; Siever, M.; McCarty, K.L.; Lott, I.T.; Doran, E.; Abner, E.L.; Schmitt, F.A.; Head, E. Microbleeds and Cerebral Amyloid Angiopathy in the Brains of People with Down Syndrome with Alzheimer’s Disease. J. Alzheimers Dis. 2019, 67, 103–112.
- Hunter, C.L.; Isacson, O.; Nelson, M.; Bimonte-Nelson, H.; Seo, H.; Lin, L.; Ford, K.; Kindy, M.S.; Granholm, A.C. Regional alterations in amyloid precursor protein and nerve growth factor across age in a mouse model of Down’s syndrome. Neurosci. Res. 2003, 45, 437–445.
- Millan Sanchez, M.; Heyn, S.N.; Das, D.; Moghadam, S.; Martin, K.J.; Salehi, A. Neurobiological elements of cognitive dysfunction in Down Syndrome: Exploring the role of APP. Biol. Psychiatry 2012, 71, 403–409.
- Chung, H.; Green, P.H.R.; Wang, T.C.; Kong, X.F. Interferon-Driven Immune Dysregulation in Down Syndrome: A Review of the Evidence. J. Inflamm. Res. 2021, 14, 5187–5200.
- Powers, R.K.; Culp-Hill, R.; Ludwig, M.P.; Smith, K.P.; Waugh, K.A.; Minter, R.; Tuttle, K.D.; Lewis, H.C.; Rachubinski, A.L.; Granrath, R.E.; et al. Trisomy 21 activates the kynurenine pathway via increased dosage of interferon receptors. Nat. Commun. 2019, 10, 4766.
- Ram, G.; Chinen, J. Infections and immunodeficiency in Down Syndrome. Clin. Exp. Immunol. 2011, 164, 9–16.
- Martini, A.C.; Helman, A.M.; McCarty, K.L.; Lott, I.T.; Doran, E.; Schmitt, F.A.; Head, E. Distribution of microglial phenotypes as a function of age and Alzheimer’s disease neuropathology in the brains of people with Down Syndrome. Alzheimers Dement. 2020, 12, e12113.
- Barone, E.; Arena, A.; Head, E.; Butterfield, D.A.; Perluigi, M. Disturbance of redox homeostasis in Down Syndrome: Role of iron dysmetabolism. Free Radic. Biol. Med. 2018, 114, 84–93.
- Hendrix, J.A.; Amon, A.; Abbeduto, L.; Agiovlasitis, S.; Alsaied, T.; Anderson, H.A.; Bain, L.J.; Baumer, N.; Bhattacharyya, A.; Bogunovic, D.; et al. Opportunities, barriers, and recommendations in Down Syndrome research. Transl. Sci. Rare Dis. 2021, 5, 99–129.
- Snyder, H.M.; Bain, L.J.; Brickman, A.M.; Carrillo, M.C.; Esbensen, A.J.; Espinosa, J.M.; Fernandez, F.; Fortea, J.; Hartley, S.L.; Head, E.; et al. Further understanding the connection between Alzheimer’s disease and Down Syndrome. Alzheimers Dement. 2020, 16, 1065–1077.
- Condello, C.; Maxwell, A.M.; Castillo, E.; Aoyagi, A.; Graff, C.; Ingelsson, M.; Lannfelt, L.; Bird, T.D.; Keene, C.D.; Seeley, W.W.; et al. Abeta and tau prions feature in the neuropathogenesis of Down Syndrome. Proc. Natl. Acad. Sci. USA 2022, 119, e2212954119.
- Rawat, P.; Sehar, U.; Bisht, J.; Selman, A.; Culberson, J.; Reddy, P.H. Phosphorylated Tau in Alzheimer’s Disease and Other Tauopathies. Int. J. Mol. Sci. 2022, 23, 2841.
- Falcon, B.; Zivanov, J.; Zhang, W.; Murzin, A.G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Newell, K.L.; Ghetti, B.; Goedert, M.; et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 2019, 568, 420–423.
- Meyer, V.; Dinkel, P.D.; Luo, Y.; Yu, X.; Wei, G.; Zheng, J.; Eaton, G.R.; Ma, B.; Nussinov, R.; Eaton, S.S.; et al. Single mutations in tau modulate the populations of fibril conformers through seed selection. Angew. Chem. Int. Ed. Engl. 2014, 53, 1590–1593.
- Siddiqua, A.; Luo, Y.; Meyer, V.; Swanson, M.A.; Yu, X.; Wei, G.; Zheng, J.; Eaton, G.R.; Ma, B.; Nussinov, R.; et al. Conformational basis for asymmetric seeding barrier in filaments of three- and four-repeat tau. J. Am. Chem. Soc. 2012, 134, 10271–10278.
- Avila, J.; Lucas, J.J.; Perez, M.; Hernandez, F. Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 2004, 84, 361–384.
- Wegmann, S.; Maury, E.A.; Kirk, M.J.; Saqran, L.; Roe, A.; DeVos, S.L.; Nicholls, S.; Fan, Z.; Takeda, S.; Cagsal-Getkin, O.; et al. Removing endogenous tau does not prevent tau propagation yet reduces its neurotoxicity. EMBO J. 2015, 34, 3028–3041.
- Weismiller, H.A.; Murphy, R.; Wei, G.; Ma, B.; Nussinov, R.; Margittai, M. Structural disorder in four-repeat Tau fibrils reveals a new mechanism for barriers to cross-seeding of Tau isoforms. J. Biol. Chem. 2018, 293, 17336–17348.
- Dujardin, S.; Commins, C.; Lathuiliere, A.; Beerepoot, P.; Fernandes, A.R.; Kamath, T.V.; De Los Santos, M.B.; Klickstein, N.; Corjuc, D.L.; Corjuc, B.T.; et al. Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease. Nat. Med. 2020, 26, 1256–1263.
- Armstrong, R.A.; McKee, A.C.; Alvarez, V.E.; Cairns, N.J. Clustering of tau-immunoreactive pathology in chronic traumatic encephalopathy. J. Neural. Transm. 2017, 124, 185–192.
- Capano, L.S.; Sato, C.; Ficulle, E.; Yu, A.; Horie, K.; Kwon, J.S.; Burbach, K.F.; Barthelemy, N.R.; Fox, S.G.; Karch, C.M.; et al. Recapitulation of endogenous 4R tau expression and formation of insoluble tau in directly reprogrammed human neurons. Cell Stem Cell 2022, 29, 918–932.e8.
- Furman, J.L.; Vaquer-Alicea, J.; White, C.L., 3rd; Cairns, N.J.; Nelson, P.T.; Diamond, M.I. Widespread tau seeding activity at early Braak stages. Acta Neuropathol. 2017, 133, 91–100.
- Guo, J.L.; Lee, V.M. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J. Biol. Chem. 2011, 286, 15317–15331.
- Garcia-Escudero, V.; Ruiz-Gabarre, D.; Gargini, R.; Perez, M.; Garcia, E.; Cuadros, R.; Hernandez, I.H.; Cabrera, J.R.; Garcia-Escudero, R.; Lucas, J.J.; et al. A new non-aggregative splicing isoform of human Tau is decreased in Alzheimer’s disease. Acta Neuropathol. 2021, 142, 159–177.
- Nothias, F.; Boyne, L.; Murray, M.; Tessler, A.; Fischer, I. The expression and distribution of tau proteins and messenger RNA in rat dorsal root ganglion neurons during development and regeneration. Neuroscience 1995, 66, 707–719.
- Nunez, J.; Fischer, I. Microtubule-associated proteins (MAPs) in the peripheral nervous system during development and regeneration. J. Mol. Neurosci. 1997, 8, 207–222.
- Condello, C.; Merz, G.E.; Aoyagi, A.; DeGrado, W.F.; Prusiner, S.B. Abeta and Tau Prions Causing Alzheimer’s Disease. Methods Mol. Biol. 2023, 2561, 293–337.
- Dinkel, P.D.; Siddiqua, A.; Huynh, H.; Shah, M.; Margittai, M. Variations in filament conformation dictate seeding barrier beween three- and four-repeat tau. Biochemistry 2011, 50, 4330–4336.
- Gerson, J.E.; Sengupta, U.; Kayed, R. Tau Oligomers as Pathogenic Seeds: Preparation and Propagation In Vitro and In Vivo. Methods Mol. Biol. 2017, 1523, 141–157.
- Goedert, M.; Eisenberg, D.S.; Crowther, R.A. Propagation of Tau Aggregates and Neurodegeneration. Annu. Rev. Neurosci. 2017, 40, 189–210.
- Gratuze, M.; Chen, Y.; Parhizkar, S.; Jain, N.; Strickland, M.R.; Serrano, J.R.; Colonna, M.; Ulrich, J.D.; Holtzman, D.M. Activated microglia mitigate Abeta-associated tau seeding and spreading. J. Exp. Med. 2021, 218, e20210542.
- Mate De Gerando, A.; Welikovitch, L.A.; Khasnavis, A.; Commins, C.; Glynn, C.; Chun, J.E.; Perbet, R.; Hyman, B.T. Tau seeding and spreading in vivo is supported by both AD-derived fibrillar and oligomeric tau. Acta Neuropathol. 2023, 146, 191–210.
- Salehi, A.; Ashford, J.W.; Mufson, E.J. The Link between Alzheimer’s Disease and Down Syndrome. A Historical Perspective. Curr. Alzheimer Res. 2016, 13, 2–6.
- McCrea, M.; Broglio, S.P.; McAllister, T.W.; Gill, J.; Giza, C.C.; Huber, D.L.; Harezlak, J.; Cameron, K.L.; Houston, M.N.; McGinty, G.; et al. Association of Blood Biomarkers With Acute Sport-Related Concussion in Collegiate Athletes: Findings From the NCAA and Department of Defense CARE Consortium. JAMA Netw. Open 2020, 3, e1919771.
- Hamlett, E.D.; Goetzl, E.J.; Ledreux, A.; Vasilevko, V.; Boger, H.A.; LaRosa, A.; Clark, D.; Carroll, S.L.; Carmona-Iragui, M.; Fortea, J.; et al. Neuronal exosomes reveal Alzheimer’s disease biomarkers in Down Syndrome. Alzheimers Dement. 2017, 13, 541–549.
- Hamlett, E.D.; LaRosa, A.; Mufson, E.J.; Fortea, J.; Ledreux, A.; Granholm, A.C. Exosome release and cargo in Down Syndrome. Dev. Neurobiol. 2019, 79, 639–655.
- Petersen, M.E.; Rafii, M.S.; Zhang, F.; Hall, J.; Julovich, D.; Ances, B.M.; Schupf, N.; Krinsky-McHale, S.J.; Mapstone, M.; Silverman, W.; et al. Plasma Total-Tau and Neurofilament Light Chain as Diagnostic Biomarkers of Alzheimer’s Disease Dementia and Mild Cognitive Impairment in Adults with Down Syndrome. J. Alzheimers Dis. 2021, 79, 671–681.
- Carmona-Iragui, M.; Alcolea, D.; Barroeta, I.; Videla, L.; Munoz, L.; Van Pelt, K.L.; Schmitt, F.A.; Lightner, D.D.; Koehl, L.M.; Jicha, G.; et al. Diagnostic and prognostic performance and longitudinal changes in plasma neurofilament light chain concentrations in adults with Down Syndrome: A cohort study. Lancet Neurol. 2021, 20, 605–614.
- Janelidze, S.; Christian, B.T.; Price, J.; Laymon, C.; Schupf, N.; Klunk, W.E.; Lott, I.; Silverman, W.; Rosas, H.D.; Zaman, S.; et al. Detection of Brain Tau Pathology in Down Syndrome Using Plasma Biomarkers. JAMA Neurol. 2022, 79, 797–807.
- Mattsson, N.; Zetterberg, H.; Janelidze, S.; Insel, P.S.; Andreasson, U.; Stomrud, E.; Palmqvist, S.; Baker, D.; Tan Hehir, C.A.; Jeromin, A.; et al. Plasma tau in Alzheimer disease. Neurology 2016, 87, 1827–1835.
- Hamlett, E.D.; Ledreux, A.; Potter, H.; Chial, H.J.; Patterson, D.; Espinosa, J.M.; Bettcher, B.M.; Granholm, A.C. Exosomal biomarkers in Down Syndrome and Alzheimer’s disease. Free Radic. Biol. Med. 2018, 114, 110–121.
- Holmes, B.B.; Furman, J.L.; Mahan, T.E.; Yamasaki, T.R.; Mirbaha, H.; Eades, W.C.; Belaygorod, L.; Cairns, N.J.; Holtzman, D.M.; Diamond, M.I. Proteopathic tau seeding predicts tauopathy in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, E4376–E4385.
- Kocisko, D.A.; Come, J.H.; Priola, S.A.; Chesebro, B.; Raymond, G.J.; Lansbury, P.T.; Caughey, B. Cell-free formation of protease-resistant prion protein. Nature 1994, 370, 471–474.
- Kraus, A.; Saijo, E.; Metrick, M.A., 2nd; Newell, K.; Sigurdson, C.J.; Zanusso, G.; Ghetti, B.; Caughey, B. Seeding selectivity and ultrasensitive detection of tau aggregate conformers of Alzheimer disease. Acta Neuropathol. 2019, 137, 585–598.
- Liu, F.; Wu, R.; Jin, N.; Chu, D.; Gu, J.; Tung, Y.C.; Hu, Z.; Gong, C.X.; Iqbal, K. Two simple assays for assessing the seeding activity of proteopathic tau. Front. Aging Neurosci. 2023, 15, 1073774.
- Jin, N.; Gu, J.; Wu, R.; Chu, D.; Tung, Y.C.; Wegiel, J.; Wisniewski, T.; Gong, C.X.; Iqbal, K.; Liu, F. Tau seeding activity in various regions of Down Syndrome brain assessed by two novel assays. Acta Neuropathol. Commun. 2022, 10, 132.
- Leuzy, A.; Chiotis, K.; Lemoine, L.; Gillberg, P.G.; Almkvist, O.; Rodriguez-Vieitez, E.; Nordberg, A. Tau PET imaging in neurodegenerative tauopathies-still a challenge. Mol. Psychiatry 2019, 24, 1112–1134.
- Rafii, M.S.; Lukic, A.S.; Andrews, R.D.; Brewer, J.; Rissman, R.A.; Strother, S.C.; Wernick, M.N.; Pennington, C.; Mobley, W.C.; Ness, S.; et al. PET Imaging of Tau Pathology and Relationship to Amyloid, Longitudinal MRI, and Cognitive Change in Down Syndrome: Results from the Down Syndrome Biomarker Initiative (DSBI). J. Alzheimers Dis. 2017, 60, 439–450.
- Lao, P.J.; Handen, B.L.; Betthauser, T.J.; Mihaila, I.; Hartley, S.L.; Cohen, A.D.; Tudorascu, D.L.; Bulova, P.D.; Lopresti, B.J.; Tumuluru, R.V.; et al. Longitudinal changes in amyloid positron emission tomography and volumetric magnetic resonance imaging in the nondemented Down Syndrome population. Alzheimers Dement. 2017, 9, 1–9.
- Zammit, M.D.; Tudorascu, D.L.; Laymon, C.M.; Hartley, S.L.; Zaman, S.H.; Ances, B.M.; Johnson, S.C.; Stone, C.K.; Mathis, C.A.; Klunk, W.E.; et al. PET measurement of longitudinal amyloid load identifies the earliest stages of amyloid-beta accumulation during Alzheimer’s disease progression in Down Syndrome. Neuroimage 2021, 228, 117728.
- Zammit, M.D.; Betthauser, T.J.; McVea, A.K.; Laymon, C.M.; Tudorascu, D.L.; Johnson, S.C.; Hartley, S.L.; Converse, A.K.; Minhas, D.S.; Zaman, S.H.; et al. Characterizing the emergence of amyloid and tau burden in Down Syndrome. Alzheimers Dement. 2024, 20, 388–398.
- Braak, H.; Braak, E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol. Aging 1995, 16, 271–278; discussion 278–284.
- Milenkovic, I.; Jarc, J.; Dassler, E.; Aronica, E.; Iyer, A.; Adle-Biassette, H.; Scharrer, A.; Reischer, T.; Hainfellner, J.A.; Kovacs, G.G. The physiological phosphorylation of tau is critically changed in fetal brains of individuals with Down Syndrome. Neuropathol. Appl. Neurobiol. 2018, 44, 314–327.
- Lemoine, L.; Ledreux, A.; Mufson, E.J.; Perez, S.E.; Simic, G.; Doran, E.; Lott, I.; Carroll, S.; Bharani, K.; Thomas, S.; et al. Regional binding of tau and amyloid PET tracers in Down Syndrome autopsy brain tissue. Mol. Neurodegener. 2020, 15, 68.
- Murugan, N.A.; Chiotis, K.; Rodriguez-Vieitez, E.; Lemoine, L.; Agren, H.; Nordberg, A. Cross-interaction of tau PET tracers with monoamine oxidase B: Evidence from in silico modelling and in vivo imaging. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1369–1382.
More
Information
Subjects:
Neurosciences; Pathology; Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Entry Collection:
Neurodegeneration
Revisions:
2 times
(View History)
Update Date:
07 Mar 2024
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No