Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Tiantian Zhang | -- | 2946 | 2024-03-06 07:30:10 | | | |
| 2 | Sirius Huang | Meta information modification | 2946 | 2024-03-08 01:43:53 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Zhang, T.; Wang, Z. Phage Display Technology in Monoclonal Antibody Production. Encyclopedia. Available online: https://encyclopedia.pub/entry/55900 (accessed on 09 August 2026).
Zhang T, Wang Z. Phage Display Technology in Monoclonal Antibody Production. Encyclopedia. Available at: https://encyclopedia.pub/entry/55900. Accessed August 09, 2026.
Zhang, Tiantian, Zhe Wang. "Phage Display Technology in Monoclonal Antibody Production" Encyclopedia, https://encyclopedia.pub/entry/55900 (accessed August 09, 2026).
Zhang, T., & Wang, Z. (2024, March 06). Phage Display Technology in Monoclonal Antibody Production. In Encyclopedia. https://encyclopedia.pub/entry/55900
Zhang, Tiantian and Zhe Wang. "Phage Display Technology in Monoclonal Antibody Production." Encyclopedia. Web. 06 March, 2024.
Copy Citation
The groundbreaking work of John McCafferty and Sir Gregory Winter in developing phage display technology revolutionized the discovery of human antibodies, paving the way for diverse applications. Since then, numerous phage-derived antibodies have been successfully developed and advanced into clinical studies, resulting in the approval of more than a dozen therapeutic antibodies. These antibodies have demonstrated efficacy across a spectrum of medical conditions, ranging from autoimmune diseases to various cancers.
phage display library
cancer treatment
antibody development
1. Introduction
Therapeutic monoclonal antibodies have emerged as transformative agents in the realm of cancer treatment, fundamentally altering the landscape of oncology. These antibodies, designed to target specific proteins associated with cancer cells, exemplify precision medicine. By leveraging the body’s immune system to identify and attack cancer cells, monoclonal antibodies have proven instrumental in the treatment of various cancers [1][2][3].
In 1985, George P. Smith pioneered the concept of phage display, unveiling a groundbreaking technique for exhibiting peptides on filamentous phages. By fusing the gene encoding the desired protein to the 5′ region of a filamentous phage’s coat protein, such as protein III or protein VIII, Smith showcased an innovative method that exhibited distinct peptides on the outer surfaces of viral clones [4]. The subsequent screening step in this process isolated peptides with the highest binding affinity, marking a pivotal advancement in molecular selection methods. The refinement of this technique continued as Stephen Parmley and George Smith introduced biopanning in 1988, demonstrating that recursive rounds of selection could effectively enrich for clones present at remarkably low frequencies, even as rare as 1 in a billion or less. In 1990, McCafferty et al. achieved a groundbreaking milestone by demonstrating the display of complete antibody variable domains on the surface of phages. This revolutionary technique enabled the selection of phages capable of binding to specific antigens [5]. Moreover, Jamie Scott and George Smith extended these capabilities by describing the creation of extensive random peptide libraries displayed on filamentous phages [6]. This methodological evolution laid the foundation for the subsequent refinement and enhancement of phage display technology. The significant contributions of George P. Smith and Greg Winter culminated in the recognition of a half share of the 2018 Nobel Prize in Chemistry for their seminal work in developing phage display.
Up to now, the impact of phage display in generating antibodies extends across diverse applications. In the field of therapeutics, it has played a pivotal role in the development of monoclonal antibodies for various diseases, including cancer, autoimmune disorders, infectious diseases, and neurological conditions. Furthermore, phage display has significantly contributed to the discovery of antibodies targeting specific viral pathogens, such as influenza and HIV, providing valuable tools for both diagnosis and treatment [7][8]. Beyond therapeutics, phage display has been instrumental in antibody engineering, epitope mapping, and the study of protein interactions. Additionally, it has facilitated the identification of novel peptide ligands for drug targeting, the development of biosensors for disease detection, and the creation of protein libraries for enzyme optimization and protein–protein interaction studies [9]. Overall, phage display continues to revolutionize biotechnology and biomedical research by enabling the rapid and efficient generation of high-affinity antibodies and protein-based therapeutics for a wide range of applications.
2. Overview of Procedures of Phage Display Technology in Monoclonal Antibody Production
2.1. Library Construction
The journey of generating antibodies through phage display technology commences with the construction of a diverse library. This process entails integrating genetic material encoding antibody fragments, such as antibodies’ heavy and light chains, single-chain variable fragments (scFvs), Fabs, or peptides, into the genome of bacteriophages, notably filamentous ones like M13 [10]. This fusion results in the expression of a multitude of unique antibody candidates on the surface of phages, forming the basis of the library. The diversity of this library, ranging from millions to billions of variants, lays the groundwork for the subsequent exploration of potential binding partners (Figure 1) [11].
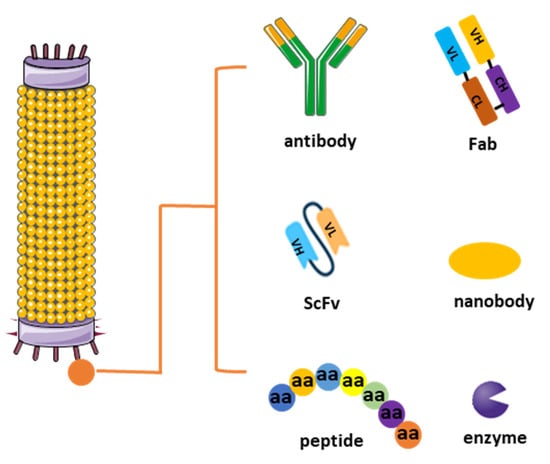
Figure 1. Different types of phage display libraries. Phage display technology enables the selection and identification of peptides, antibodies, or enzymes that bind to specific target molecules. This approach entails generating filamentous fusion phages that showcase random foreign peptides or antibodies linked to a phage coat protein. Subsequently, the filamentous phages are assembled into a phage display library comprising random peptide sequences or antibodies.
Constructing a phage display library involves several key steps aimed at generating a diverse repertoire of displayed peptides or proteins for subsequent screening against target molecules. Initially, a source of genetic material encoding the desired peptides or proteins is obtained, which can include synthetic DNA sequences, cDNA libraries, or genomic DNA. Next, this genetic material is ligated into a phagemid vector, which contains the genetic elements necessary for phage display, including a phage coat protein gene fused to the peptide or protein of interest. The ligated DNA is then introduced into competent Escherichia coli cells via transformation, where it undergoes replication and amplification. Following transformation, the bacterial cells are infected with helper phage, which provides the necessary components for packaging the phagemid DNA into phage particles. The infected cells are then cultured to allow for phage production, and the resulting phage display library is harvested by collecting the culture supernatant or lysing the bacterial cells to release the phage particles. The library can be further characterized by sequencing a subset of clones to assess diversity and by performing binding assays to confirm the display of the desired peptides or proteins (Figure 2) [12].
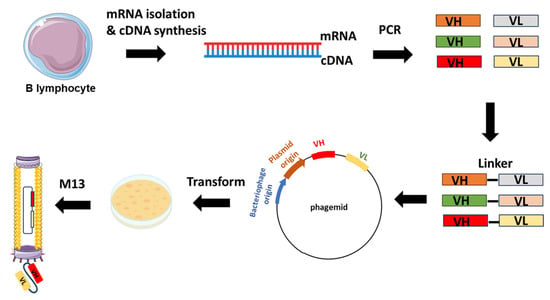
Figure 2. Example of scFv-phage display library construction. The procedure starts with the extraction of total RNA from B-lymphocytes acquired from either immunized or non-immunized healthy donors. Subsequent to RNA isolation, cDNA is synthesized through reverse transcription. The repertoire of VH and VL genes is amplified from the cDNA using forward and reverse primers designed to target the variable domains. The VH and VL genes are ligated with linker sequences. Subsequently, scFvs sequences are cloned into the phagemid vectors, leading to the generation of a phage library.
Phage construction involves the genetic modification of bacteriophages like those in the M13 group to display specific peptides or proteins on their surface. Typically, this modification targets either the tail or the main coat protein of the phage, depending on the desired outcome and the characteristics of the phage being used. Tail proteins are often chosen for peptide insertion when the aim is to display peptides for binding to specific receptors or targets on host cells. This approach allows for the display of larger peptides or proteins without compromising the structural integrity of the main coat protein. Consequently, modifying the tail protein can enhance the specificity and efficiency of phage-mediated targeting, particularly in applications such as targeted drug delivery or tissue-specific imaging.
On the other hand, main coat proteins, like the major coat protein (pVIII) in M13 phages, are commonly targeted for peptide insertion due to their abundance and accessibility on the phage surface. Genetic fusion techniques are typically employed for peptide insertion into the main coat protein, where the peptide sequence is fused to the coding sequence of the coat protein. This strategy enables the display of peptides on every copy of the coat protein, potentially leading to a higher display density compared to tail protein modifications. However, modifications to the main coat protein may impact phage assembly and stability, necessitating careful design and optimization [13].
Comparing tail and main coat protein modification reveals differences in specificity, efficiency, and structural integrity. Tail protein modification may offer higher specificity in targeting host cells or specific receptors due to the potential for more diverse peptide sequences and a larger display capacity. In contrast, main coat protein modification often results in higher display densities but may lack specificity compared to tail modifications. Furthermore, tail protein modification typically preserves the structural integrity of the main coat protein, crucial for phage assembly and stability, while main coat protein modification may disrupt these properties, potentially affecting phage viability and infectivity. Ultimately, the choice between tail and main coat protein modification depends on the specific requirements of the intended application, balancing factors such as the targeting specificity, display density, and phage stability [14].
2.2. Biopanning, Elution and Amplification
The heart of the procedure lies in the targeted selection of antibodies through a process known as panning. The phage display library is exposed to the specific target of interest, such as a protein or cell type [10][15]. Those phages displaying antibodies that bind to the target are selectively enriched through successive rounds of panning. This iterative process involves incubating the library with the target, followed by stringent washing to remove weakly bound or non-specific phages. The remaining phages, carrying antibodies with higher affinity for the target, are recovered and subjected to additional rounds of panning, amplifying the specificity of the antibody repertoire (Figure 3) [16].
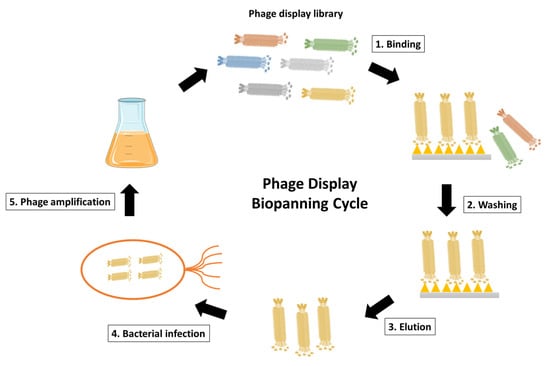
Figure 3. Schematic representation of phage display biopanning cycle. The phage display library after construction is incubated with the desired antigen, such as the surface-immobilized antigen or the cell surface antigen. Unspecific phages that fail to bind are then eliminated through washing steps. The antigen-specific phages are subsequently eluted and used to infect E. coli for phage amplification. Coinfection with a helper phage enables the production and amplification of the desired phages. The phage display biopanning cycle is typically repeated for 2–3 cycles to achieve phage enrichment.
Once the panning process is complete, the phages displaying antibodies of interest are eluted from the target, disrupting the interaction between the displayed molecules and the immobilized target. These eluted phages represent a pool enriched with potential antibody hits. Subsequently, the selected phages are amplified by infecting bacterial hosts, ensuring the preservation and proliferation of the enriched antibody candidates. This step provides a sufficient quantity of the selected phages for downstream analysis and characterization [17].
In the process of panning and elution in phage display libraries, the choice of buffers with varying pH levels can significantly influence the characteristics of the displayed peptides and the efficiency of the selection process. Buffers with high pH, such as carbonate-bicarbonate buffer (pH 9.6), are commonly employed during the panning step of phage display selections. The high pH environment helps to ensure the stability and solubility of target proteins or ligands by minimizing their aggregation or precipitation. Additionally, at higher pH levels, the surface charge of both the target molecule and the phage-displayed peptides may be altered, which can influence the strength and specificity of their interactions. Peptides displayed on the phage surface may adopt different conformations or exhibit altered electrostatic properties under high pH conditions, potentially enhancing their affinity for specific targets or enabling the recognition of unique epitopes. Conversely, buffers with low pH, such as glycine-HCl buffer (pH 2.2), are often utilized during the elution step to dissociate phage–target complexes and recover specifically bound phages. The acidic environment disrupts electrostatic interactions and weakens the binding affinity between the displayed peptides and their target molecules, thereby facilitating the release of bound phages. Low-pH elution buffers are particularly effective for eluting phages that bind tightly to their targets, as they provide a robust means of dissociating these interactions without affecting phage viability or infectivity. However, it is important to note that excessively low pH conditions can also potentially damage the phage particles or denature the displayed peptides, leading to the reduced efficacy of the selection process. By optimizing the pH conditions to suit the specific characteristics of the target molecule and the desired selection outcome, researchers can enhance the success of phage display experiments and expedite the identification of novel peptide ligands or protein–protein interactions [18][19].
2.3. DNA Sequencing and Antibody Production
Individual phage clones from the enriched pool are isolated, and the genetic sequences of the displayed peptides or antibodies are determined through DNA sequencing. This information is crucial for the subsequent production of the identified antibodies. Once the genetic sequence is known, the corresponding antibodies can be expressed and produced in various formats, such as scFv or full-length antibodies, for further analysis and characterization [20].
2.4. Characterization and Optimization
The generated antibodies undergo rigorous characterization to evaluate their binding specificity, affinity, and potential therapeutic applications. This may involve assays such as ELISA or surface plasmon resonance. Further optimization steps can be employed to enhance the desired properties of the antibodies, including improvements in the binding affinity and stability, or reduced immunogenicity. Iterative rounds of optimization may be conducted to refine the antibodies for specific therapeutic or diagnostic purposes [21]. In essence, the procedures used in phage display technology to generate antibodies encompass a strategic and iterative process, from the construction of diverse libraries to the targeted selection of high-affinity binders and the subsequent characterization and optimization of the identified antibodies. This robust methodology has been pivotal in advancing the field of antibody discovery and engineering.
3. Categories of Phage Display Libraries for Antibody Development
Phage libraries derived from rearranged V-gene repertoires are created using mRNA or RNA isolated from B cells obtained from either immunized or naïve donors. The construction process entails initially preparing the cDNA template through reverse transcription polymerase chain reaction (RT-PCR). Subsequently, the repertoire of variable heavy (VH) and variable light (VL) chain genes is amplified via PCR before being cloned into the phagemid vector [22]. While phage display technology has been widely used for novel antibody development, it is noteworthy that there are different types of phage display libraries after many years of development.
As one of the first libraries constructed, scFv libraries are commonly used for antibody selection. They consist of antibody fragments that include the VH and VL chains linked by a flexible peptide linker. This format allows the expression of functional antigen-binding sites in a single polypeptide chain. For instance, the Tomlinson I and J libraries are well-known scFv libraries derived from human germline sequences. They offer a diverse source of antibody fragments for various applications. Another well-known application is the selection of scFv antibodies against specific tumor antigens using phage display, as demonstrated by Lee et al. [23]. In contrast, Fab libraries display antibody fragments that consist of the variable regions of both the heavy and light chains, along with the constant regions of the heavy chain. Fabs maintain the antigen-binding capacity of antibodies while providing additional structural stability. The HuCAL (Human Combinatorial Antibody Library) Fab library is a widely used human-derived library for the generation of Fab fragments with therapeutic potential [24]. In addition, a study by Gram et al. utilized a Fab phage display library to isolate antibodies specific to the human epidermal growth factor receptor 2 (HER2) [25].
In addition to the libraries from human and mice, single-domain antibody (nanobody) libraries have been derived from the variable domains of the heavy-chain-only antibodies found in camelids, such as llamas and camels. Renowned for their unique structural and functional attributes, nanobodies are single polypeptide chains that retain the antigen-binding capabilities of conventional antibodies [26][27]. One remarkable feature of nanobodies is their compact size, comprising only the variable domain without the additional heavy or light chains. This simplicity grants nanobodies several advantages, including enhanced tissue penetration, increased stability, and the ability to recognize cryptic epitopes that might be challenging for larger antibodies. The absence of a light chain also simplifies production and engineering processes. The selection process from nanobody libraries often involves immunizing animals with the target antigen of interest, followed by the generation of the library and subsequent panning against the antigen. Additionally, synthetic or naïve nanobody libraries can be created without prior immunization, providing a valuable resource for discovering binders against a wide range of targets [28].
In addition, chimeric phage display libraries are a hybrid class of libraries that incorporate genetic material from multiple sources, combining elements to enhance the diversity and functionality of displayed peptides or proteins. The construction of chimeric libraries involves the fusion of genetic sequences derived from different antibody fragments or protein domains, resulting in a mosaic-like collection of variants presented on the surface of bacteriophages [29]. These libraries aim to harness the strengths of different antibody formats or functional domains to create molecules with optimized properties. One common strategy for constructing chimeric libraries is the fusion of variable regions from human antibodies with constant regions from non-human sources, such as mice. This chimerization process seeks to retain the specificity and affinity of human antibodies while benefiting from the stability and expression advantages offered by non-human constant regions. Chimeric libraries can also be generated by combining variable domains from different antibodies, allowing for the exploration of diverse binding specificities within a single library [30].
In another way, phage display antibody libraries can be classified into four distinct types based on the origin of their sequences: naive, immune, semi-synthetic, and synthetic libraries [31][32]. Naive libraries, sourced from natural entities such as primary B-cells of non-immunized donors, harness naturally rearranged variable region genes. With extensive repertoires reaching up to 1011 members, naive libraries have the capacity to generate antibodies targeting a diverse array of antigens. In contrast, immune libraries derive from B-cell antibody repertoires of immunized or immune donors, exhibiting a predisposition to a limited panel of antigens and generally being smaller in size. While adept at addressing specific antigens, immune libraries are less suited for identifying antibody fragments against a broad spectrum of antigens, particularly self-antigens [30]. Synthetic libraries leverage computational design and gene synthesis, affording precise control over the composition of complementarity-determining regions (CDRs) [28]. Designed in silico, synthetic antibody libraries enable the creation of highly diverse libraries with predetermined structural features, incorporating non-natural amino acids. For example, MorphoSys’s HuCAL PLATINUM library is a synthetic antibody library meticulously designed for optimal diversity and human-like antibody properties. Similarly, semi-synthetic libraries amalgamate CDRs from natural sources with in silico-designed elements. Early iterations of semi-synthetic libraries maintained diversity through various framework genes. For example, in 1992, Hoogenboom and Winter introduced semi-synthetic scFv-antibody phage display libraries, integrating 49 germline VH sequences and a single V_lambda 3 light chain sequence. These libraries, with a size of 1 × 107, employed the PCR-based randomization of five or eight residues in the CDR-H3 to enhance diversity [33].
References
- Stanfield, R.L.; Wilson, I.A. Antibody Structure. Microbiol. Spectr. 2014, 2, 49–62.
- Chiu, M.L.; Gilliland, G.L. Engineering antibody therapeutics. Curr. Opin. Struct. Biol. 2016, 38, 163–173.
- Shim, H. Antibody Phage Display. Adv. Exp. Med. Biol. 2017, 1053, 21–34.
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317.
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: Filamentous phage displaying antibody variable domains. Nature 1990, 348, 552–554.
- Franca, R.K.A.; Studart, I.C.; Bezerra, M.R.L.; Pontes, L.Q.; Barbosa, A.M.A.; Brigido, M.M.; Furtado, G.P.; Maranhao, A.Q. Progress on Phage Display Technology: Tailoring Antibodies for Cancer Immunotherapy. Viruses 2023, 15, 1903.
- Domm, W.; Brewer, M.; Baker, S.F.; Feng, C.; Martinez-Sobrido, L.; Treanor, J.; Dewhurst, S. Use of bacteriophage particles displaying influenza virus hemagglutinin for the detection of hemagglutination-inhibition antibodies. J. Virol. Methods 2014, 197, 47–50.
- Chikaev, A.N.; Chikaev, A.N.; Rudometov, A.P.; Merkulyeva, Y.A.; Karpenko, L.I. Phage display as a tool for identifying HIV-1 broadly neutralizing antibodies. Vavilovskii Zhurnal Genet. Sel. 2021, 25, 562–572.
- Peltomaa, R.; Benito-Pena, E.; Barderas, R.; Moreno-Bondi, M.C. Phage Display in the Quest for New Selective Recognition Elements for Biosensors. ACS Omega 2019, 4, 11569–11580.
- Jones, M.L.; Alfaleh, M.A.; Kumble, S.; Zhang, S.; Osborne, G.W.; Yeh, M.; Arora, N.; Hou, J.J.; Howard, C.B.; Chin, D.Y.; et al. Targeting membrane proteins for antibody discovery using phage display. Sci. Rep. 2016, 6, 26240.
- Parmley, S.F.; Smith, G.P. Antibody-selectable filamentous fd phage vectors: Affinity purification of target genes. Gene 1988, 73, 305–318.
- Bashir, S.; Paeshuyse, J. Construction of Antibody Phage Libraries and Their Application in Veterinary Immunovirology. Antibodies 2020, 9, 21.
- Bazan, J.; Calkosinski, I.; Gamian, A. Phage display--a powerful technique for immunotherapy: 1. Introduction and potential of therapeutic applications. Hum. Vaccin. Immunother. 2012, 8, 1817–1828.
- Anand, T.; Virmani, N.; Bera, B.C.; Vaid, R.K.; Vashisth, M.; Bardajatya, P.; Kumar, A.; Tripathi, B.N. Phage Display Technique as a Tool for Diagnosis and Antibody Selection for Coronaviruses. Curr. Microbiol. 2021, 78, 1124–1134.
- Saw, P.E.; Song, E.W. Phage display screening of therapeutic peptide for cancer targeting and therapy. Protein Cell 2019, 10, 787–807.
- Scott, J.K.; Smith, G.P. Searching for peptide ligands with an epitope library. Science 1990, 249, 386–390.
- Nascimento, A.; Mullerpatan, A.; Azevedo, A.M.; Karande, P.; Cramer, S. Development of phage biopanning strategies to identify affinity peptide ligands for kappa light chain Fab fragments. Biotechnol. Prog. 2019, 35, e2884.
- Tulika, T.; Pedersen, R.W.; Rimbault, C.; Ahmadi, S.; Rivera-de-Torre, E.; Fernandez-Quintero, M.L.; Loeffler, J.R.; Bohn, M.F.; Ljungars, A.; Ledsgaard, L.; et al. Phage display assisted discovery of a pH-dependent anti-alpha-cobratoxin antibody from a natural variable domain library. Protein Sci. 2023, 32, e4821.
- Zhang, W.; Wang, H.; Feng, N.; Li, Y.; Gu, J.; Wang, Z. Developability assessment at early-stage discovery to enable development of antibody-derived therapeutics. Antib. Ther. 2023, 6, 13–29.
- Li, W.; Caberoy, N.B. New perspective for phage display as an efficient and versatile technology of functional proteomics. Appl. Microbiol. Biotechnol. 2010, 85, 909–919.
- Ledsgaard, L.; Kilstrup, M.; Karatt-Vellatt, A.; McCafferty, J.; Laustsen, A.H. Basics of Antibody Phage Display Technology. Toxins 2018, 10, 236.
- Alfaleh, M.A.; Alsaab, H.O.; Mahmoud, A.B.; Alkayyal, A.A.; Jones, M.L.; Mahler, S.M.; Hashem, A.M. Phage Display Derived Monoclonal Antibodies: From Bench to Bedside. Front. Immunol. 2020, 11, 1986.
- Lee, C.M.; Iorno, N.; Sierro, F.; Christ, D. Selection of human antibody fragments by phage display. Nat. Protoc. 2007, 2, 3001–3008.
- Prassler, J.; Thiel, S.; Pracht, C.; Polzer, A.; Peters, S.; Bauer, M.; Norenberg, S.; Stark, Y.; Kolln, J.; Popp, A.; et al. HuCAL PLATINUM, a synthetic Fab library optimized for sequence diversity and superior performance in mammalian expression systems. J. Mol. Biol. 2011, 413, 261–278.
- Gram, H.; Marconi, L.A.; Barbas, C.F., 3rd; Collet, T.A.; Lerner, R.A.; Kang, A.S. In vitro selection and affinity maturation of antibodies from a naive combinatorial immunoglobulin library. Proc. Natl. Acad. Sci. USA 1992, 89, 3576–3580.
- Bao, G.; Tang, M.; Zhao, J.; Zhu, X. Nanobody: A promising toolkit for molecular imaging and disease therapy. EJNMMI Res. 2021, 11, 6.
- Sun, S.; Ding, Z.; Yang, X.; Zhao, X.; Zhao, M.; Gao, L.; Chen, Q.; Xie, S.; Liu, A.; Yin, S.; et al. Nanobody: A Small Antibody with Big Implications for Tumor Therapeutic Strategy. Int. J. Nanomed. 2021, 16, 2337–2356.
- Weber, M.; Bujak, E.; Putelli, A.; Villa, A.; Matasci, M.; Gualandi, L.; Hemmerle, T.; Wulhfard, S.; Neri, D. A highly functional synthetic phage display library containing over 40 billion human antibody clones. PLoS ONE 2014, 9, e100000.
- Kumar, R.; Parray, H.A.; Shrivastava, T.; Sinha, S.; Luthra, K. Phage display antibody libraries: A robust approach for generation of recombinant human monoclonal antibodies. Int. J. Biol. Macromol. 2019, 135, 907–918.
- Andre, A.S.; Moutinho, I.; Dias, J.N.R.; Aires-da-Silva, F. In vivo Phage Display: A promising selection strategy for the improvement of antibody targeting and drug delivery properties. Front. Microbiol. 2022, 13, 962124.
- Frenzel, A.; Schirrmann, T.; Hust, M. Phage display-derived human antibodies in clinical development and therapy. MAbs 2016, 8, 1177–1194.
- Chan, S.K.; Rahumatullah, A.; Lai, J.Y.; Lim, T.S. Naive Human Antibody Libraries for Infectious Diseases. Adv. Exp. Med. Biol. 2017, 1053, 35–59.
- Braunagel, M.; Little, M. Construction of a semisynthetic antibody library using trinucleotide oligos. Nucleic Acids Res. 1997, 25, 4690–4691.
More
Information
Subjects:
Medical Laboratory Technology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Revisions:
2 times
(View History)
Update Date:
08 Mar 2024
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No