Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Athanasios G. Tzioufas | -- | 4518 | 2024-03-05 18:16:04 | | | |
| 2 | Rita Xu | Meta information modification | 4518 | 2024-03-06 02:41:44 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Palamidas, D.A.; Chatzis, L.; Papadaki, M.; Gissis, I.; Kambas, K.; Andreakos, E.; Goules, A.V.; Tzioufas, A.G. Pathogenetic Mechanism in Giant Cell Arteritis. Encyclopedia. Available online: https://encyclopedia.pub/entry/55884 (accessed on 06 June 2026).
Palamidas DA, Chatzis L, Papadaki M, Gissis I, Kambas K, Andreakos E, et al. Pathogenetic Mechanism in Giant Cell Arteritis. Encyclopedia. Available at: https://encyclopedia.pub/entry/55884. Accessed June 06, 2026.
Palamidas, Dimitris Anastasios, Loukas Chatzis, Maria Papadaki, Ilias Gissis, Konstantinos Kambas, Evangelos Andreakos, Andreas V. Goules, Athanasios G. Tzioufas. "Pathogenetic Mechanism in Giant Cell Arteritis" Encyclopedia, https://encyclopedia.pub/entry/55884 (accessed June 06, 2026).
Palamidas, D.A., Chatzis, L., Papadaki, M., Gissis, I., Kambas, K., Andreakos, E., Goules, A.V., & Tzioufas, A.G. (2024, March 05). Pathogenetic Mechanism in Giant Cell Arteritis. In Encyclopedia. https://encyclopedia.pub/entry/55884
Palamidas, Dimitris Anastasios, et al. "Pathogenetic Mechanism in Giant Cell Arteritis." Encyclopedia. Web. 05 March, 2024.
Copy Citation
Giant cell arteritis (GCA) is an autoimmune disease affecting large vessels in patients over 50 years old. It is an exemplary model of a classic inflammatory disorder with IL-6 playing the leading role.
giant cell arteritis
pathogenetic mechanism
monocytes
1. Introduction
Giant cell arteritis (GCA) stands out as the most prevalent form of primary systemic vasculitis in the elderly [1]. It is an autoimmune granulomatous disease that can affect large arteries with a notable predilection of the aortic arch and its branches. The superficial cranial branches of the external carotids, along with the ophthalmic branch of the internal carotid artery, are typically involved. Despite historical recognition of the systemic repercussions, manifested through constitutional symptoms and elevated inflammatory marker levels, GCA has traditionally been considered a predominantly cranial disease [2]. However, applying advanced imaging techniques has unveiled a broader spectrum, extending beyond cranial vasculature to involve other extracranial large and medium-sized vessels, underscoring the inherently systemic nature of GCA at the tissue level [3] (Figure 1). GCA is often associated with polymyalgia rheumatica (PMR), characterized by abrupt-onset pain, aching, and morning stiffness in the shoulder and hip girdle muscles. PMR is also an independent disease, suggesting that these two disorders could represent distinct expressions of a common underlying pathological process [4].
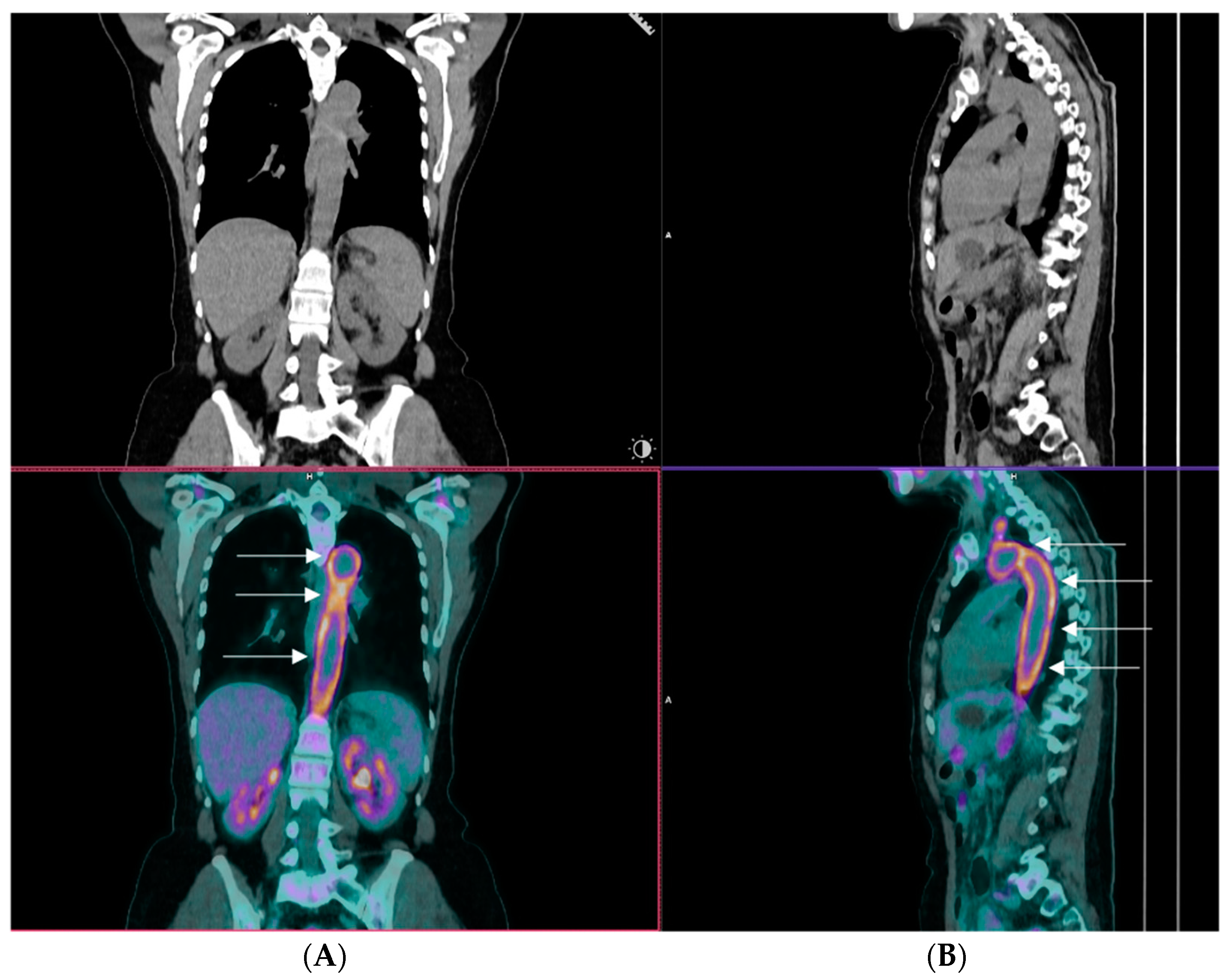
Figure 1. A representative image of an 18F-fluorodeoxyglucose positron emission tomography/computed tomography (PET/CT) of a patient with giant cell arteritis and extracranial large vessel involvement. The image displays coronal (Panel (A)) and sagittal views (Panel (B)), highlighting robust tracer uptake within the arch of the aorta, which is shown by arrows (kindly provided by our collaborator Dr CD Anagnostopoulos (BRFAA)).
1.1. Epidemiology
Despite its global prevalence, GCA exhibits geographic variability in its incidence and prevalence. Regardless of the diagnostic criteria used, several studies consistently identify people of northern European ancestry with the highest reported incidence (44 cases per 100,000 persons aged more than 50 years) and Asians, particularly in southeast Asia, as the area with the lowest (0.4 per 100,000 persons aged 50 years and above) [5]. Notably, there is a discernible north-to-south decreasing gradient even across Europe, highlighting the potential influence of ethnic origins in disease development. Females exhibit an almost threefold increased likelihood of developing GCA compared to males, with a propensity for more extracranial artery involvement and a higher prevalence of PMR [6]. Seasonal variation adds another layer to the epidemiological profile, with a notable preponderance of diagnoses during the spring and summer [7][8].
1.2. Risk Factors
Among the various risk factors associated with the development of GCA, age emerges as the single most important determinant. GCA abstains from affecting individuals under the age of 50, and after that, its incidence steadily ascents, with more than 80 percent of diagnosed patients surpassing the age of 70 [9]. This age-dependent pattern highlights the intricate interplay between aging and the pathogenesis of the disease. The etiology hangs between the age-related restructuring of the immune system and the age-related vascular remodeling and dysfunction. Immunosenescence, characterized by the reduction of the naïve T-cell pool, contraction of T-cell diversity, and accumulation of memory T-cells, is accompanied by low-grade inflammation, commonly referred to as inflammaging [10]. Vascular aging introduces pivotal changes in the structural properties of the vascular wall, leading to increased arterial stiffness and reduced compliance [11]. The interplay between these two aging-related phenomena forms a complex network that contributes to the initiation of GCA. Unraveling the most prominent driving forces in the aging process remains a critical challenge for a better understanding of the pathophysiology of GCA.
1.3. Clinical Picture
Regardless of the underlying pathogenetic origins, GCA presents with a variety of clinical features extending from constitutional symptoms to headaches, scalp tenderness, jaw claudication, and visual manifestations [12]. The onset of the disease typically follows a subacute trajectory over several weeks, although abrupt presentations are not uncommon. If left untreated, GCA poses the ominous risk of its most dreaded complication, permanent visual loss and, less frequently, stroke. Notably, recent reports indicate a decline in the incidence of visual loss, dwindling from 20% of patients to less than 10%, attributed to the advancements in awareness, timely diagnosis facilitated by rapid temporal artery color Doppler ultrasonography assessment [13], and swift medical intervention [14]. Although visual loss can occur abruptly and without warning, single or multiple episodes of transient visual loss (amaurosis fungax) may precede [15]. These brief episodes, though temporary, serve as critical warning signs, offering a window of opportunity for prompt recognition and initiation of treatment. According to the latest recommendations from EULAR in 2023, ultrasound, including assessment of the axillary arteries, is now advocated as the first-line imaging diagnostic test for GCA. If there is a high clinical suspicion coupled with a positive imaging test, a biopsy can be omitted from the diagnostic procedure [16]. However, clinical suspicion should always be heightened for GCA mimickers, with the most prominent being ANCA vasculitis, amyloidosis, and atherosclerosis [17]. Lately, COVID-19 has also been added to the list of conditions with similar presenting symptoms, such as headaches and acute phase responses. Thrombocytosis may lean more towards GCA, while lymphopenia may suggest COVID-19, aiding in the correct diagnosis [18].
2. Pathogenetic Mechanism in GCA
2.1. Initiation Phase
Similar to almost all systemic autoimmune and autoinflammatory disorders, the triggering factor(s) of GCA remains unknown. Several reports in the past have tried to elucidate the role of infections. Furthermore, tissue alterations via intrinsic mechanisms such as cell stress and accelerated atherosclerosis have also been proposed as initiating disease events. The lack of spontaneous experimental models, along with the poor understanding of tissue injury in GCA at the very early stages of the disease, are serious obstacles to understanding the initial mechanistic events. The initiating elements, when present, act synergistically upon a particular gene profile of patients with GCA that extends from antigen presentation (MHCs), adaptive and innate immunity genes (e.g., IL-2, IFN-γ), and several regulatory genes (e.g., TLRs).
2.1.1. Genetics
The genetic component has been considered an essential factor in GCA pathogenesis, based mainly on two observations: (i) reports on familial clustering of the disease [19][20][21] and (ii) an increased prevalence in Europeans of northern and Scandinavian ancestry [22]. With the utilization of molecular techniques from different scientific groups, they sought to decipher the genetic landscape of the disease. In the past two decades, genetic studies have been categorized into two types, according to the candidate-tested genes: (i) studies of targeted gene polymorphisms associated with disease susceptibility and (ii) genome-wide association studies.
In the first category, the main focus of research has included genes encoding the human leukocyte antigen (HLA) classes I and II and different molecules participating in the inflammatory response of GCA (IL-1β, IL-6, IL-10, IL-17A, IL-21, IL-23R, IL-33, TNF-α, VEGF, MMP-9, MPO, and others). Several studies in small patient cohorts interrogated the role of the HLA region in the disease. They revealed an association of GCA with the expression of HLA-DRB1*04 alleles (HLA-DRB1*0401, HLADRB1*0404, and HLADRB1*0408 haplotypes) [23][24][25][26][27][28][29][30][31][32]. Studies of genes encoding inflammatory molecules yielded various results. The rs2250889 polymorphism of the MMP-9 gene was associated with GCA susceptibility, but the study included only 30 patients [33]. Salvarani et al. demonstrated an association of the G/R 241 SNP in the ICAM-1 gene with the disease [34], but Amoli et al. reported a lack of association [35]. These studies included several weaknesses in sample size, lack of successful replicability, absence of appropriate geographical sample representation, and the use of low throughput technologies that did not allow for a large number of tested SNPs [36].
The research in this field entered the high-throughput screening era, producing more interesting results for the community. A large-scale study revealed that the HLA class II region is strongly associated with GCA [37], differentiating GCA from other systemic vasculitides that are genetically linked to HLA class I molecules (Takayasu arteritis, Behçet’s disease) [38] and supporting the implication of antigenic presentation processes in the initiation phase of the disease. A genome-wide association study (GWAS) confirmed this association, in addition to the identification of the plasminogen (PLG) gene polymorphism rs4252134 and the Prolyl 4-hydroxylase subunit alpha-2 (P4HA2) gene polymorphism rs128738 as genetic risk factors in GCA [39]. Furthermore, a meta-analysis highlighted the IL12B gene polymorphism rs755374 as a new risk SNP for GCA and a re-verification of the strongly associated HLA class II genes [40].
Overall, different studies exploring the contribution of SNPs in genes encoding proteins associated with the proinflammatory and tissue remodeling phases of GCA suggested the relevance of a plethora of innate and adaptive immunity molecules to disease pathogenesis. Additionally, GWAS introduced new genes as risk factors in GCA and reinforced the hypothesis of an unknown antigen-driven immune response initiating the pathogenesis of the disease.
2.1.2. Infections and Microbiome
The antigen-driven hypothesis of triggering the initiation of vascular inflammation observed in GCA has been a matter of debate for many years in the literature. In this direction, several studies have investigated the implication of different infectious agents in vascular dendritic cell activation and subsequent disease onset [41]. Of the infectious agents interrogated, human herpes virus (HHV)-6, HHV-7, varicella-zoster virus, and Epstein–Barr virus have not been associated with GCA. Although cytomegalovirus, parvovirus B19, herpes simplex virus, human parainfluenza 1, and chlamydia pneumonia were initially correlated with the disease, none of these results were confirmed by subsequent studies in larger cohorts [41]. A previous study reported the presence of varicella-zoster virus (VZV) antigen in 64–73% of temporal artery biopsies (TABs) of large vessel vasculitis (LVV) patients, with or without temporal artery involvement, as opposed to 22% of normal arteries tested [42]. Although this finding implied a possible disease-causing mechanism of VZV infection, the study lost its validity after Pisapia et al. reported an increased false-positive rate due to non-specific staining of the antibody used in the immunohistochemistry assay for VZV detection [43].
State-of-the-art approaches such as whole genome sequencing and 16S Ribosomal RNA gene sequencing have also been utilized in search of infectious agents implicated in GCA pathogenesis. In a shotgun sequencing-based study, none of the previously reported pathogens were detected in GCA TABs. Moreover, the microbiome did not differ between GCA cases and controls since only members of the normal skin flora were observed [44]. Two studies of another group that followed aseptic techniques during tissue collection found that the microbiome of thoracic aortic aneurysms from isolated aortitis and GCA differ substantially from the microbiome of non-inflammatory aortic aneurysms [45] and that from TABs of GCA patients [46]. These studies provided novel insights into the candidate role of the microbiome of different vascular components in health and disease. However, the translation of these findings in a comprehensive manner that could enrich the knowledge of GCA pathogenesis still has a long way to go.
2.1.3. Vascular Aging/Inflammaging
Undoubtedly, increasing age is a major etiological factor in GCA’s initiation, with different compartments of immunity being affected [11]. T-cells, in particular, exhibit a senescent phenotype due to the loss of the CD28 cell surface marker [47]. CD4+ CD28− and CD8+ CD28− cells are reported to be enriched in T-cell subsets of aged individuals [48]. There is also a marked decrease in the numbers of naïve T-cells and the T-Cell Receptor (TCR) repertoire diversity as opposed to an increase in T effector and memory cells [49]. Dendritic cells (DCs) can also be impacted by aging, displaying impaired activation and migration capacity [50]. Senescent macrophages display an altered, more inflammatory phenotype characterized by the expression of proinflammatory mediators (IL-1β, IL-6, TNF-α) [51]. It is reported that epigenetics may play a vital role during this so-called inflammaging process. Indeed, DNA methyltransferase 1 (DNMT1) levels in T-cells are decreased during the aging process [52]. DNMT1 is an enzyme that facilitates the maintenance of DNA methylation levels between cell divisions, a disturbed process during aging that leads to altered methylation levels in T-cells, a phenomenon associated with increased risk for autoimmunity and cancer. On the other hand, structural arterial changes are observed that correlate with increasing age, including calcium deposition, increased artery stiffness, wall thickening, and alterations in the extracellular matrix of the arteries [53]. The effect of inflammaging on the immune system, which is disturbed in GCA, and the vasculature, the target organ of the disease, supports the central role of senescence in GCA pathogenesis.
2.2. Perpetuation and Sustainability of the Inflammatory Response
Different studies of tissue biopsies of GCA patients reveal many types of immunocytes involved in innate and adaptive immune responses that are usually activated as attested by cellular markers and cytokines. Essentially, the perpetuation of tissue inflammatory response is performed through acute DTH mechanisms in which activated monocytes play a central role. At the same time, sustainability is implemented through chronic DTH responses with granuloma formation, which is the histological hallmark of the disease. In some instances, the local autoimmune injury is probably continuously fueled by the formation of ectopic germinal centers [54]. Disturbances in immune cell subpopulation frequencies in peripheral blood mononuclear cells (PBMCs) are also observed during active and inactive disease states [55]. An unmet need of GCA is the exact definition of cells participating in the acute inflammatory response in correlation with the cells in remission. Indeed, the type of cells and the inflammatory mediators differ in the acute and remission phases since the metabolic landscape is also different [56]. To this end, other groups and ours have suggested the investigation of pairs of biological samples in both activity and remission states in an attempt to gain clinically relevant biomarkers. Hereafter, the types and functions of major immune cell types are described.
2.2.1. Dendritic Cells and TLRs
The activation of resident DCs and subsequent activation of T-cells in the affected GCA arteries is thought to be the initial step of the intense inflammatory response. Following sensing intrinsic or extrinsic antigens, these cells induce strong innate and adaptive immune responses via TLR ligation and cell activation, which leads to chemokine and cytokine production, orchestrating tissue infiltration from T-cells. Thus, TLR differential expression and activation in the context of this pathology is of great importance [57].
Resident DCs observed in the adventitia of normal arteries display an immature phenotype [58][59] and contribute to immune surveillance. Detection of activated DCs in TABs is not exclusive to GCA but has also been described in TABs of patients with PMR. Activated DCs are located in the adventitia of PMR patient TABs as opposed to GCA patient TABs, in which activated DCs are extended in all three layers of the vascular wall [59]. Moreover, DCs are restricted to the artery and do not migrate to lymph nodes [58], a phenomenon facilitated by a positive-feedback loop of specific chemokine production (CCL19 and CCL21) and their receptor, CCR7 [59], leading to the migration of more DCs at the site of inflammation and restricting their escape to the lymph nodes. Additionally, DCs express CD83, an activation marker, and CD86, a co-stimulatory molecule that renders them capable of activating naïve T-cells. A study in a human artery–mouse chimera model of GCA showed that DC depletion with an antibody against CD83 was able to effectively reduce the inflammatory response, T-cell infiltration, and IFN-γ production [59], recapitulating the significance of DCs in the initial inflammatory cascade of GCA. It has also been demonstrated that the immunoprotective PD-1/PD-L1 immune checkpoint is defective in GCA-affected tissue DCs, thus rendering them more susceptible to activation [60]. Interestingly, a study investigating the expression profile of TLR genes of 6 GCA-affected arteries obtained during autopsy showed a distinct TLR profile for each affected vessel studied [61]. Until recently, there were no reports on DC subpopulations in the peripheral blood of GCA patients. Reitsema et al. investigated the frequencies of DC subsets in the peripheral blood of GCA and PMR patients compared to healthy individuals for the first time [62]. The authors noted that plasmacytoid DCs (pDCs) and type-2 conventional dendritic cells (cDC2) frequencies did not differ between patients and controls, whereas cDC1 frequencies were substantially reduced in patients. Additionally, in GCA/PMR patients, pDCs exhibited higher expression of the immune checkpoint CD86 and reduced expression of CD40, whereas cDC2 subsets exhibited lower activation status as shown by lower HLA-DR activation marker expression [62]. Overall, the interpretation of these results is challenging in understanding GCA pathogenesis since there are no surrogate data of DC subsets on the inflamed tissue level.
Examining the mechanism of DC activation in the context of the disease is pivotal in understanding the clinical heterogeneity in GCA, with some patients exhibiting disease restricted to temporal arteries while others display a more pronounced inflammatory response in different large vessels. A multidimensional study correlating TLR repertoire, pathogen expression (bacteria and viruses), and the activation status of different DC subpopulations in different anatomically affected arteries has never been conducted and may provide insights into understanding the etiology and initiation phase of GCA.
2.2.2. CD4 T-Cells
In GCA, a pronounced T-cell compartment participates in the underlying inflammation [63]. Indeed, activated DCs recruit CD4+ T-cells at the site of inflammation through CCL18, CCL19, CCL20, and CCL21 chemokine expression [64]. Upon arrival, activated CD4+ cells differentiate into Th1 and Th17 T-cell subsets through proinflammatory signals of the inflamed artery (IL-1β, IL-6, IL-12, IL-18, and IL23) [65][66]. In Th1 responses, CD4+ cells, under the influence of IL-12 and IL-18, differentiate into Th1 cells that secrete IFN-γ [67], a critical regulatory cytokine in the disease’s pathogenesis [68]. On the other hand, Th17 responses include the differentiation of CD4+ cells into Th17 cells under the effect of IL-1β, IL-6, and IL23 [67]. It is well established that these two T-cell axes operate in the vicious, inflammatory cycle observed in the disease. However, the order of appearance of Th1 and Th17 responses is unknown. GC treatment is highly effective in restricting Th17 responses, as proved by the reduction of both the number of Th17 cells in the peripheral blood and IL-17 serum levels [65][69]. However, IFN-γ producing Th1 responses show resistance after GC treatment in GCA patients [65][70].
Another dysregulated CD4+ subset implicated in GCA pathogenesis is T regulatory cells (Tregs). Tregs are reduced in both peripheral blood and at the tissue level in GCA [65][66][71]. Moreover, the high levels of IL-6, IL-21, and IL-23 in the inflamed tissue microenvironment lead to the restricted expression of FOXP3 transcriptional factor, which is crucial for the differentiation of Tregs, favoring the upregulation of RORγt transcriptional factor that mediates Th17 differentiation [72]. An imbalance of the Th17/Treg axis participating in the GCA pathogenetic mechanism has been observed and is restored by IL-6R blockade as shown by normalization in Treg peripheral blood frequencies and a decrease in activation status [71][73]. Miyabe et al. also highlighted that Tregs in GCA present a reduced suppressor capacity, possessing a proinflammatory phenotype with increased production of IL-17 [74].
Recent evidence has emerged for the role of Th9 cells in GCA. Th9 cells constitute a distinct lineage of T-cells differentiated from CD4+ naïve cells under the influence of IL-4, TGF-β, and thymic stromal lymphopoietin (TSLP) and are effective producers of IL-9. In GCA, Th9 cells are mainly observed in artery tissues with transmural inflammation and small vessel vasculitis, as opposed to their absence in vasa vasorum vasculitis [75]. Furthermore, patients with GCA exhibit a defective, macrophage-induced CD155-CD96 immune checkpoint that leads to Th9 expansion [76]. The same study showed increased vessel wall destruction due to increased IL-9 production in a humanized mouse model of GCA [76]. Although all groups have not demonstrated the functions and disease relevance of the Th9 subset in GCA, their implication in the pathogenic phenomena appears important and requires further research.
GCA-affected tissue may possess ectopic germinal center (EGC)-like structures that are important for perpetuating local inflammatory responses. In the formation of such structures, Follicular helper T (Tfh) cells play a central role. Tfh cells have been detected in the inflamed artery and the peripheral blood of GCA patients [77]. Tfh cells are capable of producing IL-21, which, among other functions, increases B-cell differentiation into plasmablasts and triggers germinal centers to produce immunoglobulins [78]. However, autoantibodies characterizing the disease have not been detected yet.
2.2.3. CD8 T-Cells
For years, the role of CD8+ T-cells in GCA has not attracted much attention due to their low number in both the periphery and tissue [79][80][81]. Nonetheless, CD8+ cells present oligoclonality in GCA tissue. They may infiltrate temporal arteries, producing proinflammatory cytokines such as IL-17A and IFN-γ as well as cytotoxic substances such as granzymes A and B [79]. These observations give rise, once again, to an antigen-driven theory of GCA initiation. Furthermore, a decreased number of immunosuppressive CD8+ CCR7+ FoxP3+ Tregs has been observed in the peripheral blood of GCA patients, potentially leading to a failure in the control of CD4+ T-cell proliferation and activation through a NADPH oxidase-2 dependent mechanism [82]. In addition, a decrease in CD8+ T-cells was associated with aging and GCA pathogenesis [82]. The fact that this defect was not abrogated by GC treatment in patients raises new questions about the pathogenetic role of CD8+ cells.
2.2.4. B-Cells
The role of B-cells in GCA had been overlooked for many years due to the initial reports of a low number of B-cells infiltrating the temporal artery [83][84], suggesting that humoral immunity plays a marginal role in the inflammatory process. Data on serum autoantibodies in GCA patients support a more active role for B-cells in the inflammatory process. Autoantibodies that were identified include (i) low titers of anti-cardiolipin of the IgG isotype that disappeared following GCs treatment [85][86][87][88]; (ii) anti-endothelial cell antibodies which were not specific for GCA and were also detected in other systemic vasculitides [89][90]; and (iii) anti-smooth muscle cell antibodies, but the study included a low number of patients [90]; additionally, (iv) Baerlecken et al. reported antibodies against the human ferritin heavy chain in a high percentage of GCA patients (92%) [91] while Régent et al. reported lower titers, but yet with high prevalence (71.9%) in GCA patients and 34% in patients with a diagnosis other than GCA diagnosis [92]. These results collectively support the idea that these autoantibodies are not disease-specific but an epiphenomenon of the inflammatory bulk.
In recent years, the formation of tertiary lymphoid organs (ATLOs) has been identified in the aortas of GCA patients but not in the affected temporal arteries [54][93]. Moreover, those ATLOs were found in the adventitia of inflamed aortas and were absent from GCA-positive temporal arteries, suggesting different organization according to the size of the affected artery. ATLOs consist of follicular dendritic cells located near T-cells and endothelial cells in the media layer, supporting the organization of lymphoid tissue against arterial wall-derived antigens. In the peripheral blood of treatment-naïve GCA and PMR patients, B effector cells are decreased compared to healthy controls and reach normal levels after GC treatment, exhibiting an enhanced IL-6 production capacity [94]. In contrast, serum levels of CXCL9 and CXCL13, major chemokines for the organization of ATLOs, are elevated [95][96]. The same study highlighted that B-cells were detected in the inflamed arterial tissue, and their migration pattern followed the CXCR3–CXCL9 and CXCR5–CXCL13 chemokine axes [96]. The above data indicate that B-cells in active disease are reduced in the peripheral blood due to their migration to the affected temporal arteries. In cases of large vessel involvement, ectopic lymphoid tissue is generated and is possibly connected to chronic inflammatory processes.
2.2.5. Monocytes
The abundant expression of IFN-γ in the initial steps of inflammation induces CCL2 expression by vascular smooth muscle cells (VSMCs), leading to tissue infiltration by monocytes of the classical subset, expressing the CCR2 receptor [97]. In addition, non-classical monocytes also participate in the inflammatory process and reach out to the tissue lesion under stimuli of the CX3CR1–CX3CL1 axis [98]. The predominant type of monocytes in the GCA tissue lesions is still a matter of debate, with different groups reporting either the classical monocyte subset [97] or non-classical subsets as the major monocytic type [98]. In the peripheral blood of GCA patients during diagnosis, elevated classical monocyte numbers [55][98] persisted after three months of GC treatment. Interestingly, disease remission correlated with a decrease in the number of intermediate and non-classical monocytic lineages, indicating that GCs act partly towards normalizing monocyte numbers [98]. Following integrative analysis of the methylome and transcriptome of CD14+ cells in GCA active and inactive disease, a recent study identified IL-11 as a novel cytokine pathway associated with the disease [99]. However, functional studies on the relevance of their findings to disease pathogenesis are needed.
2.2.6. Cytokines with a Significant Contribution to the Inflammatory Response of GCA
IL-6: IL-6 is considered to be the leading inflammatory cytokine in GCA’s pathogenetic mechanism. Serum levels of IL-6 appear to be elevated early during the disease course [65] and decrease rapidly after a few hours of GC initiation [100], without, however, reaching the levels of healthy individuals, even after chronic administration of GCs [101]. In addition, after GC tapering, IL-6 is among the first cytokines that arise in GCA patients’ serum [100]. IL-6 also correlates highly with acute phase reactants (erythrocyte sedimentation rate and C-reactive protein) and may serve as a biomarker of disease activity [102]. The above clinical observations made IL-6 a therapeutic target, and tocilizumab, a humanized monoclonal antibody to the IL-6 receptor (IL-6R), is now consistently used for patients with relapsing or refractory disease [103].
Although targeting of IL-6 is successful in therapy, the exact mechanism of action in GCA pathogenesis has not been elucidated, probably due to its pleiotropic effects. IL-6 is expressed by various immune and stromal cells, including activated monocytes, macrophages, B-cells, T-cells, fibroblasts, and endothelial cells [104]. IL-6 signal transduction is mediated by the complex IL-6/IL-6R/gp-130, with the latter being ubiquitously expressed, while hepatocytes, activated monocytes, macrophages, B-cells, and endothelial cells mainly express IL-6R. An established function of IL-6 is to control the balance between Th17 cells and Tregs [105], which is also evident in GCA [71][74]. The reduced frequency and the proinflammatory phenotype of Th17 expressing Tregs are reversed in GCA patients after tocilizumab treatment. There are conflicting reports on the effect of IL-6 on tissue remodeling. In a study by O’Neill et al., serum amyloid A (SAA) protein, which is triggered by IL-6 hepatocyte signaling and is increased in GCA patients’ serum, induced the protein expression of vascular endothelial growth factor (VEGF) and MMP-9 in an ex vivo culture model of temporal arteries. However, a more recent study of the same group argues a role of IL-6 in tissue remodeling since there was no effect of IL-6 treatment on myofibroblast proliferation and migration in a myofibroblast outgrowth culture model of GCA [106].
GM-CSF: Granulocyte-macrophage colony-stimulating factor (GM-CSF) has been recently suggested as a highly influential cytokine in GCA [107]. This proinflammatory cytokine is expressed by fibroblasts, endothelial, epithelial, myeloid, and T-cells upon appropriate stimulatory cues [108]. The GM-CSF heterodimeric receptor is composed of an α chain specific to GM-CSF and a signal transduction β chain, which is also found in the IL-3 and IL-5 receptors and, upon phosphorylation, triggers the activation of the JAK2/STAT5 signaling pathway [108]. Patient serum levels of GM-CSF are extremely low and comparable with healthy individuals [107]. On the contrary, GM-CSF protein levels are increased in GCA TABs [107] and PBMCs upon stimulation in vitro [66], suggesting a paracrine function of this cytokine on the inflamed tissue. The only functional study about GM-CSF in GCA was by Corbera-Bellalta et al. and highlighted the role of this cytokine in GCA. GM-CSF blockade by mavrilimumab, a fully human IgG4 monoclonal antibody to GM-CSFRα, abolished immune cell infiltration, inflammatory markers, and tissue remodeling factors, suggesting a role of GM-CSF in both the perpetuation of the inflammatory response and tissue injury phases [107]. A phase-2 clinical trial on mavrilimumab showed promising results on sustaining disease remission in week 26. However, further clinical trials are needed to determine whether this treatment modality is superior to current treatment options [109].
References
- Pugh, D.; Karabayas, M.; Basu, N.; Cid, M.C.; Goel, R.; Goodyear, C.S.; Grayson, P.C.; McAdoo, S.P.; Mason, J.C.; Owen, C.; et al. Large-vessel vasculitis. Nat. Rev. Dis. Primers 2022, 7, 93.
- Dejaco, C.; Brouwer, E.; Mason, J.C.; Buttgereit, F.; Matteson, E.L.; Dasgupta, B. Giant cell arteritis and polymyalgia rheumatica: Current challenges and opportunities. Nat. Rev. Rheumatol. 2017, 13, 578–592.
- van der Geest, K.S.M.; Sandovici, M.; van Sleen, Y.; Sanders, J.S.; Bos, N.A.; Abdulahad, W.H.; Stegeman, C.A.; Heeringa, P.; Rutgers, A.; Kallenberg, C.G.M.; et al. Review: What Is the Current Evidence for Disease Subsets in Giant Cell Arteritis? Arthritis Rheumatol. 2018, 70, 1366–1376.
- Buttgereit, F.; Matteson, E.L.; Dejaco, C. Polymyalgia Rheumatica and Giant Cell Arteritis. JAMA 2020, 324, 993–994.
- Li, K.J.; Semenov, D.; Turk, M.; Pope, J. A meta-analysis of the epidemiology of giant cell arteritis across time and space. Arthritis Res. Ther. 2021, 23, 82.
- Salvarani, C.; Crowson, C.S.; O’Fallon, W.M.; Hunder, G.G.; Gabriel, S.E. Reappraisal of the epidemiology of giant cell arteritis in Olmsted County, Minnesota, over a fifty-year period. Arthritis Rheum. 2004, 51, 264–268.
- Kønig, E.B.; Stormly Hansen, M.; Foldager, J.; Siersma, V.; Loft, A.; Terslev, L.; Møller Døhn, U.; Radmer Jensen, M.; Wiencke, A.K.; Faber, C.; et al. Seasonal variation in biopsy-proven giant cell arteritis in Eastern Denmark from 1990–2018. Acta Ophthalmol. 2021, 99, 527–532.
- Gokoffski, K.K.; Chatterjee, A.; Khaderi, S.K. Seasonal incidence of biopsy-proven giant cell arteritis: A 20-year retrospective study of the University of California Davis Medical System. Clin. Exp. Rheumatol. 2019, 37 (Suppl. 117), 90–97.
- Hoffman, G.S. Giant Cell Arteritis. Ann. Intern. Med. 2016, 165, Itc65–Itc80.
- Tu, W.; Rao, S. Mechanisms Underlying T Cell Immunosenescence: Aging and Cytomegalovirus Infection. Front. Microbiol. 2016, 7, 2111.
- Mohan, S.V.; Liao, Y.J.; Kim, J.W.; Goronzy, J.J.; Weyand, C.M. Giant cell arteritis: Immune and vascular aging as disease risk factors. Arthritis Res. Ther. 2011, 13, 231.
- Calamia, K.T.; Hunder, G.G. Giant cell arteritis (temporal arteritis) presenting as fever of undetermined origin. Arthritis Rheum. 1981, 24, 1414–1418.
- Diamantopoulos, A.P.; Haugeberg, G.; Lindland, A.; Myklebust, G. The fast-track ultrasound clinic for early diagnosis of giant cell arteritis significantly reduces permanent visual impairment: Towards a more effective strategy to improve clinical outcome in giant cell arteritis? Rheumatology 2016, 55, 66–70.
- Soriano, A.; Muratore, F.; Pipitone, N.; Boiardi, L.; Cimino, L.; Salvarani, C. Visual loss and other cranial ischaemic complications in giant cell arteritis. Nat. Rev. Rheumatol. 2017, 13, 476–484.
- Hayreh, S.S.; Podhajsky, P.A.; Zimmerman, B. Ocular manifestations of giant cell arteritis. Am. J. Ophthalmol. 1998, 125, 509–520.
- Dejaco, C.; Ramiro, S.; Bond, M.; Bosch, P.; Ponte, C.; Mackie, S.L.; Bley, T.A.; Blockmans, D.; Brolin, S.; Bolek, E.C.; et al. EULAR recommendations for the use of imaging in large vessel vasculitis in clinical practice: 2023 update. Ann. Rheum. Dis. 2023; Online ahead of print.
- Evangelatos, G.; Grivas, A.; Pappa, M.; Kouna, K.; Iliopoulos, A.; Fragoulis, G.E. Cranial giant cell arteritis mimickers: A masquerade to unveil. Autoimmun. Rev. 2022, 21, 103083.
- Mehta, P.; Sattui, S.E.; van der Geest, K.S.M.; Brouwer, E.; Conway, R.; Putman, M.S.; Robinson, P.C.; Mackie, S.L. Giant Cell Arteritis and COVID-19: Similarities and Discriminators. A Systematic Literature Review. J. Rheumatol. 2021, 48, 1053–1059.
- Kemp, A. Monozygotic twins with temporal arteritis and ophthalmic arteritis. Acta Ophthalmol. 1977, 55, 183–190.
- Fietta, P.; Manganelli, P.; Zanetti, A.; Neri, T.M. Familial giant cell arteritis and polymyalgia rheumatica: Aggregation in 2 families. J. Rheumatol. 2002, 29, 1551–1555.
- Liozon, E.; Ouattara, B.; Rhaiem, K.; Ly, K.; Bezanahary, H.; Loustaud, V.; Letellier, P.; Drouet, M.; Vidal, E. Familial aggregation in giant cell arteritis and polymyalgia rheumatica: A comprehensive literature review including 4 new families. Clin. Exp. Rheumatol. 2009, 27, S89–S94.
- Lee, J.L.; Naguwa, S.M.; Cheema, G.S.; Gershwin, M.E. The geo-epidemiology of temporal (giant cell) arteritis. Clin. Rev. Allergy Immunol. 2008, 35, 88–95.
- Cid, M.C.; Ercilla, G.; Vilaseca, J.; Sanmarti, R.; Villalta, J.; Ingelmo, M.; Urbano-Marquez, A. Polymyalgia rheumatica: A syndrome associated with HLA-DR4 antigen. Arthritis Rheum. 1988, 31, 678–682.
- Combe, B.; Sany, J.; Le Quellec, A.; Clot, J.; Eliaou, J.F. Distribution of HLA-DRB1 alleles of patients with polymyalgia rheumatica and giant cell arteritis in a Mediterranean population. J. Rheumatol. 1998, 25, 94–98.
- Jacobsen, S.; Baslund, B.; Madsen, H.O.; Tvede, N.; Svejgaard, A.; Garred, P. Mannose-binding lectin variant alleles and HLA-DR4 alleles are associated with giant cell arteritis. J. Rheumatol. 2002, 29, 2148–2153.
- Martínez-Taboda, V.M.; Bartolome, M.J.; Lopez-Hoyos, M.; Blanco, R.; Mata, C.; Calvo, J.; Corrales, A.; Rodriguez-Valverde, V. HLA-DRB1 allele distribution in polymyalgia rheumatica and giant cell arteritis: Influence on clinical subgroups and prognosis. Semin. Arthritis Rheum. 2004, 34, 454–464.
- Rauzy, O.; Fort, M.; Nourhashemi, F.; Alric, L.; Juchet, H.; Ecoiffier, M.; Abbal, M.; Adoue, D. Relation between HLA DRB1 alleles and corticosteroid resistance in giant cell arteritis. Ann. Rheum. Dis. 1998, 57, 380–382.
- Richardson, J.E.; Gladman, D.D.; Fam, A.; Keystone, E.C. HLA-DR4 in giant cell arteritis: Association with polymyalgia rheumatica syndrome. Arthritis Rheum. 1987, 30, 1293–1297.
- Salvarani, C.; Macchioni, P.; Zizzi, F.; Mantovani, W.; Rossi, F.; Castri, C.; Capozzoli, N.; Baricchi, R.; Boiardi, L.; Chiaravalloti, F.; et al. Epidemiologic and immunogenetic aspects of polymyalgia rheumatica and giant cell arteritis in northern Italy. Arthritis Rheum. 1991, 34, 351–356.
- Weyand, C.M.; Hicok, K.C.; Hunder, G.G.; Goronzy, J.J. The HLA-DRB1 locus as a genetic component in giant cell arteritis. Mapping of a disease-linked sequence motif to the antigen binding site of the HLA-DR molecule. J. Clin. Investig. 1992, 90, 2355–2361.
- Weyand, C.M.; Hunder, N.N.; Hicok, K.C.; Hunder, G.G.; Goronzy, J.J. HLA-DRB1 alleles in polymyalgia rheumatica, giant cell arteritis, and rheumatoid arthritis. Arthritis Rheum. 1994, 37, 514–520.
- Dababneh, A.; Gonzalez-Gay, M.A.; Garcia-Porrua, C.; Hajeer, A.; Thomson, W.; Ollier, W. Giant cell arteritis and polymyalgia rheumatica can be differentiated by distinct patterns of HLA class II association. J. Rheumatol. 1998, 25, 2140–2145.
- Rodríguez-Pla, A.; Beaty, T.H.; Savino, P.J.; Eagle, R.C., Jr.; Seo, P.; Soloski, M.J. Association of a nonsynonymous single-nucleotide polymorphism of matrix metalloproteinase 9 with giant cell arteritis. Arthritis Rheum. 2008, 58, 1849–1853.
- Salvarani, C.; Casali, B.; Boiardi, L.; Ranzi, A.; Macchioni, P.; Nicoli, D.; Farnetti, E.; Brini, M.; Portioli, I. Intercellular adhesion molecule 1 gene polymorphisms in polymyalgia rheumatica/giant cell arteritis: Association with disease risk and severity. J. Rheumatol. 2000, 27, 1215–1221.
- Amoli, M.M.; Shelley, E.; Mattey, D.L.; Garcia-Porrua, C.; Thomson, W.; Hajeer, A.H.; Ollier, W.E.; Gonzalez-Gay, M.A. Lack of association between intercellular adhesion molecule-1 gene polymorphisms and giant cell arteritis. J. Rheumatol. 2001, 28, 1600–1604.
- Carmona, F.D.; González-Gay, M.A.; Martín, J. Genetic component of giant cell arteritis. Rheumatology 2014, 53, 6–18.
- Carmona, F.D.; Mackie, S.L.; Martín, J.E.; Taylor, J.C.; Vaglio, A.; Eyre, S.; Bossini-Castillo, L.; Castañeda, S.; Cid, M.C.; Hernández-Rodríguez, J.; et al. A large-scale genetic analysis reveals a strong contribution of the HLA class II region to giant cell arteritis susceptibility. Am. J. Hum. Genet. 2015, 96, 565–580.
- Carmona, F.D.; Martín, J.; González-Gay, M.A. Genetics of vasculitis. Curr. Opin. Rheumatol. 2015, 27, 10–17.
- Carmona, F.D.; Vaglio, A.; Mackie, S.L.; Hernández-Rodríguez, J.; Monach, P.A.; Castañeda, S.; Solans, R.; Morado, I.C.; Narváez, J.; Ramentol-Sintas, M.; et al. A Genome-wide Association Study Identifies Risk Alleles in Plasminogen and P4HA2 Associated with Giant Cell Arteritis. Am. J. Hum. Genet. 2017, 100, 64–74.
- Carmona, F.D.; Coit, P.; Saruhan-Direskeneli, G.; Hernández-Rodríguez, J.; Cid, M.C.; Solans, R.; Castañeda, S.; Vaglio, A.; Direskeneli, H.; Merkel, P.A.; et al. Analysis of the common genetic component of large-vessel vasculitides through a meta-Immunochip strategy. Sci. Rep. 2017, 7, 43953.
- Ly, K.H.; Régent, A.; Tamby, M.C.; Mouthon, L. Pathogenesis of giant cell arteritis: More than just an inflammatory condition? Autoimmun. Rev. 2010, 9, 635–645.
- Nagel, M.A.; White, T.; Khmeleva, N.; Rempel, A.; Boyer, P.J.; Bennett, J.L.; Haller, A.; Lear-Kaul, K.; Kandasmy, B.; Amato, M.; et al. Analysis of Varicella-Zoster Virus in Temporal Arteries Biopsy Positive and Negative for Giant Cell Arteritis. JAMA Neurol. 2015, 72, 1281–1287.
- Pisapia, D.J.; Lavi, E. VZV, temporal arteritis, and clinical practice: False positive immunohistochemical detection due to antibody cross-reactivity. Exp. Mol. Pathol. 2016, 100, 114–115.
- Bhatt, A.S.; Manzo, V.E.; Pedamallu, C.S.; Duke, F.; Cai, D.; Bienfang, D.C.; Padera, R.F.; Meyerson, M.; Docken, W.P. In search of a candidate pathogen for giant cell arteritis: Sequencing-based characterization of the giant cell arteritis microbiome. Arthritis Rheumatol. 2014, 66, 1939–1944.
- Getz, T.M.; Hoffman, G.S.; Padmanabhan, R.; Villa-Forte, A.; Roselli, E.E.; Blackstone, E.; Johnston, D.; Pettersson, G.; Soltesz, E.; Svensson, L.G.; et al. Microbiomes of Inflammatory Thoracic Aortic Aneurysms Due to Giant Cell Arteritis and Clinically Isolated Aortitis Differ From Those of Non-Inflammatory Aneurysms. Pathog. Immun. 2019, 4, 105–123.
- Hoffman, G.S.; Getz, T.M.; Padmanabhan, R.; Villa-Forte, A.; Clifford, A.H.; Funchain, P.; Sankunny, M.; Perry, J.D.; Blandford, A.; Kosmorsky, G.; et al. The Microbiome of Temporal Arteries. Pathog. Immun. 2019, 4, 21–38.
- Czesnikiewicz-Guzik, M.; Lee, W.W.; Cui, D.; Hiruma, Y.; Lamar, D.L.; Yang, Z.Z.; Ouslander, J.G.; Weyand, C.M.; Goronzy, J.J. T cell subset-specific susceptibility to aging. Clin. Immunol. 2008, 127, 107–118.
- Vallejo, A.N.; Weyand, C.M.; Goronzy, J.J. T-cell senescence: A culprit of immune abnormalities in chronic inflammation and persistent infection. Trends Mol. Med. 2004, 10, 119–124.
- Sun, X.; Nguyen, T.; Achour, A.; Ko, A.; Cifello, J.; Ling, C.; Sharma, J.; Hiroi, T.; Zhang, Y.; Chia, C.W.; et al. Longitudinal analysis reveals age-related changes in the T cell receptor repertoire of human T cell subsets. J. Clin. Investig. 2022, 132, e158122.
- Grolleau-Julius, A.; Harning, E.K.; Abernathy, L.M.; Yung, R.L. Impaired dendritic cell function in aging leads to defective antitumor immunity. Cancer Res. 2008, 68, 6341–6349.
- Behmoaras, J.; Gil, J. Similarities and interplay between senescent cells and macrophages. J. Cell Biol. 2021, 220, e202010162.
- Li, Y.; Liu, Y.; Strickland, F.M.; Richardson, B. Age-dependent decreases in DNA methyltransferase levels and low transmethylation micronutrient levels synergize to promote overexpression of genes implicated in autoimmunity and acute coronary syndromes. Exp. Gerontol. 2010, 45, 312–322.
- Lee, H.Y.; Oh, B.H. Aging and arterial stiffness. Circ. J. 2010, 74, 2257–2262.
- Graver, J.C.; Boots, A.M.H.; Haacke, E.A.; Diepstra, A.; Brouwer, E.; Sandovici, M. Massive B-Cell Infiltration and Organization Into Artery Tertiary Lymphoid Organs in the Aorta of Large Vessel Giant Cell Arteritis. Front. Immunol. 2019, 10, 83.
- van Sleen, Y.; Graver, J.C.; Abdulahad, W.H.; van der Geest, K.S.M.; Boots, A.M.H.; Sandovici, M.; Brouwer, E. Leukocyte Dynamics Reveal a Persistent Myeloid Dominance in Giant Cell Arteritis and Polymyalgia Rheumatica. Front. Immunol. 2019, 10, 1981.
- Iliou, A.; Argyropoulou, O.D.; Palamidas, D.A.; Karagiannakou, M.; Benaki, D.; Tsezou, K.; Vlachoyiannopoulos, P.G.; Mikros, E.; Tzioufas, A.G. NMR-based metabolomics in giant cell arteritis and polymyalgia rheumatica sequential sera differentiates active and inactive disease. Rheumatology 2023, kead590.
- Deng, J.; Ma-Krupa, W.; Gewirtz, A.T.; Younge, B.R.; Goronzy, J.J.; Weyand, C.M. Toll-like receptors 4 and 5 induce distinct types of vasculitis. Circ. Res. 2009, 104, 488–495.
- Krupa, W.M.; Dewan, M.; Jeon, M.S.; Kurtin, P.J.; Younge, B.R.; Goronzy, J.J.; Weyand, C.M. Trapping of misdirected dendritic cells in the granulomatous lesions of giant cell arteritis. Am. J. Pathol. 2002, 161, 1815–1823.
- Ma-Krupa, W.; Jeon, M.S.; Spoerl, S.; Tedder, T.F.; Goronzy, J.J.; Weyand, C.M. Activation of arterial wall dendritic cells and breakdown of self-tolerance in giant cell arteritis. J. Exp. Med. 2004, 199, 173–183.
- Zhang, H.; Watanabe, R.; Berry, G.J.; Vaglio, A.; Liao, Y.J.; Warrington, K.J.; Goronzy, J.J.; Weyand, C.M. Immunoinhibitory checkpoint deficiency in medium and large vessel vasculitis. Proc. Natl. Acad. Sci. USA 2017, 114, E970–E979.
- Pryshchep, O.; Ma-Krupa, W.; Younge, B.R.; Goronzy, J.J.; Weyand, C.M. Vessel-specific Toll-like receptor profiles in human medium and large arteries. Circulation 2008, 118, 1276–1284.
- Reitsema, R.D.; Hesselink, B.C.; Abdulahad, W.H.; van der Geest, K.S.M.; Brouwer, E.; Heeringa, P.; van Sleen, Y. Aberrant phenotype of circulating antigen presenting cells in giant cell arteritis and polymyalgia rheumatica. Front. Immunol. 2023, 14, 1201575.
- Brack, A.; Geisler, A.; Martinez-Taboada, V.M.; Younge, B.R.; Goronzy, J.J.; Weyand, C.M. Giant cell vasculitis is a T cell-dependent disease. Mol. Med. 1997, 3, 530–543.
- Weyand, C.M.; Goronzy, J.J. Medium- and large-vessel vasculitis. N. Engl. J. Med. 2003, 349, 160–169.
- Samson, M.; Audia, S.; Fraszczak, J.; Trad, M.; Ornetti, P.; Lakomy, D.; Ciudad, M.; Leguy, V.; Berthier, S.; Vinit, J.; et al. Th1 and Th17 lymphocytes expressing CD161 are implicated in giant cell arteritis and polymyalgia rheumatica pathogenesis. Arthritis Rheum. 2012, 64, 3788–3798.
- Terrier, B.; Geri, G.; Chaara, W.; Allenbach, Y.; Rosenzwajg, M.; Costedoat-Chalumeau, N.; Fouret, P.; Musset, L.; Benveniste, O.; Six, A.; et al. Interleukin-21 modulates Th1 and Th17 responses in giant cell arteritis. Arthritis Rheum. 2012, 64, 2001–2011.
- Kaiko, G.E.; Horvat, J.C.; Beagley, K.W.; Hansbro, P.M. Immunological decision-making: How does the immune system decide to mount a helper T-cell response? Immunology 2008, 123, 326–338.
- Weyand, C.M.; Younge, B.R.; Goronzy, J.J. IFN-γ and IL-17: The two faces of T-cell pathology in giant cell arteritis. Curr. Opin. Rheumatol. 2011, 23, 43–49.
- Corbera-Bellalta, M.; García-Martínez, A.; Lozano, E.; Planas-Rigol, E.; Tavera-Bahillo, I.; Alba, M.A.; Prieto-González, S.; Butjosa, M.; Espígol-Frigolé, G.; Hernández-Rodríguez, J.; et al. Changes in biomarkers after therapeutic intervention in temporal arteries cultured in Matrigel: A new model for preclinical studies in giant-cell arteritis. Ann. Rheum. Dis. 2014, 73, 616–623.
- Deng, J.; Younge, B.R.; Olshen, R.A.; Goronzy, J.J.; Weyand, C.M. Th17 and Th1 T-cell responses in giant cell arteritis. Circulation 2010, 121, 906–915.
- Samson, M.; Greigert, H.; Ciudad, M.; Gerard, C.; Ghesquière, T.; Trad, M.; Corbera-Bellalta, M.; Genet, C.; Ouandji, S.; Cladière, C.; et al. Improvement of Treg immune response after treatment with tocilizumab in giant cell arteritis. Clin. Transl. Immunol. 2021, 10, e1332.
- Kimura, A.; Kishimoto, T. IL-6: Regulator of Treg/Th17 balance. Eur. J. Immunol. 2010, 40, 1830–1835.
- Samson, M.; Audia, S.; Janikashvili, N.; Ciudad, M.; Trad, M.; Fraszczak, J.; Ornetti, P.; Maillefert, J.F.; Miossec, P.; Bonnotte, B. Brief report: Inhibition of interleukin-6 function corrects Th17/Treg cell imbalance in patients with rheumatoid arthritis. Arthritis Rheum. 2012, 64, 2499–2503.
- Miyabe, C.; Miyabe, Y.; Strle, K.; Kim, N.D.; Stone, J.H.; Luster, A.D.; Unizony, S. An expanded population of pathogenic regulatory T cells in giant cell arteritis is abrogated by IL-6 blockade therapy. Ann. Rheum. Dis. 2017, 76, 898–905.
- Ciccia, F.; Rizzo, A.; Guggino, G.; Cavazza, A.; Alessandro, R.; Maugeri, R.; Cannizzaro, A.; Boiardi, L.; Iacopino, D.G.; Salvarani, C.; et al. Difference in the expression of IL-9 and IL-17 correlates with different histological pattern of vascular wall injury in giant cell arteritis. Rheumatology 2015, 54, 1596–1604.
- Ohtsuki, S.; Wang, C.; Watanabe, R.; Zhang, H.; Akiyama, M.; Bois, M.C.; Maleszewski, J.J.; Warrington, K.J.; Berry, G.J.; Goronzy, J.J.; et al. Deficiency of the CD155-CD96 immune checkpoint controls IL-9 production in giant cell arteritis. Cell Rep. Med. 2023, 4, 101012.
- Watanabe, R.; Maeda, T.; Zhang, H.; Berry, G.J.; Zeisbrich, M.; Brockett, R.; Greenstein, A.E.; Tian, L.; Goronzy, J.J.; Weyand, C.M. MMP (Matrix Metalloprotease)-9-Producing Monocytes Enable T Cells to Invade the Vessel Wall and Cause Vasculitis. Circ. Res. 2018, 123, 700–715.
- Nurieva, R.I.; Chung, Y. Understanding the development and function of T follicular helper cells. Cell. Mol. Immunol. 2010, 7, 190–197.
- Samson, M.; Ly, K.H.; Tournier, B.; Janikashvili, N.; Trad, M.; Ciudad, M.; Gautheron, A.; Devilliers, H.; Quipourt, V.; Maurier, F.; et al. Involvement and prognosis value of CD8(+) T cells in giant cell arteritis. J. Autoimmun. 2016, 72, 73–83.
- Banks, P.M.; Cohen, M.D.; Ginsburg, W.W.; Hunder, G.G. Immunohistologic and cytochemical studies of temporal arteritis. Arthritis Rheum. 1983, 26, 1201–1207.
- Schaufelberger, C.; Andersson, R.; Nordborg, E.; Hansson, G.K.; Nordborg, C.; Wahlström, J. An uneven expression of T cell receptor V genes in the arterial wall and peripheral blood in giant cell arteritis. Inflammation 2008, 31, 372–383.
- Wen, Z.; Shimojima, Y.; Shirai, T.; Li, Y.; Ju, J.; Yang, Z.; Tian, L.; Goronzy, J.J.; Weyand, C.M. NADPH oxidase deficiency underlies dysfunction of aged CD8+ Tregs. J. Clin. Investig. 2016, 126, 1953–1967.
- Cid, M.C.; Campo, E.; Ercilla, G.; Palacin, A.; Vilaseca, J.; Villalta, J.; Ingelmo, M. Immunohistochemical analysis of lymphoid and macrophage cell subsets and their immunologic activation markers in temporal arteritis. Influence of corticosteroid treatment. Arthritis Rheum. 1989, 32, 884–893.
- Lavignac, C.; Jauberteau-Marchan, M.O.; Liozon, E.; Vidal, E.; Catanzano, G.; Liozon, F. . Rev. Med. Interne 1996, 17, 814–820.
- Cid, M.C.; Cervera, R.; Font, J.; Lopez-Soto, A.; Pallarés, L.; Navarro, M.; Ingelmo, M. Late thrombotic events in patients with temporal arteritis and anticardiolipin antibodies. Clin. Exp. Rheumatol. 1990, 8, 359–363.
- Duhaut, P.; Berruyer, M.; Pinede, L.; Demolombe-Rague, S.; Loire, R.; Seydoux, D.; Dechavanne, M.; Ninet, J.; Pasquier, J. Anticardiolipin antibodies and giant cell arteritis: A prospective, multicenter case-control study. Groupe de Recherche sur l’Artérite à Cellules Géantes. Arthritis Rheum. 1998, 41, 701–709.
- Liozon, E.; Roblot, P.; Paire, D.; Loustaud, V.; Liozon, F.; Vidal, E.; Jauberteau, M.O. Anticardiolipin antibody levels predict flares and relapses in patients with giant-cell (temporal) arteritis. A longitudinal study of 58 biopsy-proven cases. Rheumatology 2000, 39, 1089–1094.
- Manna, R.; Latteri, M.; Cristiano, G.; Todaro, L.; Scuderi, F.; Gasbarrini, G. Anticardiolipin antibodies in giant cell arteritis and polymyalgia rheumatica: A study of 40 cases. Br. J. Rheumatol. 1998, 37, 208–210.
- Alard, J.E.; Hillion, S.; Guillevin, L.; Saraux, A.; Pers, J.O.; Youinou, P.; Jamin, C. Autoantibodies to endothelial cell surface ATP synthase, the endogenous receptor for hsp60, might play a pathogenic role in vasculatides. PLoS ONE 2011, 6, e14654.
- Régent, A.; Dib, H.; Ly, K.H.; Agard, C.; Tamby, M.C.; Tamas, N.; Weksler, B.; Federici, C.; Broussard, C.; Guillevin, L.; et al. Identification of target antigens of anti-endothelial cell and anti-vascular smooth muscle cell antibodies in patients with giant cell arteritis: A proteomic approach. Arthritis Res. Ther. 2011, 13, R107.
- Baerlecken, N.T.; Linnemann, A.; Gross, W.L.; Moosig, F.; Vazquez-Rodriguez, T.R.; Gonzalez-Gay, M.A.; Martin, J.; Kötter, I.; Henes, J.C.; Melchers, I.; et al. Association of ferritin autoantibodies with giant cell arteritis/polymyalgia rheumatica. Ann. Rheum. Dis. 2012, 71, 943–947.
- Régent, A.; Ly, K.H.; Blet, A.; Agard, C.; Puéchal, X.; Tamas, N.; Le-Jeunne, C.; Vidal, E.; Guillevin, L.; Mouthon, L. Contribution of antiferritin antibodies to diagnosis of giant cell arteritis. Ann. Rheum. Dis. 2013, 72, 1269–1270.
- Ciccia, F.; Rizzo, A.; Maugeri, R.; Alessandro, R.; Croci, S.; Guggino, G.; Cavazza, A.; Raimondo, S.; Cannizzaro, A.; Iacopino, D.G.; et al. Ectopic expression of CXCL13, BAFF, APRIL and LT-β is associated with artery tertiary lymphoid organs in giant cell arteritis. Ann. Rheum. Dis. 2017, 76, 235–243.
- van der Geest, K.S.; Abdulahad, W.H.; Chalan, P.; Rutgers, A.; Horst, G.; Huitema, M.G.; Roffel, M.P.; Roozendaal, C.; Kluin, P.M.; Bos, N.A.; et al. Disturbed B cell homeostasis in newly diagnosed giant cell arteritis and polymyalgia rheumatica. Arthritis Rheumatol. 2014, 66, 1927–1938.
- van der Geest, K.S.; Abdulahad, W.H.; Rutgers, A.; Horst, G.; Bijzet, J.; Arends, S.; Roffel, M.P.; Boots, A.M.; Brouwer, E. Serum markers associated with disease activity in giant cell arteritis and polymyalgia rheumatica. Rheumatology 2015, 54, 1397–1402.
- Graver, J.C.; Abdulahad, W.; van der Geest, K.S.M.; Heeringa, P.; Boots, A.M.H.; Brouwer, E.; Sandovici, M. Association of the CXCL9-CXCR3 and CXCL13-CXCR5 axes with B-cell trafficking in giant cell arteritis and polymyalgia rheumatica. J. Autoimmun. 2021, 123, 102684.
- Cid, M.C.; Hoffman, M.P.; Hernández-Rodríguez, J.; Segarra, M.; Elkin, M.; Sánchez, M.; Vilardell, C.; García-Martínez, A.; Pla-Campo, M.; Grau, J.M.; et al. Association between increased CCL2 (MCP-1) expression in lesions and persistence of disease activity in giant-cell arteritis. Rheumatology 2006, 45, 1356–1363.
- van Sleen, Y.; Wang, Q.; van der Geest, K.S.M.; Westra, J.; Abdulahad, W.H.; Heeringa, P.; Boots, A.M.H.; Brouwer, E. Involvement of Monocyte Subsets in the Immunopathology of Giant Cell Arteritis. Sci. Rep. 2017, 7, 6553.
- Estupiñán-Moreno, E.; Ortiz-Fernández, L.; Li, T.; Hernández-Rodríguez, J.; Ciudad, L.; Andrés-León, E.; Terron-Camero, L.C.; Prieto-González, S.; Espígol-Frigolé, G.; Cid, M.C.; et al. Methylome and transcriptome profiling of giant cell arteritis monocytes reveals novel pathways involved in disease pathogenesis and molecular response to glucocorticoids. Ann. Rheum. Dis. 2022, 81, 1290–1300.
- Roche, N.E.; Fulbright, J.W.; Wagner, A.D.; Hunder, G.G.; Goronzy, J.J.; Weyand, C.M. Correlation of interleukin-6 production and disease activity in polymyalgia rheumatica and giant cell arteritis. Arthritis. Rheum. 1993, 36, 1286–1294.
- García-Martínez, A.; Hernández-Rodríguez, J.; Espígol-Frigolé, G.; Prieto-González, S.; Butjosa, M.; Segarra, M.; Lozano, E.; Cid, M.C. Clinical relevance of persistently elevated circulating cytokines (tumor necrosis factor alpha and interleukin-6) in the long-term followup of patients with giant cell arteritis. Arthritis Care Res. 2010, 62, 835–841.
- Weyand, C.M.; Fulbright, J.W.; Hunder, G.G.; Evans, J.M.; Goronzy, J.J. Treatment of giant cell arteritis: Interleukin-6 as a biologic marker of disease activity. Arthritis Rheum. 2000, 43, 1041–1048.
- Villiger, P.M.; Adler, S.; Kuchen, S.; Wermelinger, F.; Dan, D.; Fiege, V.; Bütikofer, L.; Seitz, M.; Reichenbach, S. Tocilizumab for induction and maintenance of remission in giant cell arteritis: A phase 2, randomised, double-blind, placebo-controlled trial. Lancet 2016, 387, 1921–1927.
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295.
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238.
- O’Neill, L.; McCormick, J.; Gao, W.; Veale, D.J.; McCarthy, G.M.; Murphy, C.C.; Fearon, U.; Molloy, E.S. Interleukin-6 does not upregulate pro-inflammatory cytokine expression in an ex vivo model of giant cell arteritis. Rheumatol. Adv. Pract. 2019, 3, rkz011.
- Corbera-Bellalta, M.; Alba-Rovira, R.; Muralidharan, S.; Espígol-Frigolé, G.; Ríos-Garcés, R.; Marco-Hernández, J.; Denuc, A.; Kamberovic, F.; Pérez-Galán, P.; Joseph, A.; et al. Blocking GM-CSF receptor α with mavrilimumab reduces infiltrating cells, pro-inflammatory markers and neoangiogenesis in ex vivo cultured arteries from patients with giant cell arteritis. Ann. Rheum. Dis. 2022, 81, 524–536.
- Becher, B.; Tugues, S.; Greter, M. GM-CSF: From Growth Factor to Central Mediator of Tissue Inflammation. Immunity 2016, 45, 963–973.
- Cid, M.C.; Unizony, S.H.; Blockmans, D.; Brouwer, E.; Dagna, L.; Dasgupta, B.; Hellmich, B.; Molloy, E.; Salvarani, C.; Trapnell, B.C.; et al. Efficacy and safety of mavrilimumab in giant cell arteritis: A phase 2, randomised, double-blind, placebo-controlled trial. Ann. Rheum. Dis. 2022, 81, 653–661.
More
Information
Subjects:
Rheumatology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
471
Revisions:
2 times
(View History)
Update Date:
06 Mar 2024
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No