 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | xufeng chen | -- | 2707 | 2024-03-05 18:15:01 | | | |
| 2 | Peter Tang | Meta information modification | 2707 | 2024-03-06 06:55:01 | | |
Video Upload Options
Pathological pain emerges from nociceptive system dysfunction, resulting in heightened pain circuit activity. Various forms of circuitry plasticity, such as central sensitization, synaptic plasticity, homeostatic plasticity, and excitation/inhibition balance, contribute to the malfunction of neural circuits during pain pathogenesis. A new form of plasticity in the spinal dorsal horn (SDH), named neural circuit polarization (NCP), was discovered in pain models induced by HIV-1 gp120 and chronic morphine administration. NCP manifests as an increase in excitatory postsynaptic currents (EPSCs) in excitatory neurons and a decrease in EPSCs in inhibitory neurons, presumably facilitating hyperactivation of pain circuits. The expression of NCP is associated with astrogliosis. Ablation of reactive astrocytes or suppression of astrogliosis blocks NCP and, concomitantly, the development of gp120- or morphine-induced pain.
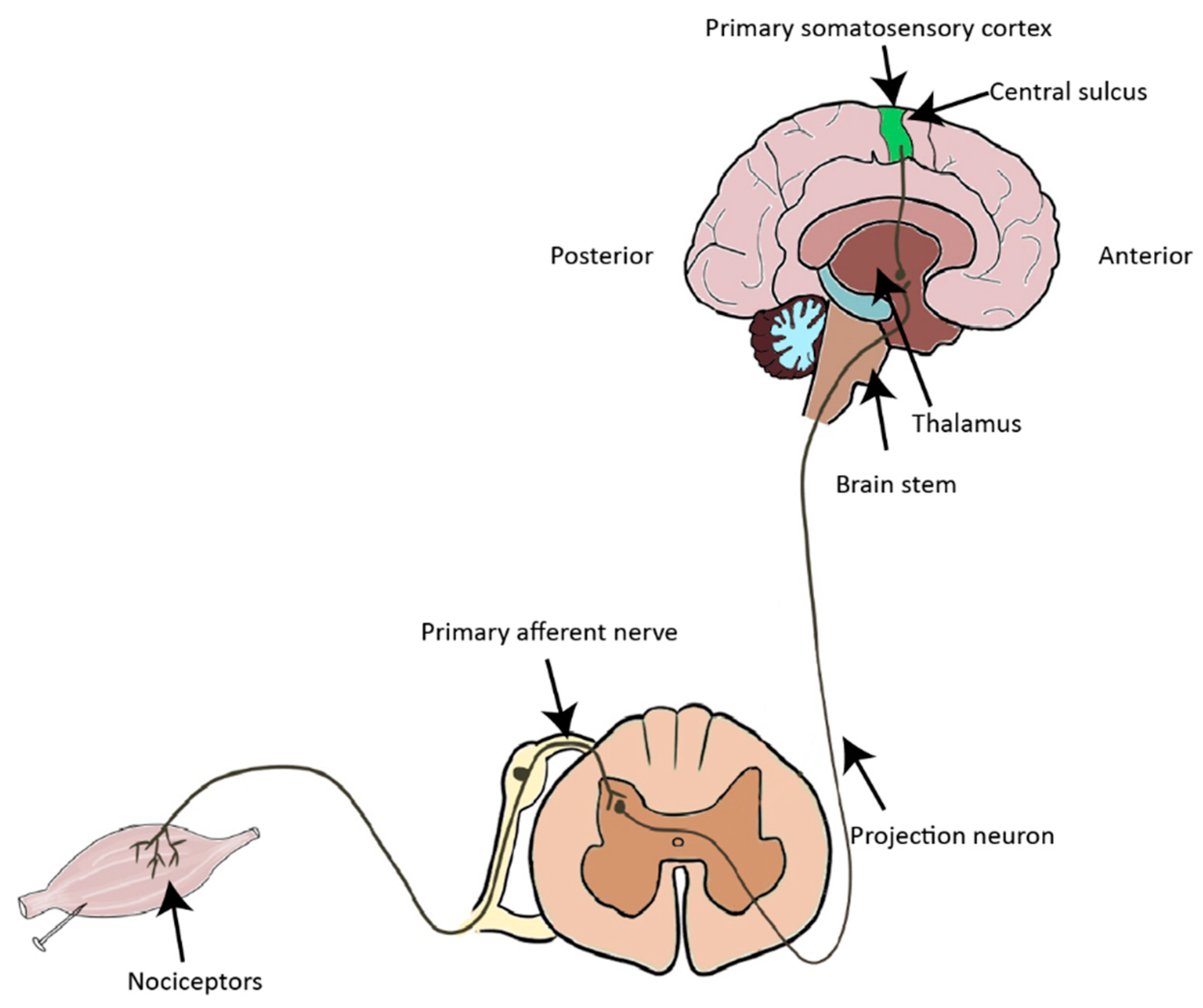
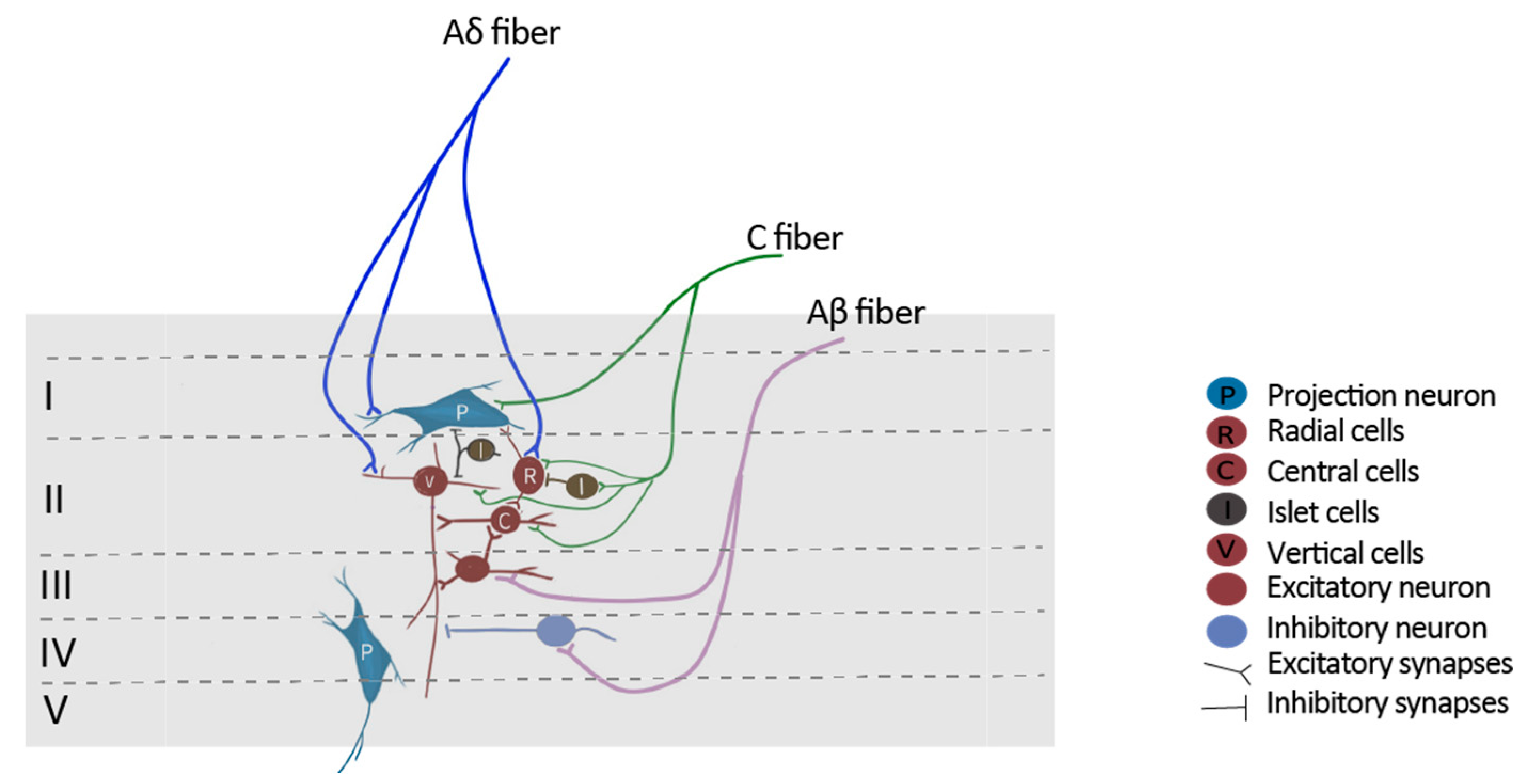
1. Introduction to the Nociceptive Pathway
Microglia and astrocytes are deeply involved in chronic pain. Microglia are macrophage-like cells in the central nervous system, which modulates synaptic transmission via releasing cytokine, chemokine and trophic factors in response to nerve injury (Figure 3). Astrocytes have extensive contacts with synapses, enabling them to regulate the local interstitial ionic and chemical environment during synaptic transmission. In addition, reactive astrocytes also release chemokines and cytokines which modulates synaptic transmission under the situation of pathogenic pain (Figure 3).
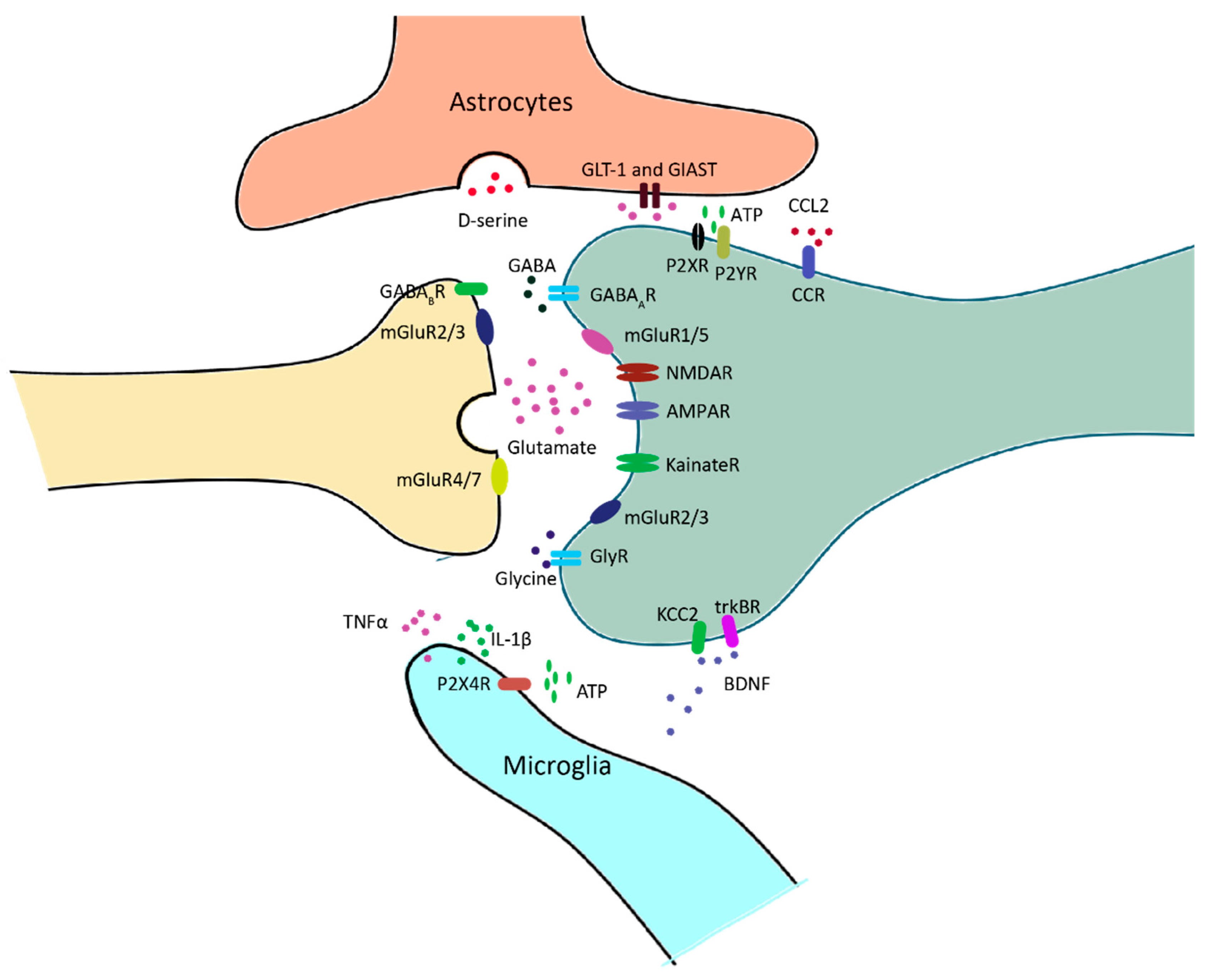
Figure 3. Neuron–glia interactions at nociceptive synapses in the spinal dorsal horn (SDH). Glutamate or GABA/Glycine is released upon the arrival of action potential at the presynaptic fiber. GABA and glycine receptors mediate inhibitory transmission, and glutamatergic receptors mediate excitatory transmission. Ionotropic glutamate receptors, including AMPA receptors, NMDA receptors and kainate receptors, are mainly located in the postsynaptic domain. Metabotropic glutamate receptors are located in both the pre- and postsynaptic domains. Excessive glutamate is taken up by astrocytes and converted to glutamine for the synthesis of glutamate. In response to neuropathy, microglia release a series of inflammatory factors, such as TNFα and IL-1β, which modulate the activity of neurons via a neuro-immune interaction. BDNF is also released by microglia, and it affects neuronal excitability by regulating Cl- homeostasis.
2. Central Sensitization
3. Short-Term Synaptic Plasticity
4. Long-Term Synaptic Plasticity
5. Homeostatic Plasticity/Synaptic Scaling
6. Excitation/Inhibition Balance
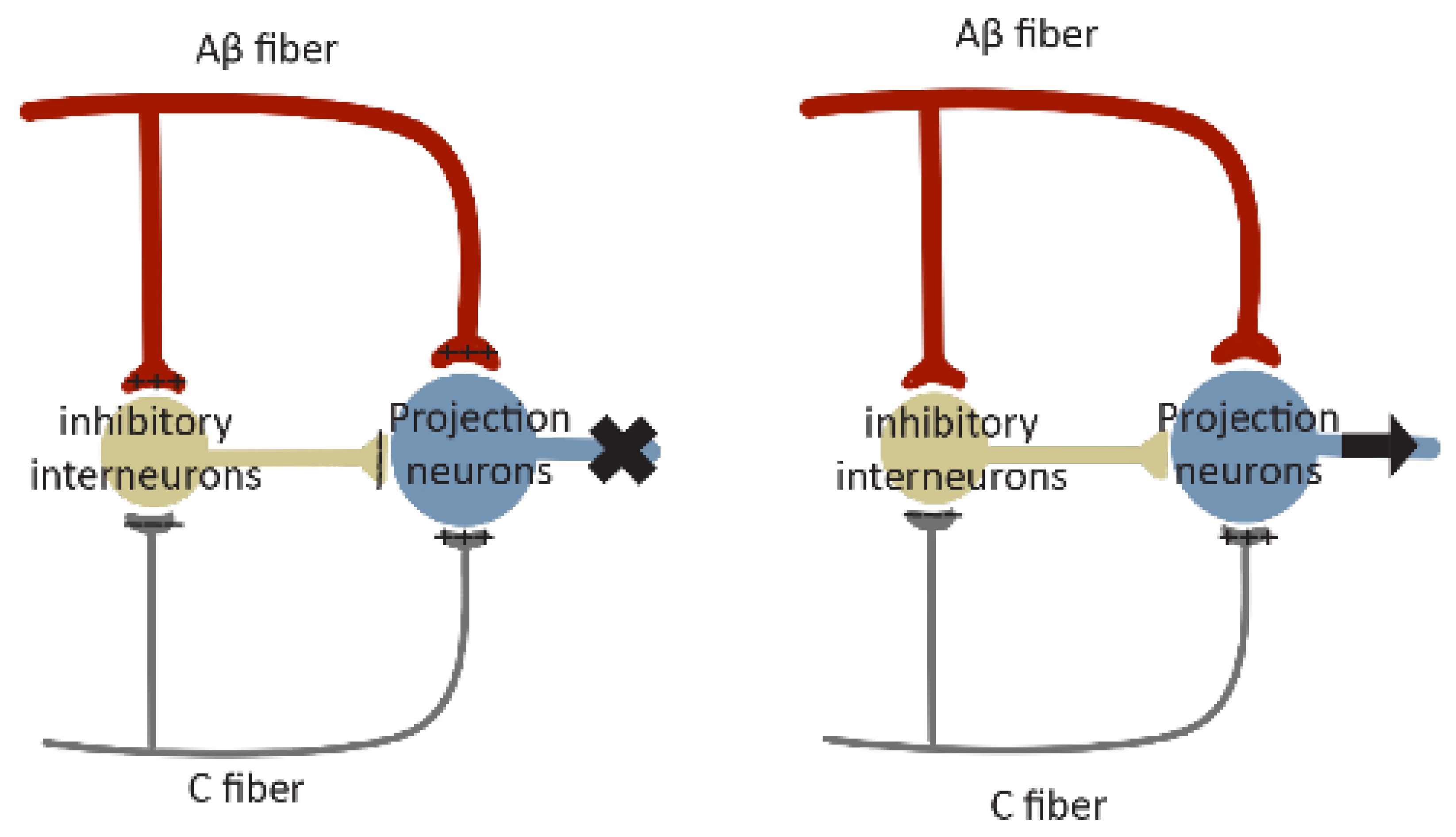
7. Contribution of NCP in Circuits Plasticities of Pain
NCP probably integrates with different types of plasticity in the pain circuits and facilitates their expression. It is tempting to speculate, for instance, that at the initial stage of NCP expression, it may contribute to the development of short-term facilitation, leading to Wind-up. The continuous expression of NCP may contribute to the development of long-lasting synaptic plasticity in pain circuits, such as LTP at excitatory synapses on excitatory neurons and LTD at excitatory synapses on inhibitory neurons. Meanwhile, NCP expressed at both its early and maintenance stages may cause an E/I imbalance, resulting in central sensitization of the pain circuitry. If these conceived integrations are validated, NCP may provide a general circuitry mechanism to support the expression of various forms of circuitry plasticity underlying pain pathogenesis and contribute to specific aspects of pain circuitry malfunction mediated by these forms of plasticity.
References
- Belmonte, C.; Viana, F. Molecular and cellular limits to somatosensory specificity. Mol. Pain 2008, 4, 14.
- Pope, J.E.; Deer, T.R.; Kramer, J. A systematic review: Current and future directions of dorsal root ganglion therapeutics to treat chronic pain. Pain Med. 2013, 14, 1477–1496.
- Jessell, T.M.; Kandel, E.R.; Schwartz, J.H. Principles of Neural Science; McGraw-Hill: New York, NY, USA, 2000; Volume 4, pp. 1227–1246.
- Todd, A.J. Neuronal circuitry for pain processing in the dorsal horn. Nat. Rev. Neurosci. 2010, 11, 823–836.
- Todd, A.J.; Puskar, Z.; Spike, R.C.; Hughes, C.; Watt, C.; Forrest, L. Projection neurons in lamina I of rat spinal cord with the neurokinin 1 receptor are selectively innervated by substance p-containing afferents and respond to noxious stimulation. J. Neurosci. 2002, 22, 4103–4113.
- McCarthy, P.W.; Lawson, S.N. Cell type and conduction velocity of rat primary sensory neurons with substance P-like immunoreactivity. Neuroscience 1989, 28, 745–753.
- Lawson, S.N.; Perry, M.J.; Prabhakar, E.; McCarthy, P.W. Primary sensory neurones: Neurofilament, neuropeptides, and conduction velocity. Brain. Res. Bull. 1993, 30, 239–243.
- Naim, M.; Spike, R.C.; Watt, C.; Shehab, S.A.; Todd, A.J. Cells in laminae III and IV of the rat spinal cord that possess the neurokinin-1 receptor and have dorsally directed dendrites receive a major synaptic input from tachykinin-containing primary afferents. J. Neurosci. 1997, 17, 5536–5548.
- Woolf, C.J. Evidence for a central component of post-injury pain hypersensitivity. Nature 1983, 306, 686–688.
- Latremoliere, A.; Woolf, C.J. Central sensitization: A generator of pain hypersensitivity by central neural plasticity. J. Pain 2009, 10, 895–926.
- Mannion, R.J.; Costigan, M.; Decosterd, I.; Amaya, F.; Ma, Q.P.; Holstege, J.C.; Ji, R.R.; Acheson, A.; Lindsay, R.M.; Wilkinson, G.A.; et al. Neurotrophins: Peripherally and centrally acting modulators of tactile stimulus-induced inflammatory pain hypersensitivity. Proc. Natl. Acad. Sci. USA 1999, 96, 9385–9390.
- Neumann, S.; Doubell, T.P.; Leslie, T.; Woolf, C.J. Inflammatory pain hypersensitivity mediated by phenotypic switch in myelinated primary sensory neurons. Nature 1996, 384, 360–364.
- Baba, H.; Doubell, T.P.; Woolf, C.J. Peripheral inflammation facilitates Abeta fiber-mediated synaptic input to the substantia gelatinosa of the adult rat spinal cord. J. Neurosci. 1999, 19, 859–867.
- Coull, J.A.; Boudreau, D.; Bachand, K.; Prescott, S.A.; Nault, F.; Sik, A.; De Koninck, P.; De Koninck, Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature 2003, 424, 938–942.
- Zieglgansberger, W. Substance P and pain chronicity. Cell Tissue Res. 2019, 375, 227–241.
- Matayoshi, S.; Jiang, N.; Katafuchi, T.; Koga, K.; Furue, H.; Yasaka, T.; Nakatsuka, T.; Zhou, X.F.; Kawasaki, Y.; Tanaka, N.; et al. Actions of brain-derived neurotrophic factor on spinal nociceptive transmission during inflammation in the rat. J. Physiol. 2005, 569, 685–695.
- Lekan, H.A.; Carlton, S.M.; Coggeshall, R.E. Sprouting of A beta fibers into lamina II of the rat dorsal horn in peripheral neuropathy. Neurosci. Lett. 1996, 208, 147–150.
- Mannion, R.J.; Doubell, T.P.; Coggeshall, R.E.; Woolf, C.J. Collateral sprouting of uninjured primary afferent A-fibers into the superficial dorsal horn of the adult rat spinal cord after topical capsaicin treatment to the sciatic nerve. J. Neurosci. 1996, 16, 5189–5195.
- Shortland, P.; Kinman, E.; Molander, C. Sprouting of A-fibre primary afferents into lamina II in two rat models of neuropathic pain. Eur. J. Pain 1997, 1, 215–227.
- Woolf, C.J.; Shortland, P.; Coggeshall, R.E. Peripheral nerve injury triggers central sprouting of myelinated afferents. Nature 1992, 355, 75–78.
- Feng, T. Studies on the neuromuscular junction XXVI. The changes of the end-plate potential during and after prolonged stimulation. Chin. J. Physiol. 1941, 16, 341–372.
- Eccles, J.C.; O’Connor, W.J. Abortive impulses at the neuro-muscular junction. J Physiol 1941, 100, 318–328.
- Stevens, C.F.; Wang, Y. Facilitation and depression at single central synapses. Neuron 1995, 14, 795–802.
- Abbott, L.F.; Varela, J.A.; Sen, K.; Nelson, S.B. Synaptic depression and cortical gain control. Science 1997, 275, 220–224.
- Tsodyks, M.V.; Markram, H. The neural code between neocortical pyramidal neurons depends on neurotransmitter release probability. Proc. Natl. Acad. Sci. USA 1997, 94, 719–723.
- Chen, C.; Blitz, D.M.; Regehr, W.G. Contributions of receptor desensitization and saturation to plasticity at the retinogeniculate synapse. Neuron 2002, 33, 779–788.
- Zucker, R.S.; Regehr, W.G. Short-term synaptic plasticity. Annu. Rev. Physiol. 2002, 64, 355–405.
- Chen, X.; Aslam, M.; Gollisch, T.; Allen, K.; von Engelhardt, J. CKAMP44 modulates integration of visual inputs in the lateral geniculate nucleus. Nat. Commun. 2018, 9, 261.
- LaMotte, R.H.; Campbell, J.N. Comparison of responses of warm and nociceptive C-fiber afferents in monkey with human judgments of thermal pain. J. Neurophysiol. 1978, 41, 509–528.
- Greffrath, W.; Nemenov, M.I.; Schwarz, S.; Baumgartner, U.; Vogel, H.; Arendt-Nielsen, L.; Treede, R.D. Inward currents in primary nociceptive neurons of the rat and pain sensations in humans elicited by infrared diode laser pulses. Pain 2002, 99, 145–155.
- Peng, Y.B.; Ringkamp, M.; Meyer, R.A.; Campbell, J.N. Fatigue and paradoxical enhancement of heat response in C-fiber nociceptors from cross-modal excitation. J. Neurosci. 2003, 23, 4766–4774.
- Shyu, B.C.; Vogt, B.A. Short-term synaptic plasticity in the nociceptive thalamic-anterior cingulate pathway. Mol. Pain 2009, 5, 51.
- Luo, C.; Kuner, T.; Kuner, R. Synaptic plasticity in pathological pain. Trends Neurosci. 2014, 37, 343–355.
- Bliss, T.V.; Lomo, T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol. 1973, 232, 331–356.
- Zhou, L.-J.; Peng, J.; Xu, Y.-N.; Zeng, W.-J.; Zhang, J.; Wei, X.; Mai, C.-L.; Lin, Z.-J.; Liu, Y.; Murugan, M.; et al. Microglia Are Indispensable for Synaptic Plasticity in the Spinal Dorsal Horn and Chronic Pain. Cell Rep. 2019, 27, 3844–3859.e6.
- Ikeda, H.; Stark, J.; Fischer, H.; Wagner, M.; Drdla, R.; Jager, T.; Sandkuhler, J. Synaptic amplifier of inflammatory pain in the spinal dorsal horn. Science 2006, 312, 1659–1662.
- Azkue, J.J.; Liu, X.G.; Zimmermann, M.; Sandkuhler, J. Induction of long-term potentiation of C fibre-evoked spinal field potentials requires recruitment of group I, but not group II/III metabotropic glutamate receptors. Pain 2003, 106, 373–379.
- Zhang, X.C.; Zhang, Y.Q.; Zhao, Z.Q. Involvement of nitric oxide in long-term potentiation of spinal nociceptive responses in rats. Neuroreport 2005, 16, 1197–1201.
- Klein, T.; Magerl, W.; Hopf, H.C.; Sandkuhler, J.; Treede, R.D. Perceptual correlates of nociceptive long-term potentiation and long-term depression in humans. J. Neurosci. 2004, 24, 964–971.
- Wei, F.; Li, P.; Zhuo, M. Loss of synaptic depression in mammalian anterior cingulate cortex after amputation. J. Neurosci. 1999, 19, 9346–9354.
- Kang, S.J.; Liu, M.G.; Chen, T.; Ko, H.G.; Baek, G.C.; Lee, H.R.; Lee, K.; Collingridge, G.L.; Kaang, B.K.; Zhuo, M. Plasticity of metabotropic glutamate receptor-dependent long-term depression in the anterior cingulate cortex after amputation. J. Neurosci. 2012, 32, 11318–11329.
- Rottmann, S.; Jung, K.; Ellrich, J. Electrical low-frequency stimulation induces homotopic long-term depression of nociception and pain from hand in man. Clin. Neurophysiol. 2008, 119, 1895–1904.
- Turrigiano, G. Homeostatic synaptic plasticity: Local and global mechanisms for stabilizing neuronal function. Cold Spring Harb. Perspect. Biol. 2012, 4, a005736.
- Del Rosario, J.S.; McIlvried, L.A.; Pullen, M.Y.; Wangzhou, A.; Sheahan, T.D.; Shepherd, A.J.; Slivicki, R.A.; Price, T.J.; Copits, B.A.; Bertels, Z. Sustained Depolarization Induces Homeostatic Plasticity in Mouse and Human Sensory Neurons. J. Pain 2022, 23, 6.
- Aguilar, J.; Humanes-Valera, D.; Alonso-Calvino, E.; Yague, J.G.; Moxon, K.A.; Oliviero, A.; Foffani, G. Spinal cord injury immediately changes the state of the brain. J. Neurosci. 2010, 30, 7528–7537.
- Cha, M.H.; Kim, D.S.; Cho, Z.H.; Sohn, J.H.; Chung, M.A.; Lee, H.J.; Nam, T.S.; Lee, B.H. Modification of cortical excitability in neuropathic rats: A voltage-sensitive dye study. Neurosci. Lett. 2009, 464, 117–121.
- Boord, P.; Siddall, P.J.; Tran, Y.; Herbert, D.; Middleton, J.; Craig, A. Electroencephalographic slowing and reduced reactivity in neuropathic pain following spinal cord injury. Spinal Cord 2008, 46, 118–123.
- Xiong, W.; Ping, X.; Ripsch, M.S.; Chavez, G.S.C.; Hannon, H.E.; Jiang, K.; Bao, C.; Jadhav, V.; Chen, L.; Chai, Z.; et al. Enhancing excitatory activity of somatosensory cortex alleviates neuropathic pain through regulating homeostatic plasticity. Sci. Rep. 2017, 7, 12743.
- Galhardoni, R.; Correia, G.S.; Araujo, H.; Yeng, L.T.; Fernandes, D.T.; Kaziyama, H.H.; Marcolin, M.A.; Bouhassira, D.; Teixeira, M.J.; de Andrade, D.C. Repetitive transcranial magnetic stimulation in chronic pain: A review of the literature. Arch. Phys. Med. Rehabil. 2015, 96, S156–S172.
- Fregni, F.; Boggio, P.S.; Lima, M.C.; Ferreira, M.J.; Wagner, T.; Rigonatti, S.P.; Castro, A.W.; Souza, D.R.; Riberto, M.; Freedman, S.D.; et al. A sham-controlled, phase II trial of transcranial direct current stimulation for the treatment of central pain in traumatic spinal cord injury. Pain 2006, 122, 197–209.
- Cao, B.; Scherrer, G.; Chen, L. Spinal cord retinoic acid receptor signaling gates mechanical hypersensitivity in neuropathic pain. Neuron 2022, 110, 4108–4124 e6.
- Melzack, R.; Wall, P.D. Pain mechanisms: A new theory. Science 1965, 150, 971–979.
- Antal, M.; Petko, M.; Polgar, E.; Heizmann, C.W.; Storm-Mathisen, J. Direct evidence of an extensive GABAergic innervation of the spinal dorsal horn by fibres descending from the rostral ventromedial medulla. Neuroscience 1996, 73, 509–518.
- Harvey, R.J.; Depner, U.B.; Wässle, H.; Ahmadi, S.; Heindl, C.; Reinold, H.; Smart, T.G.; Schütz, B.; Abo-Salem, O.M.; Zimmer, A.; et al. GlyR alpha3, an essential target for spinal PGE2-mediated inflammatory pain sensitization. Science 2004, 304, 884–887.
- Moore, K.A.; Kohno, T.; Karchewski, L.A.; Scholz, J.; Baba, H.; Woolf, C.J. Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. J. Neurosci. 2002, 22, 6724–6731.
- Scholz, J.; Broom, D.C.; Youn, D.H.; Mills, C.D.; Kohno, T.; Suter, M.R.; Moore, K.A.; Decosterd, I.; Coggeshall, R.E.; Woolf, C.J. Blocking caspase activity prevents transsynaptic neuronal apoptosis and the loss of inhibition in lamina II of the dorsal horn after peripheral nerve injury. J. Neurosci. 2005, 25, 7317–7323.
- Drew, G.M.; Siddall, P.J.; Duggan, A.W. Mechanical allodynia following contusion injury of the rat spinal cord is associated with loss of GABAergic inhibition in the dorsal horn. Pain 2004, 109, 379–388.
- Yaksh, T.L. Behavioral and autonomic correlates of the tactile evoked allodynia produced by spinal glycine inhibition: Effects of modulatory receptor systems and excitatory amino acid antagonists. Pain 1989, 37, 111–123.
- Torsney, C.; MacDermott, A.B. Disinhibition opens the gate to pathological pain signaling in superficial neurokinin 1 receptor-expressing neurons in rat spinal cord. J. Neurosci. 2006, 26, 1833–1843.
- Malan, T.P.; Mata, H.P.; Porreca, F. Spinal GABA(A) and GABA(B) receptor pharmacology in a rat model of neuropathic pain. Anesthesiology 2002, 96, 1161–1167.
- Tucker, A.P.; Mezzatesta, J.; Nadeson, R.; Goodchild, C.S. Intrathecal midazolam II: Combination with intrathecal fentanyl for labor pain. Anesth. Analg. 2004, 98, 1521–1527.
- Lee, K.Y.; Ratte, S.; Prescott, S.A. Excitatory neurons are more disinhibited than inhibitory neurons by chloride dysregulation in the spinal dorsal horn. eLife 2019, 8, e49753.


