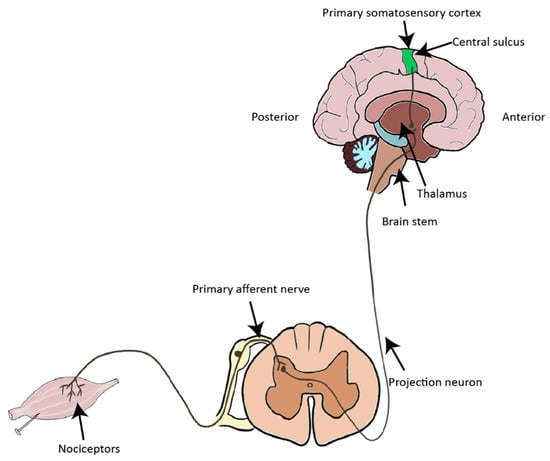
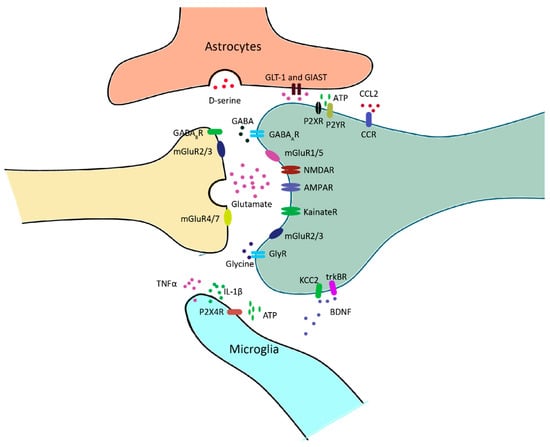
2. Central Sensitization
Although the enlargement of the receptive field of dorsal horn neurons after peripheral nerve injury has long been observed, it is generally thought to be the enhancement of input from silent or ineffective synapses in periphery but not central neuronal plasticity. Clifford J. Woolf first demonstrated a central component of pain hypersensitivity
[9][109]. Instead of recording dorsal horn neurons, he measured the activity of single biceps femoris α-motoneuron axons as the output of the nociceptive signal since the firing of these neurons in response to nociceptive stimulus explicitly leads to the flex or reflex withdrawal response, and the threshold of the withdrawal response is identical to that of activating pain. Thus, the withdrawal responses are used as a surrogate for pain. This study reveals that, under basal conditions, biceps femoris α-motoneurons display a high firing threshold, and their receptive field is restricted to certain regions such as the toes or hind paws. However, repeated peripheral noxious heat stimuli lead to a reduction in threshold and enlargement of the cutaneous receptive fields from the same neurons, and this effect cannot be reversed by local anesthetic block of the peripheral injury site. These data strongly indicate that noxious heat stimulation induced a central plasticity of the nociceptive system (now termed central sensitization), making it respond to stimuli outside of the injury area and to low-threshold afferents that previously carried innocuous stimuli.
Nowadays, central sensitization is defined as a nociceptive neuronal response in the CNS to normal stimulus or excessive activity in response to painful insults. It is usually caused by increased neuronal excitability, synaptic efficacy, and reduced inhibition. The overall effects of central sensitization include hypersensitivity to stimuli (hyperalgesia), responsiveness to non-noxious stimuli (allodynia), and increased pain response evoked by stimuli outside the area of injury.
In the spinal cord, SDH neurons subject to central sensitization show one or more of the following alterations: increases in spontaneous activity, reduction in the threshold for activation, increased responses to suprathreshold stimulation, and enlargement of their receptive fields
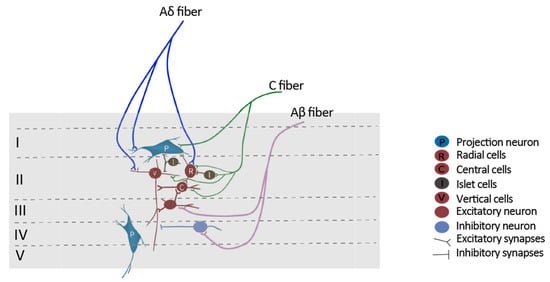
[10][23]. SDH neurons generally receive effective inputs from small, unmyelinated nociceptors as well as ineffective or subthreshold inputs from large, myelinated fibers. In response to peripheral inflammation, large myelinated fibers, such as Aβ-fibers, begin to release substance P and BDNF, which substantially increases the activities of Aβ-fibers as well as the Aβ-mediated synaptic input to superficial dorsal horn neurons
[11][12][13][110,111,112]. Substance P can cause a long-lasting membrane depolarization when binding to NK1 receptors, whereas BDNF can switch inhibitory GABAA receptor-mediated inputs to excitatory
[14][15][72,113]. Continuous stimulation of these fibers will drive central sensitization. Consequently, the previous subthreshold stimulation from mechanic receptors is able to generate the action potential of superficial dorsal horn neurons, therefore leading to allodynia
[16][114]. In addition, peripheral nerve injury might lead to a degeneration of C fiber terminals in lamina II together with regeneration of injured neurons, including Aβ fibers, providing an opportunity for Aβ fibers to sprout from deep lamina into laminae I-II and impinge on nociceptive-specific neurons. This process will lead to the pain response by the stimuli outside the area of injury
[17][18][19][20][115,116,117,118].
Central sensitization is pivotal in the study of chronic pain pathogenesis. However, the ambiguity needs to be clarified. Central sensitization is a phenomenon that can be explained by a series of cellular or molecular mechanisms. In addition, it is not a universal phenomenon for all the neurons in the nociceptive system. For example, it cannot explain the behavior of inhibitory neurons. The activities of inhibitory neurons, as well as inhibitory synapses, are shown to be reduced during chronic pain. Thus, a more general mechanism underlying chronic pain should be investigated.
3. Short-Term Synaptic Plasticity
Short-term plasticity was concurrently recognized by a Chinese and a German group about 80 years ago
[21][22][119,120]. It was first reported to modulate electrical transmission in the neuromuscular junction, but virtually all types of synapses are regulated by a variety of short-lived and long-lasting processes. Short-term plasticity refers to the change in synaptic strength if a synapse is activated repeatedly within a time scale of several milliseconds to a few seconds. Synaptic strength increases during short-term facilitation and decreases during short-term depression
[23][121]. Short-term plasticity is regulated by both pre- and postsynaptic mechanisms
[24][25][26][27][28][122,123,124,125,126].
In the nociceptive system, repetitive heat or mechanical stimuli in a few hundred milliseconds resulted in nociceptor sensitization
[29][30][31][127,128,129]. Moreover, short-term facilitation is also observed in the synapses from the midline and intralaminar thalamic nuclei (MITN) to the anterior cingulate cortex (ACC)
[32][130]. In the spinal cord, Wind Up is one of the hallmarks of neuronal plasticity during persistent pain. It is characterized by an increased excitability of SDH neurons in response to a low-frequency stimulation train of C fibers. Short-term facilitation might lead to the temporal summation of synaptic potentials, which contributes to the formation of Wind Up
[33][131]. However, since short-term plasticity is recovered in a few seconds, this might not account for the maintenance of chronic pain.
4. Long-Term Synaptic Plasticity
Different from short-term plasticity, long-term plasticity reflects the synaptic efficacy change in the range from hours to days. It leads to an enhancement in synaptic efficacy (long-term potentiation, LTP) or a decrease in synaptic efficacy (long-term depression, LTD).
LTP was first observed in the rabbit hippocampus in 1973
[34][132]. Spinal LTP is reported and regarded as a potential mechanism of pain amplification. For example, high-frequency stimulation (HFS) of the sciatic nerve induced LTP at C-fiber synapses and produced chronic pain-like behavior in mice, with microglia being a particularly important contributor
[35][133]. In addition, low-frequency stimulation (LFS), which is closer to the physiological firing rate of C fibers, is also able to induce LTP on NK1-positive neurons projecting to the periaqueductal grey
[36][134]. In either case, fiber stimulation induces the release of substance P, which binds NK1 receptors on projection neurons in SHD. This process directly potentiates NMDA receptors and leads to a substantial rise in postsynaptic Ca
2+—an essential process for the development of LTP
[37][135].
LTP has been shown to be closely associated with chronic pain. For example, spinal LTP induces hyperalgesia in rats
[38][136]. Using a similar stimulation protocol, the pain perception of human beings is potentiated
[39][137]. On the other hand, the role of LTD in chronic pain is also investigated in rodents and humans. LTD in ACC was substantially impaired in the rat with single-paw digit amputation as well as tail amputation
[40][41][138,139]. Applying low-frequency stimulation at 1 Hz—the protocol that is used to induce LTD in the spinal cord or cortex—reduces pain perception in humans
[39][42][137,140].
Long-term plasticity can last for hours or up to days and months, which fits the time course of chronic pain. However, long-term plasticity is homosynaptic, meaning that synaptic strength is selectively enhanced in those synapses receiving repetitive stimuli, whereas in central sensitization, both activated synapses and non-activated neighboring synapses are enhanced. In addition, continuous long-term plasticity results in the endless augmentation of synaptic strength, which makes the synapses unstable.
5. Homeostatic Plasticity/Synaptic Scaling
LTP and LTD are the two forms of Hebbian plasticity, which refers to a form of activity-dependent synaptic plasticity where concurrent activation of pre- and postsynaptic neurons leads to the strengthening (or weakening) of the connection between the two neurons. However, as mentioned above, continuous involvement of Hebbian plasticity leads to an indefinite change in synaptic strength, resulting in instability. To keep neuronal activity at healthy levels, plasticity must incorporate certain homeostatic control mechanisms. Homeostatic plasticity acts globally in a negative feedback manner to counter the change in synaptic strength
[43][141]. Homeostatic plasticity is not synaptic-specific, and all the synapses on the affected neurons will be regulated.
Homeostatic plasticity is an important mechanism underlying chronic pain. It has been shown that sustained depolarization of nociceptors strongly reduces the intrinsic excitability of mouse and human DRG neurons
[44][142]. Moreover, spinal cord injury results in an initial activity loss and subsequent hyperexcitability of cortical neurons, consistent with homeostatic activity regulation
[45][46][47][143,144,145]. Enhancement of cortical activity diminished injury-induced behavioral hypersensitivity in mice with neuropathic pain
[48][146]. In addition, cortical stimulation techniques, which might restore the initial loss of neuronal activity, have been used for patients with refractory neuropathic pain
[49][50][147,148]. In the spinal cord, it is recently reported that nerve injury triggers the homeostatic reduction in inhibitory inputs to excitatory interneurons, causing mechanical hypersensitivity and neuropathic pain
[51][149].
6. Excitation/Inhibition Balance
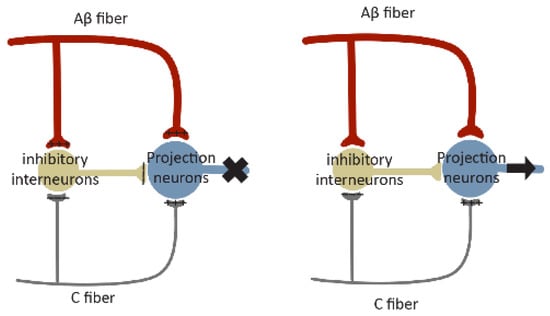
Gate control theory (
Figure 43), formulated in 1965 by Melzack and Wall
[52][150], proposed a role of the ‘gate’ of the spinal cord that controls pain signals to the brain. According to this theory, nociceptors and mechanoreceptors project to the same interneurons and projection neurons in SDH. Both inputs excite projection neurons, but they have opposite effects on inhibitory interneuron activity. Painful signals reduce the activity of inhibitory interneurons, whereas non-painful signals increase their activity. These inhibitory interneurons themselves reduce synaptic transmission between primary afferent and projection neurons. Under normal conditions, mechanoreceptors produce stronger and faster signals than nociceptive stimuli since they are low threshold and their axons are myelinated. In this case, the inhibitory interneurons driven by mechanical signals will block the painful signal, leading to a closed ‘gate’. Loss or reduction of synaptic inhibition will result in an open ‘gate’, allowing pain sensation (
Figure 43).
Figure 43. Schematic diagram of gate control theory. Projection neurons receive excitatory inputs (indicated by ‘+’ signs) from large Aβ fibers and small C fibers, and inhibitory inputs (indicated by ‘−‘ signs) from inhibitory interneurons. Interneurons are excited by Aβ fibers but inhibited by C fibers. The balance of large Aβ fibers and small C fibers determines the output of inhibitory interneurons. When inputs from Aβ fibers are stronger, the inhibitory interneurons are excited, resulting in a closed ‘gate’ to inhibit projection neuron activation (indicated by ‘X’ in the
left panel). When the inhibitory inputs from C fibers are stronger, the activity of inhibitory interneurons will be reduced, leading to an open ‘gate’ to permit projection neuron activation (indicated by the arrow in the
right panel).
The spinal cord receives inhibitory inputs, mainly from inhibitory interneurons and the supraspinal regions
[53][151]. Disruption of synaptic inhibition is a common feature of chronic pain resulting from inflammation
[54][152] and nerve injury
[14][55][56][57][42,72,153,154]. For example, GABAergic synaptic transmission is diminished in the spinal cord of neuropathic rats
[55][42]. In addition, peripheral nerve injury results in apoptosis of inhibitory interneurons, which substantially reduce GABAergic and glycinergic inhibitory currents, leading to a state of disinhibition
[56][153]. Intrathecal administration of the GABAA receptor antagonist bicuculline or the glycine receptor antagonist strychnine induces pain behavior
[58][59][155,156], which is partially due to the facilitated low-threshold input in response to a reduced inhibition. Accordingly, the application of benzodiazepines—agonists of GABAA receptors—exerts clear analgesic actions in animal models of hyperalgesia, as well as in human patients
[56][60][61][153,157,158]. Interestingly, although both excitatory and inhibitory neurons receive inhibitory inputs, excitatory neurons rely more heavily on inhibition. They are, therefore, more affected by disinhibition
[62][159].
7. Contribution of NCP in Ccircuits Pplasticities of Ppain
NCP probably integrates with different types of plasticity in the pain circuits and facilitates their expression. It is tempting to speculate, for instance, that at the initial stage of NCP expression, it may contribute to the development of short-term facilitation, leading to Wind-up. The continuous expression of NCP may contribute to the development of long-lasting synaptic plasticity in pain circuits, such as LTP at excitatory synapses on excitatory neurons and LTD at excitatory synapses on inhibitory neurons. Meanwhile, NCP expressed at both its early and maintenance stages may cause an E/I imbalance, resulting in central sensitization of the pain circuitry. If these conceived integrations are validated, NCP may provide a general circuitry mechanism to support the expression of various forms of circuitry plasticity underlying pain pathogenesis and contribute to specific aspects of pain circuitry malfunction mediated by these forms of plasticity.