1. Severe Acute Respiratory Syndrome SARS Proteins
Coronavirus Disease 2019 (COVID-19) is a highly transmissible viral infection caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The global impact of COVID-19 has been devastating, resulting in over 6 million deaths worldwide. Initially reported in Wuhan, Hubei Province, China in late December 2019, SARS-CoV-2 swiftly spread across the globe, prompting the World Health Organization to declare it a global pandemic on 11 March 2020. In 2020, COVID-19 ranked as the third leading cause of death in the United States after heart disease and cancer, accounting for approximately 375,000 deaths
[1].
Few glycoproteins have captured as much attention or undergone more detailed investigations than the SARS proteins. Studies have unveiled how glycans can function as shields preventing recognition of viral proteins by the immune system
[2] and how they may serve as activators for the lectin pathway
[3].
In terms of structure and phylogeny, SARS-CoV-2 exhibits similarities to both SARS-CoV and Middle East Respiratory Syndrome Coronavirus (MERS-CoV). It comprises four primary structural proteins: spike (S), envelope (E) glycoprotein, nucleocapsid (N), and membrane (M) protein, along with 16 nonstructural proteins and 5–8 accessory proteins
[4]. The surface spike glycoprotein, resembling a crown, is a threefold symmetric homo-trimer
[5], where each protein contains approximately 1200 residues (
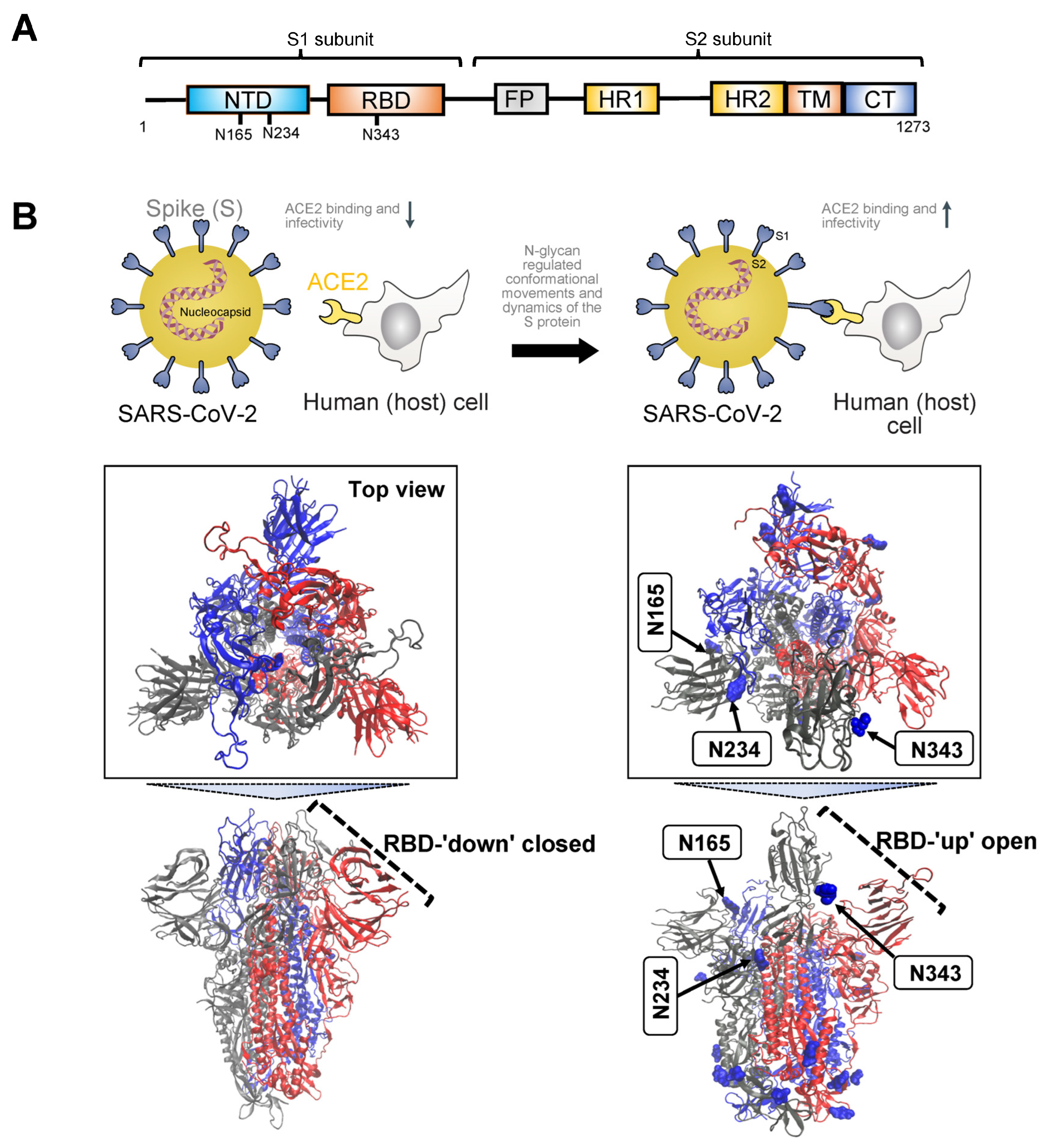
Figure 1A). The spike is positioned on the virion’s outer surface. It undergoes cleavage into an amino N-terminal S1 subunit, facilitating virus incorporation into the host cell. The carboxyl C-terminal S2 subunit includes a fusion peptide (FP), a transmembrane domain (TM), and a cytoplasmic domain crucial for virus–cell membrane fusion
[6]. The S1 subunit is further divided into a receptor-binding domain (RBD) and an N-terminal domain (NTD), playing roles in viral entry and serving as a potential target for neutralization by antisera or vaccines
[7]. The RBD is pivotal in infection pathogenesis as it binds to the human angiotensin-converting enzyme 2 (ACE2) receptors in the respiratory epithelium. Following viral attachment, the spike protein S2 subunit is primed by the host transmembrane serine protease 2, facilitating cell entry and subsequent viral replication
[8].
Figure 1. (A). Schematic illustrating the primary structure of the full-length SARS-CoV-2 spike (S) protein with color-coded domains: N-terminal domain (NTD), receptor binding domain (RBD), fusion peptide (FP), heptad repeat 1 and 2 (HR1, HR2) domain, transmembrane domain (TM), and cytoplasmic tail (CT). The S protein comprises two subunits, S1 and S2. Asparagine residues crucial for N-glycosylation regulation of S binding to the angiotensin-converting enzyme 2 (ACE2) receptor on the host cell are highlighted based on their sequence position. (B). Illustration depicting the mechanism of trimeric S glycoprotein binding to human ACE2, initiated by at least one protomer’s RBD switching from a ‘down’ (closed) state (unfavorable for ACE2 binding) to an ‘up’ (open) state (favorable for ACE2 binding). The S protein is represented by cartoons, with individual protomers colored in cyan, red, and gray. Both the ‘down’ closed state (PDB: 7ZH2) and ‘up’ open state (PDB: 7ZH5) of the S protein RBD are shown. Glycans are depicted in blue using Van der Waals representation. N-glycans at positions N165 and N234 are identified as essential structural elements for maintaining the SARS-CoV-2 spike protein in a conformation conducive to ACE2 recognition, facilitating subsequent viral entry into host cells. The glycan at N343 lifts and stabilizes the RBD throughout the opening transition. Visualizations of the S protein were generated using Visual Molecular Dynamics (v1.9.4a55).
Initiation of the binding process between the trimeric S glycoprotein and human ACE2 is marked by the transition of at least one protomer’s RBD from a ‘down’ (closed) to an ‘up’ (open) state
[9][10][11] (
Figure 1B). This dynamic conformational change involves the transient interconversion of states through a hinge-like motion exposing the receptor binding motif (RBM), which is composed of RBD residues S438 to Q506
[12]. The RBM is buried in the inter-protomer interface of the down S protein; therefore, binding to ACE2 relies on the stochastic interconversion between the ‘down’ and ‘up’ states. The spike S has 22 predicted N-glycosylation sites per protomer, with at least 17 experimentally demonstrated to be occupied
[2][13]. Among these, the S1 subunit features 13 putative N-glycosites (N17, N61, N74, N122, N149, N165, N234, N282, N331, N343, N603, N616, and N657), each with the N-X-S/T (X ≠ P) sequon, and one putative N-glycosite (N334) with the N-X-C (X ≠ P) sequon. The S2 subunit, on the other hand, includes nine putative N-glycosites (N709, N717, N801, N1074, N1098, N1134, N1158, N1173, and N1194), all exhibiting the N-X-S/T (X ≠ P) sequon
[14]. Several of these sites play a crucial role in regulating the conformational movements of the protein as well as its dynamics, and, in turn, they have an impact on binding to ACE2 and on infectivity
[2][12][15][16][17][18][19][20] (
Figure 1B).
MD simulations have revealed detailed information about the structural stability and the role of glycosylation for both the ‘down’ and ‘up’ states, as well as for inter-residue interactions and details of binding to ACE2. A study investigating the SARS-CoV-2 spike protein has emphasized the functional significance of glycans at N165 and N234 in regulating the ‘up’ and ‘down’ conformational states of the spike
[2]. Through multiple microsecond-long, all-atom, explicitly solvated MD simulations of the full-length SARS-CoV-2 S glycoprotein with a complete glycosylation profile consistent with glycomic data, the study has unveiled a crucial structural role of N-glycans linked to N165 and N234 in modulating the conformational transitions of the RBD. When the RBD transitioned to the ‘up’ state, the glycan at N234 rotated into the resulting void, stabilizing the ‘up’ conformation. Simulating the deletion of these glycans via N165A and N234A mutations resulted in a destabilizing effect on the RBD, prompting a conformational shift toward the ‘down’ state (i.e., a state unfavorable for ACE2 receptor binding). This altered RBD conformation substantially reduced binding to ACE2, as confirmed by biolayer interferometry experiments. Consequently, the specific N-glycans at positions N165 and N234 have been identified as essential structural elements for maintaining the SARS-CoV-2 spike protein in a conformation conducive to ACE2 recognition, facilitating subsequent viral entry into host cells.
To explore the pathway of the fully glycosylated SARS-CoV-2 spike protein opening its RBD, a study employed the weighted ensemble (WE) path-sampling strategy, allowing for atomistic simulations of the spike-opening process
[21]. WE, as a path-sampling strategy, directs computational resources toward the transitions between stable states rather than the stable states themselves
[22]. This is achieved by running multiple trajectories concurrently and periodically duplicating trajectories that transition between previously and newly visited regions of configurational space. This minimizes the time spent waiting in the initial stable state for transitions over the free energy barrier. The extensive WE MD simulations of the glycosylated SARS-CoV-2 spike head, characterizing the transition from the ‘down’ to ‘up’ conformation of the RBD, revealed a significant gating role for the glycan at N343. This glycan lifted and stabilized the RBD throughout the opening transition. The study also identified an ‘open’ state of the spike RBD, where the N165 glycan of chain B remained the last contact with the RBD on the route to further opening of S1. In conjunction with prior studies by Casalino et al.
[2], the research underscored the crucial role of N343 in gating and facilitating the RBD-opening process, emphasizing the necessity of sampling functional transitions for a comprehensive understanding of mechanistic details.
Pang et al. elucidated two-dimensional free-energy landscapes depicting the opening and closing transitions of the SARS-CoV-2 S protein, considering both glycosylated and un-glycosylated forms
[23]. The study emphasized the influence of glycans on each state and their role in modifying the kinetics of spike opening. It introduced a nuanced perspective on the role of glycans, suggesting a more intricate impact than previously recognized, especially regarding the glycans at N165 and N343. According to the research, these glycans may affect both the ‘down’ and ‘up’ states, creating a local energy minimum for each. Specifically, the study proposed that these glycans could wrap around the RBM when the RBD is in the ‘down’ state, effectively maintaining it in that configuration. Consequently, these two glycans at N165 and N343 were identified as contributors to stabilizing both ‘down’ and ‘up’ states, establishing a local energy minimum for each.
Glycans also contribute to infectivity, specifically the fusion peptide’s ability to capture the host cell. In the process of spike-protein-mediated fusion, the fusion peptides need to be released from the protein core and associate with the host membrane. Successful infection depends on the transition between pre-fusion and post-fusion conformations. To mechanistically describe this pre-to-post rearrangement and understand the impact of glycans, a study conducted thousands of simulations using an all-atom model with simplified energetics
[24]. These simulations revealed that the steric composition of the glycans can induce a pause during the conformational change of the spike protein. This glycan-induced delay presents a crucial opportunity for fusion peptides to effectively capture the host cell, a process that would be inefficient in the absence of glycans. Therefore, the steric composition of both the spike protein and glycans may guide the overall dynamics of host–membrane capture.
In sum, the SARS-CoV-2 S protein undergoes conformational changes crucial for host cell entry (Figure 1). Key N-glycosylation sites stabilize the ‘up’ conformation, facilitating ACE2 recognition and viral entry. Select N-glycans act in gating and stabilizing the S protein during conformational transitions. Furthermore, N-glycans actively contribute to SARS-CoV-2 infectivity by influencing the dynamics of the fusion peptide. This peptide is instrumental in capturing the host cell during the spike-protein-mediated fusion process. The steric composition of glycans induces strategic pauses in conformational changes, creating vital windows for efficient host cell capture. To sum up, strategically positioned N-glycans intricately regulate the conformation and dynamic movements of the S protein. This regulation, in turn, dictates its ability to attach to the ACE2 receptor, enter the cell, and initiate infection in the host.
2. Prion Protein
Prion diseases, or transmissible spongiform encephalopathies (TSE), are a class of fatal, infectious neurodegenerative illnesses that affect both humans and animals. Among human prion diseases are Creutzfeldt–Jakob Disease (CJD), Variant Creutzfeldt–Jakob Disease (vCJD), Gerstmann–Straussler–Scheinker Syndrome, Fatal Familial Insomnia, and Kuru. Most prevalent in animals are bovine spongiform encephalopathy (BSE) (or Mad Cow Disease) in cattle, chronic wasting disease (CWD) in deer, elk, and moose, and scrapie in sheep
[25][26][27]. These diseases occur when a normally occurring protein, the prion protein, undergoes a pathogenic transformation, where the key factor driving the pathology is a conformational change.
Prion diseases are caused by the conformational rearrangement of the endogenous cellular prion protein (PrP
C) into an abnormal, toxic form (PrP
Sc)
[28]. PrP
C, an
N-glycosylated protein, is tethered to the outer leaflet of the plasma membrane through a glycosyl phosphatidylinositol (GPI) moiety
[29][30]. While expressed across various tissues, it is particularly enriched in neurons, making them the primary sites susceptible to TSE-related degeneration
[31]. N-Glycosylation influences the conformational stability of PrP
[32][33][34][35][36]. Changes in glycosylation patterns can impact the folding kinetics of PrP and may contribute to the transition from the normal, cellular form to the disease-associated form. Different glycoforms may exhibit variations in their susceptibility to conversion into the pathological PrP
Sc form, affecting the progression of prion diseases. The impact of the glycans, or the lack of, however, may be strain-dependent, and the conversion of PrP
C into PrP
Sc may be sustained through several pathways depending on the origin of the disease
[32][33][34][35][36]. In the context of infectious TSE, it is proposed that exogenous PrP
Sc interacts with PrP
C, acting as a template for the conversion of the latter into an additional PrP
Sc molecule. In familial prion diseases, mutations within the prion protein gene are suggested to facilitate the folding of PrP into a pathogenic conformation
[32][37][38][39].
As anticipated for a membrane protein, the newly synthesized 254-amino acid PrP
C undergoes cleavage of its hydrophobic N-terminal signal peptide (residues 1–23) to enable its targeting to the rough endoplasmic reticulum, the initial compartment of the secretory pathway. Within this compartment, co- and post-translational modifications of PrP
C take place, including the addition of high mannose-type N-glycans (Glc
3–Man
9–GlcNAc
2) at positions Asn181 and Asn197 in human
(huPr
P) and Syrian hamster (
ShPr
P) PrP
C (corresponding to Asn180 and Asn196 in mouse,
MoPr
P), formation of a unique disulfide bond, and attachment of a GPI moiety following cleavage of the hydrophobic C-terminal signal peptide. The glycosylation sites of PrP
C exhibit variable occupancy, resulting in non-, mono-, or di-glycosylated forms
[30][32][33][36]. The N-terminal segment (residues 23–125, after removal of the signal peptide) exhibits significant flexibility. This segment encompasses an octapeptide repeat domain capable of binding divalent ions, such as Cu
2+ and Zn
2+. In contrast, the C-terminal domain (residues 126–230), housing the glycan attachment sites, adopts a distinctive structure characterized by three α-helices, labeled as helices one through three (one through three or A through C) and two anti-parallel β-strands flanking helix 1 (
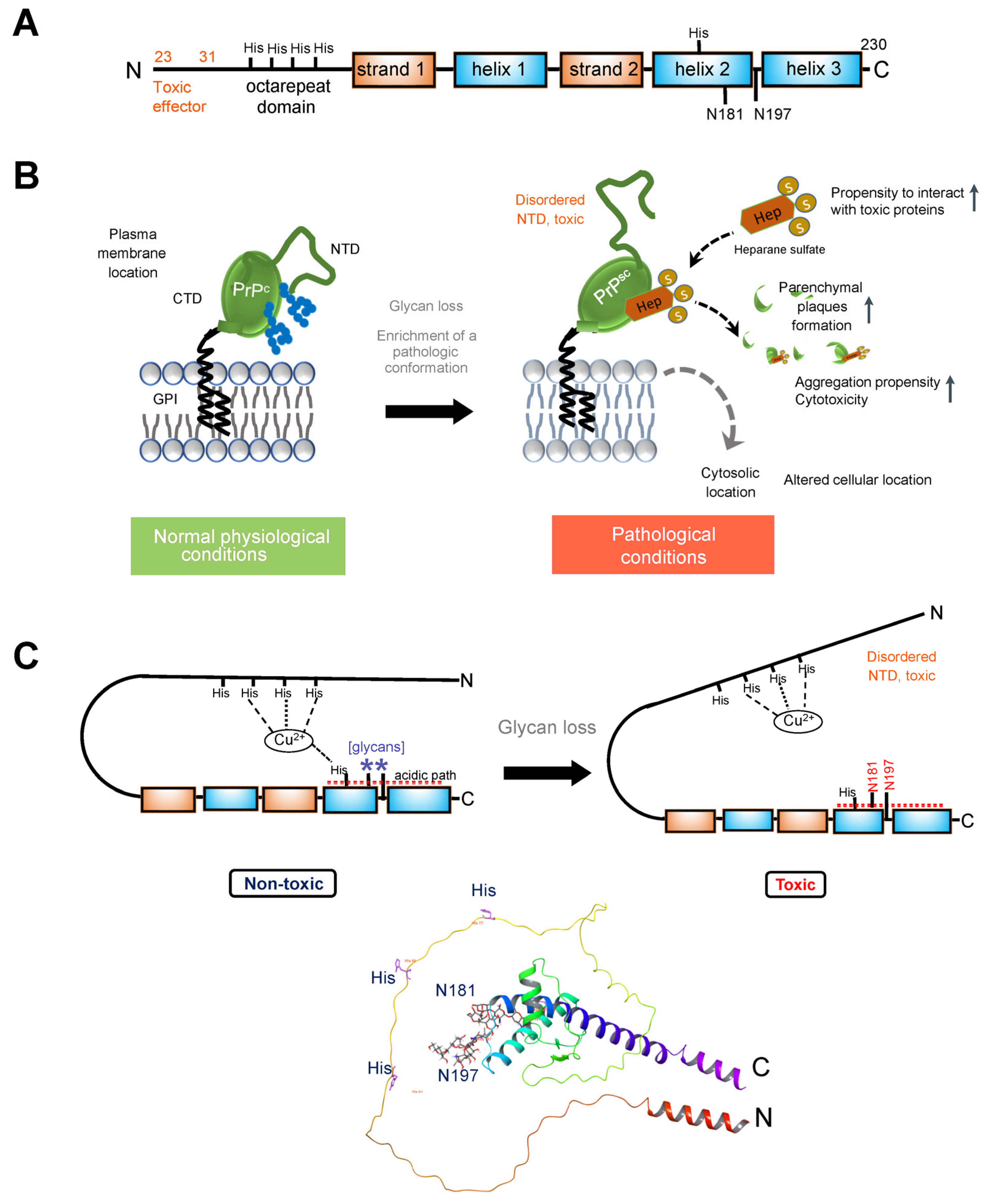
Figure 2A).
Figure 2. (A). Schematic illustrating the primary structure of the full-length prion (PrPc) protein. The N-terminal segment (residues 23–125, after removal of the signal peptide) exhibits a high degree of flexibility. Within this segment is an octapeptide repeat domain that binds divalent ions. The C-terminal domain (residues 126–230) folds to a characteristic structure composed of three α-helices, numbered one through three (or A through C), and two anti-parallel β-strands flanking helix 1. PrPc is modified by N-glycans at positions Asn181 and Asn197 in the human protein (corresponding to Asn180 and Asn196 in the mouse protein). Several histidine residues implicated in regulating PrPc stability are also indicated. (B). PrPC is attached to the outer leaflet of the plasma membrane through a glycosyl phosphatidylinositol (GPI) moiety. Loss of glycans destabilizes the prion protein structure, enriching it in a conformation that enables pathologic interactions (such as with heparane sulfate). The fewer glycosylation modifications PrP undergoes, the more likely it is to be located in the cytoplasm and the stronger its proteolytic resistance, toxicity, and aggregation ability. (C). Top, cartoon showing how glycans stabilize the PrP protein. Glycosylation at Asn197 may have an allosteric effect, with impact on participation in stabilizing a stable conformation of the protein. N181 may act in concert with other residues to anchor the disordered NTD to its regulatory CDT. Specifically, Histidine residues, acting through Cu2+ coordination, glycans at Asn181, and acidic residues (see acidic patch) may act in concert to anchor the toxic effector N-terminal domain to its regulatory site on the C-terminal domain. Bottom, ribbon representation of the PrPC showing the position of the two glycans (sticks). AlphaFold-generated structure using the human prion protein sequence (UNIPROT ID: P04156).
In cellulo and in vivo studies support the role of these glycans in modulating a pathologic conformation and function of PrP
[32][40][41][42] (
Figure 2B). For example, since
ShPrP contains complex-type oligosaccharides attached to Asn residues 181 and 197 (as in
HuPrP), an early study mutated the Thr residues to Ala within the NXT consensus sites
[43]. This substitution disrupts the specific motif required for N-glycosylation, hindering the proper attachment of glycan moieties to the protein. Single and double glycosylation site mutations were expressed in transgenic mice deficient for mouse
MoPrP, and the brains were analyzed for the distribution of mutant
ShPrP
C. The analysis focused on the hippocampal region in each case. Wild-type
ShPrP
C was predominantly found in the dendritic trees of the CA1 to CA4 regions of Ammon’s horn and the dentate gyrus. It was notably absent from the cell bodies of pyramidal and granule cell layers in these regions and largely absent from white matter tracts like the corpus callosum. In contrast, mutation in either one or both glycosylation consensus sites had a significant impact on the anatomical distribution of
ShPrP
C. Mutation of the first glycosylation site alone or in combination with the second site led to low levels of mutated
ShPrP
C, its accumulation in nerve cell bodies, and limited presence in dendritic trees. When the second glycosylation site was mutated,
ShPrP
C levels were comparable to wild-type
ShPrP
C. This mutant protein, however, was observed throughout all neuronal compartments, including the cell body, dendritic tree, and axons in the white matter. Transgenic mice with inactivation at the second Asn197 site (T199A) supported prion replication upon infection, while mice mutated at the first site appeared resistant
[29][43].
A recent study employed knock-in mouse models expressing cell surface PrP
C with zero or two N-glycans and several complementary approaches to address the impact of glycosylation on prion protein localization and function
[36]. Mice expressing PrP
C without glycosylation were generated through the introduction of two-point mutations at the endogenous
Prnp locus using a single guide RNA. These mutations, corresponding to the substitution of asparagine to glutamine at positions 180 and 196 (in accordance with mouse PrP numbering), led to alterations in the N-glycosylation sequons. PrP(180Q196Q)) mice exhibited normal expression and trafficking of PrP
C with no evidence of spontaneous prion disease. However, a significant difference in susceptibility to prion infection was observed between PrP(180Q196Q) mice and wild-type mice. Notably, the PrP(180Q196Q) mice consistently showed more severe spongiform degeneration across all strains compared to wild-type mice. Additionally, upon prion infection, these mice displayed marked atrophy of the hippocampus due to severe neuronal loss, including complete loss of CA1 pyramidal neurons and the presence of numerous gemistocytic astrocytes, reflecting the enlarged and filled appearance of the cell. Gemistocytic astrocytes are often associated with certain pathological conditions. This effect persisted upon second passage. In contrast, wild-type mice exhibited moderate loss of hippocampal neurons. Furthermore, the cerebellum of all infected PrP(180Q196Q) mice lacked PrP
Sc, a notable distinction from wild-type mice where all three strains were present in the cerebellum. Importantly, the absence of PrP
Sc in the cerebellum of PrP(180Q196Q) mice was not attributed to a lack of PrP
C expression, as PrP(180Q196Q) was expressed in the cerebellum at levels similar to wild-type PrP. The morphology of PrP
Sc in PrP(180Q196Q) brains differed from wild-type mice, as most PrP(180Q196Q) brains displayed either plaque-like deposits or dense parenchymal plaques. Consequently, PrP(180Q196Q) mice when exposed to subfibrillar prion strains manifested distinct disease characteristics, including an elevated presence of plaques and plaque-like structures, more pronounced cortical spongiosis, and a conspicuous absence of prions in the cerebellum.
A study by Yi and colleagues
[41] explored the impact of glycosylation on various aspects of PrP using cultured cells expressing wild-type PrP and glycosylation mutants. The study found that glycosylation significantly influenced the subcellular localization, resistance to proteolytic digestion, and aggregation ability of human PrP. Wild-type PrP and monoglycosylated mutants—N181D, N197D, and T199N/N181D/N197D—were primarily attached to the plasma membrane, while pathological mutants with altered glycosylation sites (i.e., PrP F198S) and unglycosylated PrP mutants (i.e., N181D/N197D) were mainly present in the cytoplasm. Furthermore, the study revealed that the degree of glycosylation correlated with the protein’s proteolytic resistance and aggregation ability. PrP with fewer glycosylation modifications exhibited higher aggregation propensity and a higher degree of abnormal conformers, as measured by its resistance to protease digestion. Additionally, glycosylation deficiency increased the vulnerability of the protein to stressors and enhanced its cytotoxicity.
In the context of understanding the impact of N-glycans, it is crucial to underscore the role of protein conformational changes and their resulting pathologic functions, which are intricately linked to how the affected protein assembles and interacts with other proteins. Glycosaminoglycans, especially heparan sulfate and heparin, are considered crucial molecules for prion conversion and infection. For instance, in prion diseases, heparan sulfate, a prevalent polyanion in the brain, accelerates disease progression by facilitating the conversion and assembly of extracellular, ADAM10-cleaved PrP into parenchymal plaques
[44]. In mice, di-glycosylated PrP
Sc demonstrated the least heparan sulfate binding, while unglycosylated PrP
Sc exhibited the highest heparan sulfate binding
[36]. Similarly, glycans also disrupt PrP binding to heparin, with di-glycosylated PrP
C exhibiting the lowest affinity for heparin binding
[32]. Notably, PrP
C with two to three glycans displayed low heparin affinity, while unglycosylated PrP
C showed high affinity, with this affinity progressively decreasing with each additional glycan. The heparin-binding affinity of PrP
C from age-matched wild-type and PrP(180Q196Q) mouse brain homogenates followed a similar pattern—unglycosylated PrP
C exhibited higher heparin-binding affinity than glycosylated wild-type PrP
C, a trend also observed in their ADAM10-cleaved counterparts
[36].
Structural and biochemical studies have provided explanations for how these glycans may affect the prion protein. From early NMR structures of the full-length and N-terminally-truncated forms of recombinant
MoPrP,
ShPrP, and
HuPrP, it became known that the entire N-terminal segment (residues 23–126) is flexibly disordered and that only the C-terminal part (residues 127–231) possesses a defined 3D structure
[45][46]. MD simulations on the C-terminal region of
HuPrP (residues 90–230), with and without the glycans, suggested the structured part of the
HuPrP protein (residues 127–227) was stabilized overall from addition of the glycans, specifically by extensions of Helix-B (i.e., helix 2) and Helix-C (i.e., helix 3) and reduced flexibility of the linking turn containing Asn197. The stabilization appeared indirect and not from specific interactions, such as H bonds or ion pairs. Thus, glycosylation at Asn197 has an allosteric effect, with an impact on stabilizing a conformation of the protein
[47].
Recent biochemical studies have provided additional insights into the role of glycans in the prion protein. NMR and electron paramagnetic resonance spectroscopy studies suggest that the two N-glycans play a crucial role in maintaining an intramolecular interaction, effectively bringing together the C- and the N-terminal domains, as elaborated further
[42][48][49]. This interaction is mediated through a Cu
2+–histidine tether, bringing the C- and N-terminal domains into proximity and likely stabilizing the overall structure while reducing dynamic motion (
Figure 2C). Additionally, PrP glycans at N181 and N197 actively promote the interaction between the N-terminal and C-terminal domains, synergizing with the effect of His-Cu
2+ coordination
[42][50]. A patch of negatively charged amino acids on the same protein surface as the histidines and glycans serves as a third contributor to these interactions. These interactions play a functional role in suppressing the neurotoxic activity of PrP
C, as demonstrated by studies on the PrP mutant N180Q/N196Q. This mutant, where Asn residues at the glycan attachment sites were replaced with Gln residues to prevent glycosylation while preserving the polar carboxamide side chain common to both Asn and Gln, exhibited effects similar to a highly toxic deletion mutant of PrP
[42][50].
Together, these studies indicate that the loss of glycans destabilizes the prion protein structure, enriching for a conformation that enables pathologic interactions. Loss of glycosylation could increase the affinity of PrPC for a particular conformer of PrPSc and of other pathologic interactors (such as heparan sulfate) determining the rate of nascent PrPSc formation and the specific patterns of PrPSc deposition (Figure 2). Intriguingly, distinct brain regions, and presumably cell types, demonstrated distinct vulnerability to these pathologic conformers. Also, the lack of glycans increased the vulnerability of the protein to additional stressors, increasing its pathogenicity.
3. Glucose-Regulated Protein 94 (GRP94)
Glucose-regulated protein 94 (GRP94), also known as endoplasmin and gp96, serves as a crucial molecular chaperone located in the ER of eukaryotic cells
[51]. Its primary functions involve ensuring proper folding, maturation, and quality control of client proteins within the ER. Beyond its role in protein folding, GRP94 actively participates in various cellular processes, contributing to cellular homeostasis and overall cell function
[52][53]. GRP94 forms a homodimer, and each chain comprises three domains: the N-terminal (NTD), middle, and C-terminal (CTD) (
Figure 3A). Mechanistically, the chaperone undergoes ATP-driven conformational changes associated with the folding of a client protein. ATP binding at the NTD induces a shift in the chaperone to a closed conformation, inducing changes in regions critical for protein client binding. Following ATP hydrolysis, rearrangements occur in the residues of the client-binding site, leading to mechanical translation into conformational changes in the bound client
[52][53].
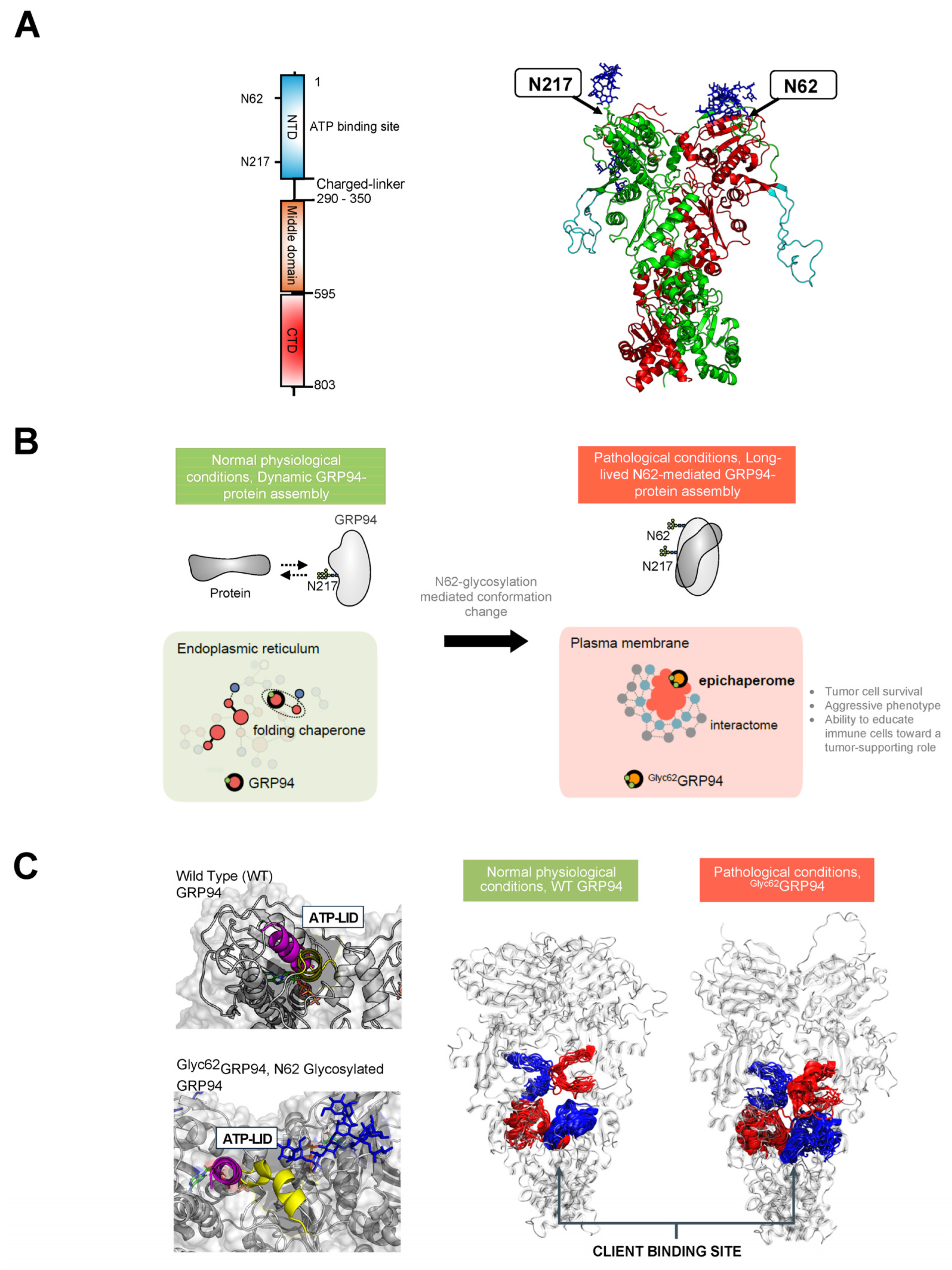
Figure 3. (A). Schematic illustrating the primary structure of the full-length GRP94 protein with color-coded domains: N-terminal domain (NTD), middle domain, and C-terminal domain (CTD). The right-side figure depicts the 3D structure of the GRP94 dimer, with individual protomers colored in green and red, respectively. An unstructured loop is colored in light blue. The location of two key regulatory N-glycans, on asparagine residues N217 and N62, is indicated by an arrow. (B). GRP94 glycosylation at N62 is a pivotal factor turning GRP94 from a folding chaperone to a pathologic protein. Primarily, N-glycosylation at N62 serves as a structural mediator, inducing the conversion of the GRP94 chaperone into epichaperomes—hetero-oligomeric forms tightly composed of chaperones, co-chaperones, and other factors. In these epichaperomes, GRP94 adopts scaffolding functions not observed in normal cells, where GRP94 primarily participates in protein control and folding. Through this scaffolding function, GRP94 influences the assembly and connectivity of proteins crucial for maintaining a malignant phenotype, enhancing their activity. Ultimately, the N62 glycosylation affects GRP94’s structural ensembles and its chaperone cycle kinetics, leading to a remodeling of the interactome at a much larger scale than one might hypothesize based on a simple local covalent modification. This malfunction in GRP94 amplifies its impact beyond immediate interactors, extending to the remodeling of cellular phenotypes. (C). The figure illustrates the structural impact of N62 glycan on GRP94. In the absence of the N62 glycan, GRP94 adopts a conformation favorable for its folding function, as evidenced by the lid of the ATP binding site (depicted in purple) and a more open, flexible protein client binding site (depicted as red or blue ribbons for protomer A or B, respectively). In the N62 glycosylated GRP94, the glycan pulls the ATP-lid into an open conformation (yellow), favoring a more closed and inaccessible protein client binding site that is thus unfavorable for folding. Through its impact on the ATP binding site’s efficiency and by perturbing GRP94’s structural dynamics, N62 glycosylation thus actively shifts GRP94 from a foldase to a protein-assembly platform.
While primarily localized to the ER, GRP94 is also found in the cytosol, at the cell surface, and extracellularly
[52][54][55]. This altered distribution is often associated with and intensified in disease-related scenarios
[56][57] (
Figure 3B). For instance, pathogens utilize surface GRP94 to infect host cells
[58][59][60]. In autoimmune diseases, overexpression of cell-surface GRP94 enhances toll-like receptor function and downstream signaling through MyD88
[61]. In cancer cells, surface GRP94 imparts an aggressive phenotype by regulating the stability of receptor tyrosine kinases (RTKs), such as HER2 and EGFR, inhibiting their internalization and enhancing their aberrant downstream signaling
[62][63][64][65]. In inflammatory diseases, surface GRP94 induced a pro-inflammatory profile in macrophages
[66][67]. Similarly, an extracellular GRP94 complexed with immunoglobulin Gs (IgGs) contributes to the pathogenesis of type 1 diabetes
[68].
N-glycosylation plays an important role in the generation of such pathologic surface-expressed and extracellular GRP94 forms (
Figure 3C). GRP94 contains six potential
N-glycan acceptor sites, namely, N62, N107, N217, N445, N481, and N502, yet under normal conditions, the protein is predominantly monoglycosylated at N217
[69][70]. Cherepanova and colleagues
[69] demonstrated that GRP94 can become glycosylated at all sites in cells that are exposed to oligosaccharyltransferase inhibitors or low doses of ER stress-inducing agents and in cells with partial or complete loss of oligosaccharyltransferase complex activity. In a later study, Wen and colleagues demonstrated that glycosylation of silent sites was heterogenous. Some sites, such as N62, were enriched with mannosylated N-glycans, such as Man
9GlcNAc
2 and Glc
1Man
9GlcNAc
2 (N2H9 and N2H10), whereas N107 and N445 had a varied content of mannosylated N-glycans but also fucosylated or sialofucosylated complex-type N-glycan structures
[71].
The regulation of GRP94 glycosylation, promoting the consistent omission of silent sites in non-stressed cells and facilitating swift and efficient glycosylation of silent sites in stressed cells, poses intriguing questions. None of the GRP94 sequons have a negative flanking sequence score (i.e., surrounding amino acid sequences do not hinder glycan addition), indicating that these sites are not skipped due to suboptimal conditions. Cherepanova et al.
[69] proposed instead that the existence of a mechanism restricting nascent GRP94 access to the STT3A active site, blocking cotranslational glycosylation of the silent sites, is responsible for the lack of glycosylation on the silent sites. In this proposed scenario, the N62 and N107 sites in GRP94 could enter the STT3A active site before the normal glycosylation site (N217) is incorporated into the nascent chain. However, factors associated with cellular stress may saturate pathways responsible for blocking glycosylation of the silent sites in GRP94 by oligosaccharyltransferase. Consequently, the N62 and N107 silent sites could become glycosylated by oligosaccharyltransferase complexes in cells exposed to stressors, potentially contributing to disease-related glycosylation patterns. One cannot exclude, however, that other (probably context-specific) alterations in the glycosylation machinery may exist that shape the occupancy of individual silent sites, with each possibly impacting the conformation and function of the GRP94 protein.
Supportive of this notion, N-glycan occupancy at these silent sites was indeed reported in disease. In OVCAR-3 ovarian cancer cells, which exhibit high levels of EGFR at the plasma membrane
[72], a study employed an unbiased, large-scale MS method to determine N-glycosylation site occupancy by comparing tunicamycin treated to inhibit the overall N-glycosylation occupancies of the cells and untreated cells
[73]. Using this method, they determined that GRP94 had four of the sites—N62, N107, N217, and N481—occupied in OVCAR-3 cells. Out of these sites, N217 had a high occupancy (~100%) and N62 occupancy was moderate (~50%), followed by N107 (<50%) and N481 (minor, ~10%). Yan and colleagues performed glycosylation site mapping through mass spectrometry and identified N62, N217, and N502 as putative N-glycosylated sites on a GRP94 variant enriched on the cell surface of MDA-MB-468 cells, an EGFR-overexpressing triple negative breast cancer cell line
[64]. Here, too, N217 was a high-occupancy site, with N62 and N502 being partially occupied.
Biochemical, functional, and structural investigations unequivocally affirm the pathological implications of N-glycan occupancy at specific silent sites
[60][64][66][67][74]. Within certain breast cancer cell subtypes, the glycosylation event at N62 emerges as a pivotal factor contributing to their aggressive phenotype, resistance to therapy, and immune evasion
[64][66][75]. This transformation leads to the stabilization of a unique conformation of GRP94, with significant repercussions
[64][74]. Primarily, N-glycosylation at N62 serves as a structural mediator, inducing the conversion of the GRP94 chaperone into epichaperomes—hetero-oligomeric forms tightly composed of chaperones, co-chaperones, and other factors
[64][76][77]. In these epichaperomes, GRP94 adopts scaffolding functions not observed in normal cells, where GRP94 primarily participates in protein control and folding. Through this scaffolding function, GRP94 influences the assembly and connectivity of proteins crucial for maintaining a malignant phenotype, enhancing their activity. Consequently, the functions of these proteins are markedly enhanced, leading to the aberrant remodeling of dependent cellular protein networks. This provides a survival advantage to cancer cells and tumor-supporting cells within the tumor microenvironment. While the precise composition of the glycan at N62 remains unknown, evidence suggests that the modified residue, rather than the specific sugar structure, is crucial for mediating the cancer-supporting conformation of GRP94 in the N62 glycoform
[64][74][78]. Despite this uncertainty, its susceptibility to deglycosylation by EndoH suggests a high mannose glycan characteristic
[64]. This notion is also supported by glycoproteomics studies from Wen and colleagues, which found N62 to be enriched in mannosylated N-glycans, such as Man
9GlcNAc
2 and Glc
1Man
9GlcNAc
2 (N2H9 and N2H10)
[71].
Among the proteins affected by this pathologic GRP94 glycoform are RTKs. In breast tumors characterized by the overexpression of RTKs like HER2 and EGFR, GRP94 with the N62 occupied site becomes enriched at the cell surface
[64]. This enrichment plays a pivotal role in reducing RTK internalization and maintaining the RTK in a state conducive to constitutively enhanced downstream signaling. To validate these findings, Yan and colleagues utilized CRISPR-Cas9 to manipulate endogenous GRP94, generating homozygous clones—N62Q, N217A, and N62Q/N217A—from the MDA-MB-468 breast cancer cell line
[64]. Clones expressing GRP94(N62Q) exhibited a significant absence of EGFR at the plasma membrane, accompanied by a lack of EGFR-signaling activity. Conversely, the GRP94(N217A) mutants, representing the folding form of GRP94, demonstrated no deleterious effects on EGFR levels or signaling
[64]. This underscores the pathologic gain-of-function nature of N62 glycosylated GRP94 in the context of cancer. Specifically for GRP94, at the plasma membrane (where
Glyc62GRP94 is located),
Glyc62GRP94 may promote cancer as, by forming epichaperome platforms
[64][76][78], it provides a backbone upon which oncogenic proteins and protein assemblies cluster, augmenting their pathologic function and leading to an aggressive phenotype in
Glyc62GRP94-expressing cancer cells (
Figure 3B).
To understand how N-glycans at N62 and N217 influence GRP94 conformation and dynamics, Castelli et al. employed MD simulation studies
[74]. Their study revealed dynamic modulation of GRP94’s conformation and interactions by these glycans, impacting protein interaction mode and ATP processing essential for folding (
Figure 3C). In the fully glycosylated state, sugars obstructed the N-terminal domain (NTD), impeding ATP binding. The N62 glycan favored an open, ATPase-incompetent NTD lid conformation, influencing the charged linker between the NTD and M-Domain, favoring a more closed and inaccessible protein client binding site that is thus unfavorable for folding. Therefore, the N62 glycan-induced alterations in conformational ensembles significantly impact the efficiency of translating nucleotide-encoded signals. Conversely, the N217 glycan had little impact on these factors. The results suggest that N62 glycosylation actively shifts GRP94 from a foldase to a protein-assembly platform, impacting the ATP-binding site’s efficiency and perturbing the charged linker’s dynamics. Ultimately, glycosylation induces GRP94 malfunction, disrupting its structural ensembles and chaperone cycle kinetics and leading to interactome remodeling at a much larger scale than the simple local covalent modification that might lead to hypothesize, amplifying dysfunction, and remodeling cellular phenotypes.
In summary, the N-glycosylation of silent sites in GRP94 during disease profoundly influences the protein’s conformation, assembly, and function (Figure 3). Under normal physiological conditions, GRP94 is glycosylated at N217 and localized to the ER, and it facilitates client protein folding through transient interactions (Figure 3A–C). However, N-glycosylation at N62 disrupts GRP94’s ER confinement, leading to a conformational shift that promotes stable interactions with proteins at the plasma membrane, enhancing their functions and inducing aberrant remodeling of cellular protein pathways. This glycosylation transforms GRP94 from a folding chaperone to a scaffolding protein, consequently reshaping protein assembly and connectivity, resulting in systems-level dysfunction. Consequently, alterations in N-glycosylation at N62 generate a distinct protein with unique conformational, dynamic, and functional characteristics compared to normal GRP94 in healthy cells. Thus, GRP94 is a unique example where N-glycosylation increases the pathologic properties of a protein indirectly by modulating its complexation.
 +1 credit
+1 credit