Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marta Lualdi | -- | 2462 | 2024-03-04 16:18:35 | | | |
| 2 | Peter Tang | + 1 word(s) | 2463 | 2024-03-05 04:28:05 | | | | |
| 3 | Marta Lualdi | Meta information modification | 2463 | 2024-03-05 17:07:59 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Lualdi, M.; Alberio, T.; Fasano, M. Proteostasis and Proteotoxicity in Network Medicine Era. Encyclopedia. Available online: https://encyclopedia.pub/entry/55838 (accessed on 06 July 2026).
Lualdi M, Alberio T, Fasano M. Proteostasis and Proteotoxicity in Network Medicine Era. Encyclopedia. Available at: https://encyclopedia.pub/entry/55838. Accessed July 06, 2026.
Lualdi, Marta, Tiziana Alberio, Mauro Fasano. "Proteostasis and Proteotoxicity in Network Medicine Era" Encyclopedia, https://encyclopedia.pub/entry/55838 (accessed July 06, 2026).
Lualdi, M., Alberio, T., & Fasano, M. (2024, March 04). Proteostasis and Proteotoxicity in Network Medicine Era. In Encyclopedia. https://encyclopedia.pub/entry/55838
Lualdi, Marta, et al. "Proteostasis and Proteotoxicity in Network Medicine Era." Encyclopedia. Web. 04 March, 2024.
Copy Citation
Neurodegenerative proteinopathies are complex diseases that share some pathogenetic processes. One of these is the failure of the proteostasis network (PN), which includes all components involved in the synthesis, folding, and degradation of proteins, thus leading to the aberrant accumulation of toxic protein aggregates in neurons. The single components that belong to the three main modules of the PN are highly interconnected and can be considered as part of a single giant network.
neurodegeneration
proteostasis
systems biology
network medicine
network pharmacology
1. Introduction
Proteins are major players in the maintenance of cellular homeostasis and display an almost endless variety of functions. Protein function is tightly dependent on the ability of a protein to acquire and maintain a specific structure, which results from the folding of the polypeptide chain in a process mainly guided by its primary aminoacidic sequence [1]. However, the vast majority of proteins display a complex structure and need assistance to obtain the final correct conformation. Thus, in physiologic conditions, several proteins (i.e., the molecular chaperones) assist the folding process in order to avoid inappropriate interactions leading to misfolded states [2]. Their activity is even more crucial when facing cellular stress conditions, which favour protein misfolding and aggregation. Mistakes in protein folding can indeed result in protein loss-of-function and/or aberrant aggregation, with detrimental consequences for the whole cell homeostasis.
The continual maintenance of an active pool of functional proteins in a cellular system is called “proteostasis” [3]. Cellular proteostasis involves many pathways: (i) protein synthesis, (ii) protein folding, (iii) refolding of partially unfolded proteins, and (iv) sequestration and disposal of irreversibly unfolded/unneeded proteins. These pathways include hundreds of enzymes and specialized proteins.
Failures in the correct proteostasis dramatically affect cellular functions, leading to the development of many diseases. In this frame, post-mitotic cells, such as neurons, are particularly vulnerable to improper cellular proteostasis. The accumulation of toxic protein aggregates in the form of extra- and/or intra-neuronal inclusions, indeed, represents a pathogenetic hallmark in neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) [4].
The proteostasis network can be therapeutically targeted in different ways in order to partially restore the correct protein homeostasis and to reduce cell death; for instance, molecular chaperone activity can be directly modulated by small molecules [5]. This targeted pharmacological approach represents one of the possible strategies to ameliorate and slow down neurodegeneration.
2. The Proteostasis Network
It has been estimated that human ribosomes synthesize the bulk of the cellular proteome at a rate of five to six amino acids per second, producing more than a billion proteins per single human cell [6]. This process is not error-free, and approximately one in twenty newly translated proteins contain a sequence error, which can cause misfolding and/or reduced stability [7]. Aberrant protein products can also result from posttranscriptional errors (e.g., splicing) and from the production of defective mRNAs, which elude the RNA surveillance quality control pathways [8]. Moreover, the rate of protein synthesis, which can be regulated in response to a variety of stimuli (e.g., stress conditions), is also a crucial aspect, and the ribosome is emerging as a central quality control organelle by checking the conformation of the nascent protein and recruiting protein folding and translocation machineries. During protein translation, the so-called “optimal codons” are recognized by highly available tRNAs, which speeds up the translation process. By contrast, “non-optimal” codons are recognized by less abundant tRNAs, thus slowing down translation at key structural motifs in order to facilitate proper protein folding [9]. Moreover, reduced tRNAs availability may lower the translation rate, thus favouring protein aggregation [10]. The protein folding process, which is both co- and post-translational, is also crucial in determining protein fate, such as the maintenance of the correct conformational state and the disposal of unwanted protein products.
The proteostasis network (PN) comprises three major modules (Figure 1), which govern the three main processes involved in protein homeostasis: synthesis, folding, and degradation. Indeed, protein products must be translated from their corresponding mRNAs, reach their final conformation (linked to their function), and eventually be degraded, based on their programmed lifespan. A central mechanism, which encompasses all steps in proteostasis, is the ability of proteins to create either stable or transient interactions, thus forming specific complex structures (e.g., cytoskeleton proteins) and/or multi-protein complexes with specific activities (e.g., enzymes). Thus, proteins are physiologically prone to aggregate with one another in a controlled way, while their aberrant aggregation leads to the accumulation of toxic products.
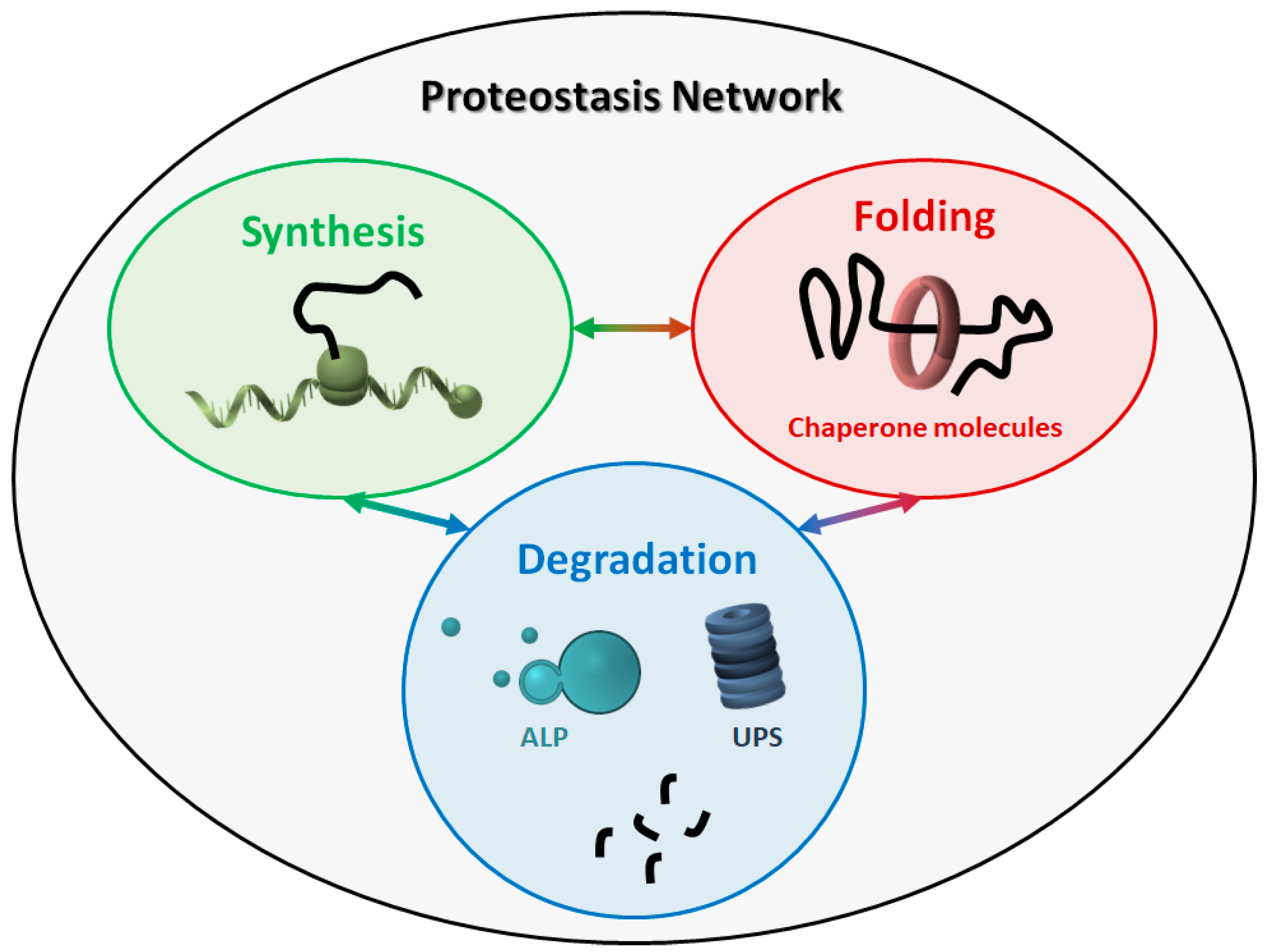
Figure 1. The proteostasis network (PN). The PN includes three modules, namely protein synthesis, folding, and degradation. The three modules are tightly connected to one another, and all components work together to maintain the correct protein homeostasis. ALP: autophagy-lysosome pathway. UPS: ubiquitin-proteasome system.
3. The Failure of the PN in Neurodegeneration
Age represents the most important risk factor for the development of many neurodegenerative disorders. One of the reasons for this is the unavoidable decline in the ability of human cells to maintain the correct proteostasis. Why the PN deteriorates with aging is still not completely clear, however, the lack of evolutionary pressure for proteome maintenance beyond the reproductive age surely contributes.
Independently of the causative mechanisms, a hallmark of the aging proteome is decreased protein solubility, accompanied by the accumulation of aggregates. This process has been extensively studied in Caenorhabditis elegans, where it has been demonstrated that low abundant proteins display higher aggregation propensities than the highly abundant ones. However, even though highly abundant proteins have greater intrinsic solubility, they actually contribute the most to the total aggregates [11]. Reasonably, their solubility is not sufficient to protect them from aggregation when the PN is deregulated and proteins exceed their critical concentration within the cells.
PN deregulation and decline dramatically affect neurons. Indeed, misfolded and/or oxidized proteins mainly accumulate in non-dividing, long-lived cells [12]. Moreover, in the human brain, the expression of ATP-dependent chaperones is reduced with age, thus promoting misfolding and aggregation [13]. When the efficiency of the PN falls below a critical level, the aggregation-prone proteins cannot be maintained in a soluble state. This threshold level can also be lowered by additional stress conditions or in the presence of mutations that affect specific proteins, thus further promoting aggregation in a positive feedback loop.
Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS) are examples of neurodegenerative proteinopathies [14][15]. During disease progression, the accumulation of toxic protein aggregates is favoured by PN malfunction, and the activity of PN components, in turn, is affected at several levels by the presence of the aggregates. This triggers a vicious cycle that leads to proteostasis disruption and eventually cell death.
Even though different pathogenetic mechanisms underlie the progression of different neurodegenerative proteinopathies, some shared features linked to PN failure can be identified. Indeed, chaperone molecules and degradation machineries can be sequestered into the pathological aggregates [16][17][18], the UPS is usually overwhelmed [19][20], and the general cellular ability to cope with stress conditions is hampered [21][22]. These mechanisms could be targeted in an attempt to ameliorate and to lower proteinopathies progression.
4. Targeting the PN in Neurodegeneration: “Classical” Pharmacological Approaches
Due to the role of PN in maintaining the correct proteostasis, the easiest pharmacological approach that can be explored in neurodegenerative proteinopathies is to boost the system. This can be achieved by either modulating the activity of individual components or acting broadly on the master regulators of the PN. These targeted pharmacological approaches proved to be effective in ameliorating some proteinopathies, as extensively revisedelsewhere [23]. Here, the researchers discuss some of these strategies, with the aim of also highlighting the limitations of such approaches.
The upregulation of HSP40s represents a viable strategy, since mutations in some of these proteins have been linked to neurodegeneration [24]. Three HSP40 family members are of particular interest, namely DNAJB2, DNAJB6, and DNAJB9, which bind polyQ-containing proteins, α-synuclein, and β-amyloid, respectively. One small compound has been described which increases HSP40s activity, thus improving the function of the HSP70s system [25]. Also, the enhancement of HIP function, thus increasing HSP70s activity, seems to inhibit the aggregation of misfolded proteins [26][27].
By contrast, the inhibition of HSP90s has been proposed as a therapeutic strategy in neurodegeneration. Indeed, when the activity of HSP90s is reduced, HSP70s levels are increased in turn, thus enhancing the degradation of toxic protein aggregates (e.g., tau and polyQ-containing proteins) [28]. Moreover, both α-synuclein and tau are HSP90s client proteins, and it has been proposed that HSP90s contribute to the stabilization of the toxic intermediates [29]. However, the systemic inhibition of HSP90s is toxic and the use of brain penetrant inhibitors, originally developed as antineoplastic drugs, is also neurotoxic [30][31].
The activity of chaperonins can also be targeted as a therapeutic strategy in neurodegeneration. As an example, the mHtt protein, which forms toxic fibrils in Huntington’s disease, is a known client protein of TRiC, a cytoplasmic 1 MDa complex composed of members of the HSP60 family [32]. Also, in this case, an indirect approach to increase the levels of TRiC has been proposed, namely, the use of small molecules for the inhibition of the VRK2 kinase, which normally inhibits the activity of the USP25 deubiquitinase, which deubiquitinates TRiC. Thus, the inhibition of VRK2 results in increased levels of TRiC, which is no longer degraded via the UPS. It has been demonstrated that such inhibitors are effective in reducing the aggregation of mHtt [33].
Since a general upregulation of the chaperone system is expected to ameliorate neurodegeneration, some efforts have focused on the HSF1 protein, which is recognised as the master transcription factor that regulates the activity of the PN [34]. HSF1 is normally responsive to stress; thus, small molecules that induce HSF1 act through non-specific stressful mechanisms, which makes them of little use as therapeutics. A small molecule has been recently identified, namely HSF1A, which activates HSF1 without causing cellular stress and increases the levels of HSP70s, thus reducing protein aggregates in various mammalian cells and fruit fly models [35][36].
Even though molecular chaperones seem to be promising targets, the tuning of the other modules of the PN (i.e., protein synthesis and degradation) also represents a good strategy. For instance, the use of small molecules which boost the UPS and/or ALP system has been proposed [37][38][39] in order to improve protein clearance, thus ameliorating neuron cells viability. On the other hand, reducing the translational activity represents an equally promising strategy. Indeed, reducing the levels of newly synthesized proteins results in the unburdening of the downstream PN modules. A general reduction of protein translation is always observed in response to stress when the cellular energy needs to be saved to cope with the adverse conditions. In this frame, the pharmacological lengthening of stress-induced translational attenuation has been proved to be effective in proteinopathies [40][41], even though it cannot be envisioned as an actual therapeutic strategy.
5. Novel Strategies to Ameliorate Proteinopathies: The “Network Medicine” Approach
As already described, the PN comprises around 2000 components functionally grouped into three main modules. These modules are deeply interconnected, and all components work together and compensate one another to maintain proper protein homeostasis. The malfunction of even a single or a small group of components unavoidably impacts the whole PN, with consequences that are difficult to predict if every single component is considered as a standalone.
5.1. Complex Systems and Diseases
Biological organisms are complex systems made of individual components which interact at different levels, thus creating a complex network. In this frame, any possible alterations which impact the function of a single component (e.g., a specific gene mutation) will introduce a perturbation in the entire network. Typically, complex systems are capable of reacting to both internal and external changes by the reorganization of their individual components, which results in the acquisition of novel properties. These properties of the system can be explained only in the frame of the systems theory [42][43]. Indeed, an “emergent property” is a characteristic of the system that is not present in its individual components but arises from the collaboration among the elements of the system.
The concept of “emergence” is perfectly suitable to multifactorial diseases, in which multiple factors can be associated with the pathogenetic process. Neurodegenerative diseases belong to this category, since the diseased state is usually the result of multiple genetic and environmental causes. Even in familiar forms (e.g., PARK2-mutated PD patients), where a clear genetic alteration is recognized as the main etiological driver, the clinical phenotype can vary, based on the co-occurrence of additional factors. Moreover, complex diseases always display nonlinear correlation between genotype and phenotype, which means that the same genotype can result in different phenotypes and also that the same disease phenotype can arise from different genotypes. One example is the GGGGCC hexanucleotide repeat expansion in the C9orf72 gene, which is associated with both ALS and frontotemporal dementia (FTD) [44]. On the other hand, several gene mutations (i.e., the so-called PARK PD-associated loci) are associated with familiar PD as a clearly defined clinical phenotype [45].
5.2. Network Medicine
The “network medicine” approach has been proposed to overcome the main limitations of the classical targeted approach in medicine [46]. This novel approach is founded on the observation that a disease is almost never the result of a single protein dysregulation but usually reflects alterations of a complex intracellular network. Thus, targeting a single component of a complex network based on its involvement in a disease can represent an effective therapeutic strategy, but also entails several limitations. First, the outcomes are not completely predictable. For instance, the inhibition/boosting of a chaperone molecule can trigger downstream events, such as compensation mechanisms from other chaperones. Second, there is never only one real target. Every single component of the PN displays at least a small group of direct interactors (either physical or functional), whose activity is obviously also affected. Targeting a single component that plays a central role in the network can disrupt the entire network. For instance, the inhibition of HSP90s, which are able to increase the levels of HSP70s, resulting in beneficial effects on protein aggregation, is actually not feasible due to the fact that HSP90s are central highly connected nodes (so-called “hubs”) in the PN.
The classical reductionist view of the “one-target” pharmacology usually fails to identify novel therapeutics for complex diseases. In this frame, “network pharmacology” represents a way to innovate drug discovery. It takes advantage of the networks generated from the integration of “omics” data through the tools of systems biology. This network-centric approach is able to (i) suggest novel targets, (ii) identify hidden pathways involved in the disease, (iii) predict the effects of a treatment considering the entire system, (iv) suggest the dosing of a drug, based, for instance, on metabolic profiling, and (v) identify the causes of drug resistance and/or toxicity based on the “shape” (e.g., robustness, connectivity, fragility) of the network.
References
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230.
- Ellis, R.J. Roles of molecular chaperones in protein folding. Curr. Opin. Struct. Biol. 1994, 4, 117–122.
- Sontag, E.M.; Samant, R.S.; Frydman, J. Mechanisms and functions of spatial protein quality control. Annu. Rev. Biochem. 2017, 86, 97–122.
- Schulz, J.B.; Dichgans, J. Molecular pathogenesis of movement disorders: Are protein aggregates a common link in neuronal degeneration? Curr. Opin. Neurol. 1999, 12, 433–439.
- Newton, T.M.; Duce, J.A.; Bayle, E.D. The proteostasis network provides targets for neurodegeneration. Br. J. Pharm. 2019, 176, 3508–3514.
- Milo, R. What is the total number of protein molecules per cell volume? A call to rethink some published values. BioEssays News Rev. Mol. Cell. Dev. Biol. 2013, 35, 1050–1055.
- Zaher, H.S.; Green, R. Fidelity at the molecular level: Lessons from protein synthesis. Cell 2009, 136, 746–762.
- Isken, O.; Maquat, L.E. Quality control of eukaryotic mRNA: Safeguarding cells from abnormal mRNA function. Genes Dev. 2007, 21, 1833–1856.
- Pechmann, S.; Willmund, F.; Frydman, J. The ribosome as a hub for protein quality control. Mol. Cell 2013, 49, 411–421.
- Nedialkova, D.D.; Leidel, S.A. Optimization of codon translation rates via tRNA modifications maintains proteome integrity. Cell 2015, 161, 1606–1618.
- Walther, D.M.; Kasturi, P.; Zheng, M.; Pinkert, S.; Vecchi, G.; Ciryam, P.; Morimoto, R.I.; Dobson, C.M.; Vendruscolo, M.; Mann, M.; et al. Widespread proteome remodeling and aggregation in aging C. elegans. Cell 2015, 161, 919–932.
- Sala, A.J.; Bott, L.C.; Morimoto, R.I. Shaping proteostasis at the cellular, tissue, and organismal level. J. Cell Biol. 2017, 216, 1231–1241.
- Brehme, M.; Voisine, C.; Rolland, T.; Wachi, S.; Soper, J.H.; Zhu, Y.; Orton, K.; Villella, A.; Garza, D.; Vidal, M.; et al. A chaperome sub-network safeguards proteostasis in aging and neurodegenerative disease. Cell Rep. 2014, 9, 1135–1150.
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68.
- Iadanza, M.G.; Jackson, M.P.; Hewitt, E.W.; Ranson, N.A.; Radford, S.E. A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 755–773.
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786.
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 2018, 9, 1097.
- Yu, A.; Shibata, Y.; Shah, B.; Calamini, B.; Lo, D.C.; Morimoto, R.I. Protein aggregation can inhibit clathrin-mediated endocytosis by chaperone competition. Proc. Natl. Acad. Sci. USA 2014, 111, E1481–E1490.
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001, 292, 1552–1555.
- Hipp, M.S.; Park, S.-H.; Hartl, F.U. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 2014, 24, 506–514.
- Olzscha, H.; Schermann, S.M.; Woerner, A.C.; Pinkert, S.; Hecht, M.H.; Tartaglia, G.G.; Vendruscolo, M.; Hayer-Hartl, M.; Hartl, F.U.; Vabulas, R.M. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 2011, 144, 67–78.
- Roth, D.M.; Hutt, D.M.; Tong, J.; Bouchecareilh, M.; Wang, N.; Seeley, T.; Dekkers, J.F.; Beekman, J.M.; Garza, D.; Drew, L.; et al. Modulation of the maladaptive stress response to manage diseases of protein folding. PLoS Biol. 2014, 12, e1001998.
- Eisele, Y.S.; Monteiro, C.; Fearns, C.; Encalada, S.E.; Wiseman, R.L.; Powers, E.T.; Kelly, J.W. Targeting protein aggregation for the treatment of degenerative diseases. Nat. Rev. Drug Discov. 2015, 14, 759–780.
- Zarouchlioti, C.; Parfitt, D.A.; Li, W.; Gittings, L.M.; Cheetham, M.E. DNAJ Proteins in neurodegeneration: Essential and protective factors. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373.
- Wisén, S.; Bertelsen, E.B.; Thompson, A.D.; Patury, S.; Ung, P.; Chang, L.; Evans, C.G.; Walter, G.M.; Wipf, P.; Carlson, H.A.; et al. Binding of a small molecule at a protein-protein interface regulates the chaperone activity of Hsp70–Hsp40. ACS Chem. Biol. 2010, 5, 611–622.
- Wang, A.M.; Miyata, Y.; Klinedinst, S.; Peng, H.-M.; Chua, J.P.; Komiyama, T.; Li, X.; Morishima, Y.; Merry, D.E.; Pratt, W.B.; et al. Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nat. Chem. Biol. 2013, 9, 112–118.
- Miyata, Y.; Li, X.; Lee, H.-F.; Jinwal, U.K.; Srinivasan, S.R.; Seguin, S.P.; Young, Z.T.; Brodsky, J.L.; Dickey, C.A.; Sun, D.; et al. Synthesis and initial evaluation of YM-08, a blood-brain barrier permeable derivative of the heat shock protein 70 (Hsp70) inhibitor MKT-077, which reduces tau levels. ACS Chem. Neurosci. 2013, 4, 930–939.
- Luo, W.; Sun, W.; Taldone, T.; Rodina, A.; Chiosis, G. Heat shock protein 90 in neurodegenerative diseases. Mol. Neurodegener. 2010, 5, 24.
- Luo, W.; Dou, F.; Rodina, A.; Chip, S.; Kim, J.; Zhao, Q.; Moulick, K.; Aguirre, J.; Wu, N.; Greengard, P.; et al. Roles of heat-shock protein 90 in maintaining and facilitating the neurodegenerative phenotype in tauopathies. Proc. Natl. Acad. Sci. USA 2007, 104, 9511–9516.
- Blair, L.J.; Sabbagh, J.J.; Dickey, C.A. Targeting Hsp90 and its co-chaperones to treat Alzheimer’s disease. Expert Opin. Ther. Targets 2014, 18, 1219–1232.
- Spreafico, A.; Delord, J.-P.; De Mattos-Arruda, L.; Berge, Y.; Rodon, J.; Cottura, E.; Bedard, P.L.; Akimov, M.; Lu, H.; Pain, S.; et al. A first-in-human phase I, dose-escalation, multicentre study of HSP990 administered orally in adult patients with advanced solid malignancies. Br. J. Cancer 2015, 112, 650–659.
- Kitamura, A.; Kubota, H.; Pack, C.-G.; Matsumoto, G.; Hirayama, S.; Takahashi, Y.; Kimura, H.; Kinjo, M.; Morimoto, R.I.; Nagata, K. Cytosolic chaperonin prevents polyglutamine toxicity with altering the aggregation state. Nat. Cell Biol. 2006, 8, 1163–1170.
- Kim, S.; Lee, D.; Lee, J.; Song, H.; Kim, H.-J.; Kim, K.-T. Vaccinia-related kinase 2 controls the stability of the eukaryotic chaperonin TRiC/CCT by inhibiting the deubiquitinating enzyme USP25. Mol. Cell Biol. 2015, 35, 1754–1762.
- Neef, D.W.; Jaeger, A.M.; Thiele, D.J. Heat shock transcription factor 1 as a therapeutic target in neurodegenerative diseases. Nat. Rev. Drug Discov. 2011, 10, 930–944.
- Neef, D.W.; Turski, M.L.; Thiele, D.J. Modulation of heat shock transcription factor 1 as a therapeutic target for small molecule intervention in neurodegenerative disease. PLoS Biol. 2010, 8, e1000291.
- Neef, D.W.; Jaeger, A.; Gomez-Pastor, R.; Willmund, F.; Frydman, J.; Thiele, D.J. A direct regulatory interaction between chaperonin TRiC and stress responsive transcription factor HSF1. Cell Rep. 2014, 9, 955–966.
- Leestemaker, Y.; de Jong, A.; Witting, K.F.; Penning, R.; Schuurman, K.; Rodenko, B.; Zaal, E.A.; van de Kooij, B.; Laufer, S.; Heck, A.J.R.; et al. Proteasome activation by small molecules. Cell Chem. Biol. 2017, 24, 725–736.e7.
- Barmada, S.J.; Serio, A.; Arjun, A.; Bilican, B.; Daub, A.; Ando, D.M.; Tsvetkov, A.; Pleiss, M.; Li, X.; Peisach, D.; et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat. Chem. Biol. 2014, 10, 677–685.
- Kuo, S.-Y.; Castoreno, A.B.; Aldrich, L.N.; Lassen, K.G.; Goel, G.; Dančík, V.; Kuballa, P.; Latorre, I.; Conway, K.L.; Sarkar, S.; et al. Small-molecule enhancers of autophagy modulate cellular disease phenotypes suggested by human genetics. Proc. Natl. Acad. Sci. USA 2015, 112, E4281–E4287.
- Tsaytler, P.; Harding, H.P.; Ron, D.; Bertolotti, A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 2011, 332, 91–94.
- Das, I.; Krzyzosiak, A.; Schneider, K.; Wrabetz, L.; D’Antonio, M.; Barry, N.; Sigurdardottir, A.; Bertolotti, A. Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 2015, 348, 239–242.
- Ma’ayan, A. Complex systems biology. J. R. Soc. Interface 2017, 14.
- Lualdi, M.; Fasano, M. Statistical analysis of proteomics data: A review on feature selection. J. Proteom. 2019, 198, 18–26.
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558.
- Deng, H.; Wang, P.; Jankovic, J. The genetics of Parkinson disease. Ageing Res. Rev. 2018, 42, 72–85.
- Barabási, A.-L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.5K
Entry Collection:
Neurodegeneration
Revisions:
3 times
(View History)
Update Date:
05 Mar 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No