 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Judit Oláh | -- | 2754 | 2024-02-29 15:31:42 | | | |
| 2 | Peter Tang | Meta information modification | 2754 | 2024-03-04 03:47:02 | | |
Video Upload Options
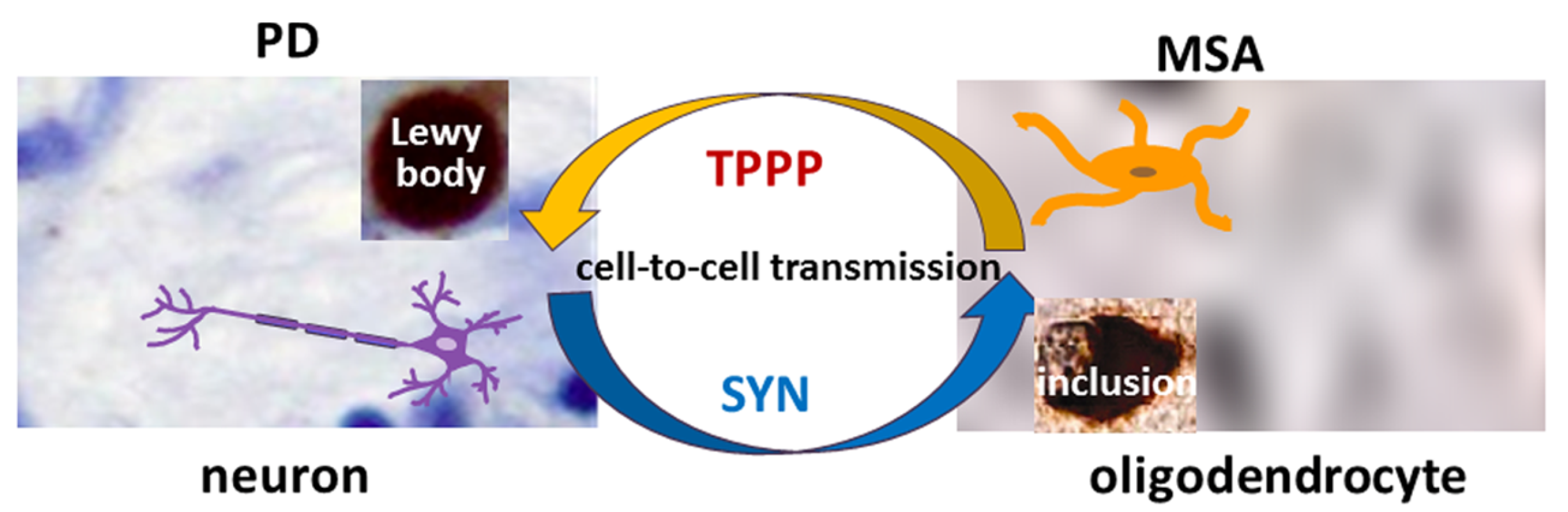
Neurological disorders such as Parkinsonism cause serious socio-economic problems as there are only therapies that treat their symptoms. The well-established hallmark alpha-synuclein (SYN) is enriched in the inclusion bodies characteristic of Parkinsonism. A prominent partner of SYN was discovered, termed Tubulin Polymerization Promoting Protein (TPPP), which has important physiological and pathological activities such as the regulation of the microtubule network and the promotion of SYN aggregation. The role of TPPP in Parkinsonism is often neglected in research. In the normal brain, SYN and TPPP are expressed endogenously in neurons and oligodendrocytes, respectively, whilst, at an early stage of Parkinsonism, soluble hetero-associations of these proteins are found in both cell types. The cell-to-cell transmission of these proteins, which is central to disease progression, provides a unique situation for specific drug targeting.
1. Introduction
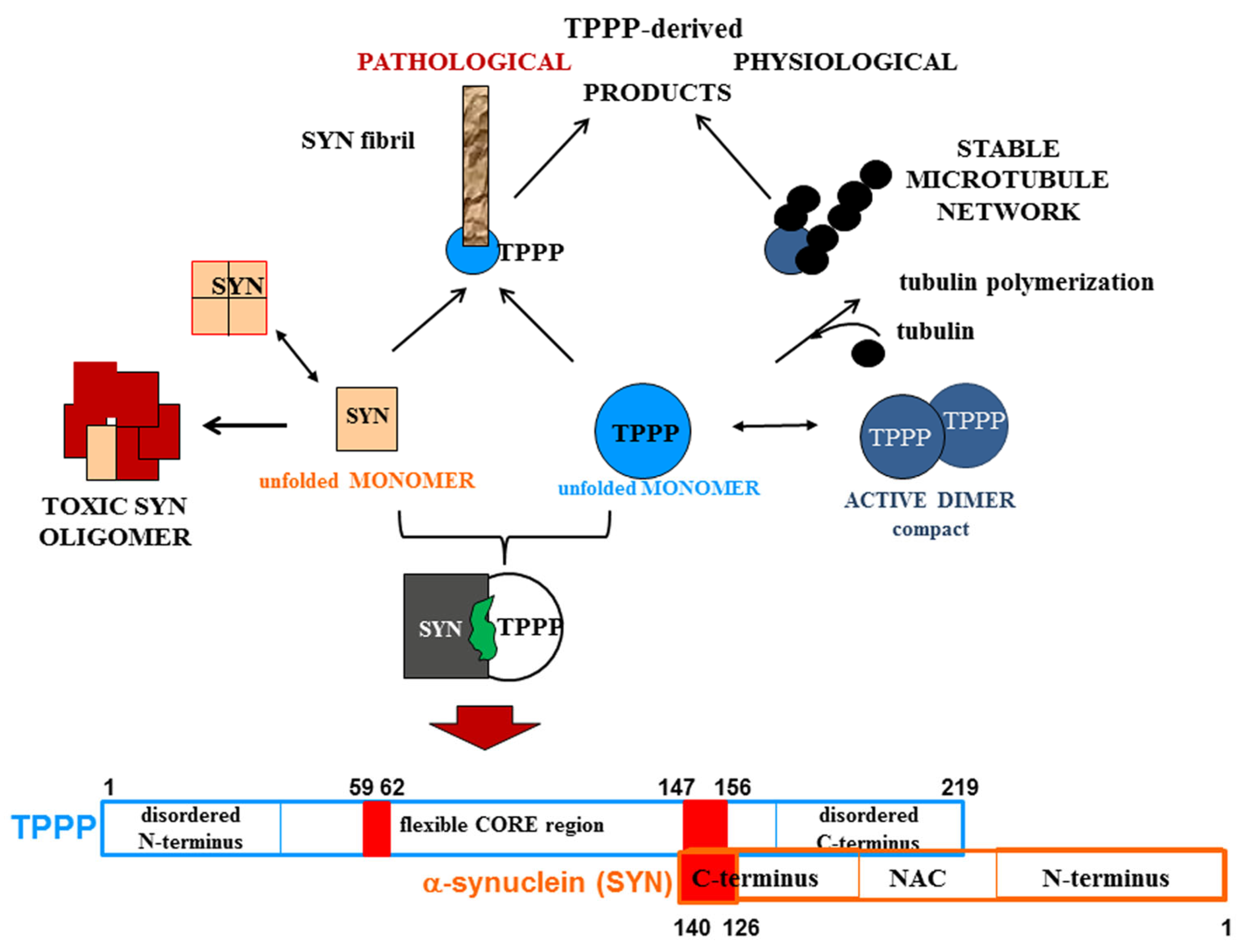
2. Pathological Interaction between TPPP and SYN
3. The Role of TPPP in the Pathomechanism of Synucleinopathies: Protein Aggregation
4. The Role of TPPP in the Dysregulation of Protein Degradation in Parkinsonism
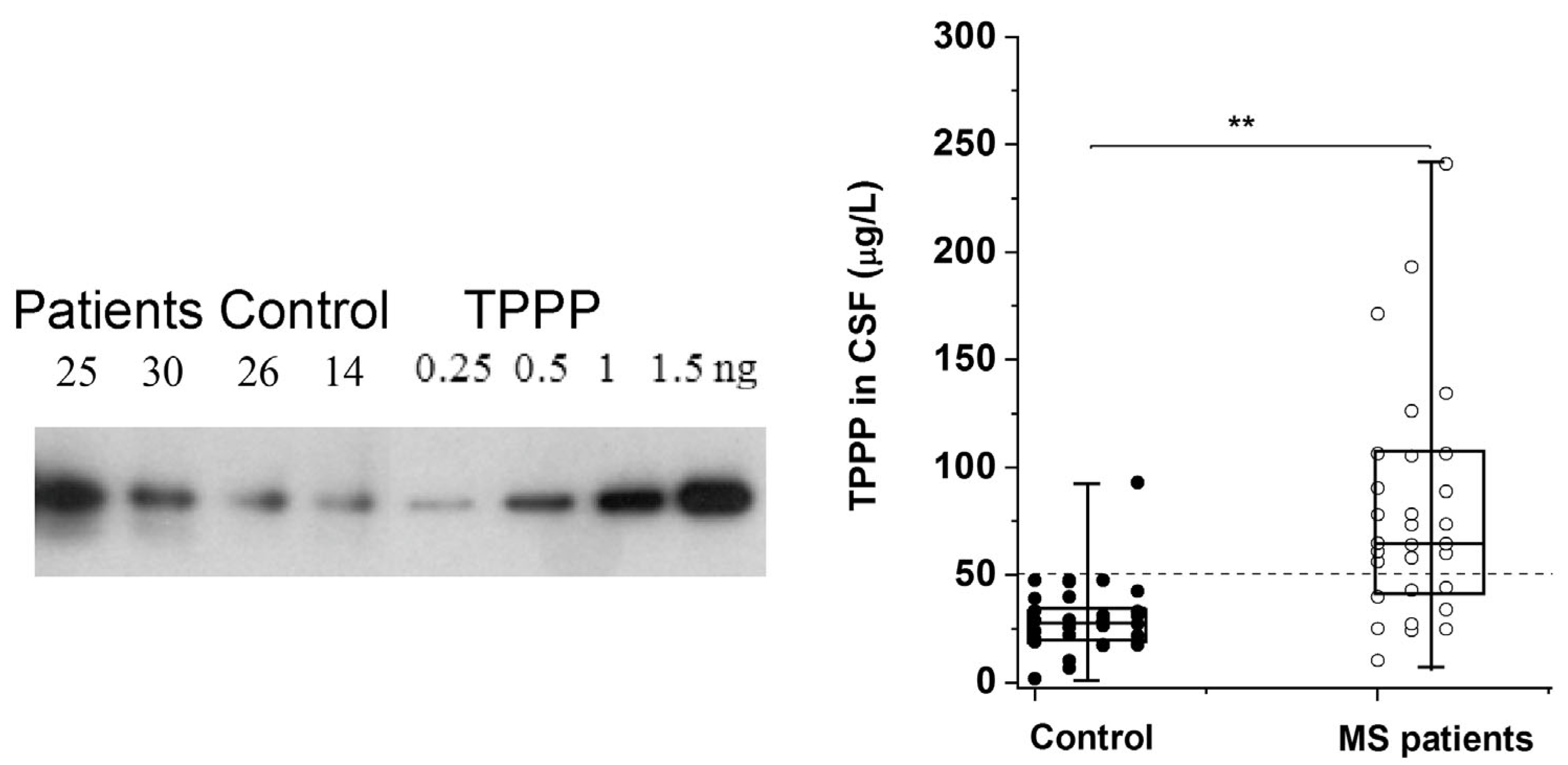
5. SYN and TPPP as Biomarkers
6. Targeting the Interface of the Pathological SYN-TPPP Complex
References
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912.
- Soni, R.; Delvadia, P.; Joharapurkar, A.; Shah, J. Uncovering Novel Therapeutic Targets for Parkinson’s Disease. ACS Chem. Neurosci. 2023, 14, 1935–1949.
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840.
- Wakabayashi, K.; Yoshimoto, M.; Tsuji, S.; Takahashi, H. Alpha-synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci. Lett. 1998, 249, 180–182.
- Koga, S.; Sekiya, H.; Kondru, N.; Ross, O.A.; Dickson, D.W. Neuropathology and molecular diagnosis of Synucleinopathies. Mol. Neurodegener. 2021, 16, 83.
- Graves, N.J.; Gambin, Y.; Sierecki, E. alpha-Synuclein Strains and Their Relevance to Parkinson’s Disease, Multiple System Atrophy, and Dementia with Lewy Bodies. Int. J. Mol. Sci. 2023, 24, 12134.
- Stefanova, N.; Wenning, G.K. Multiple system atrophy: At the crossroads of cellular, molecular and genetic mechanisms. Nat. Rev. Neurosci. 2023, 24, 334–346.
- Hill, D.R.; Huters, A.D.; Towne, T.B.; Reddy, R.E.; Fogle, J.L.; Voight, E.A.; Kym, P.R. Parkinson’s Disease: Advances in Treatment and the Syntheses of Various Classes of Pharmaceutical Drug Substances. Chem. Rev. 2023, 123, 13693–13712.
- Stott, S.R.W.; Wyse, R.K.; Brundin, P. Novel approaches to counter protein aggregation pathology in Parkinson’s disease. Prog. Brain Res. 2020, 252, 451–492.
- Morris, H.R.; Spillantini, M.G.; Sue, C.M.; Williams-Gray, C.H. The pathogenesis of Parkinson’s disease. Lancet 2024, 403, 293–304.
- Peng, C.; Gathagan, R.J.; Covell, D.J.; Medellin, C.; Stieber, A.; Robinson, J.L.; Zhang, B.; Pitkin, R.M.; Olufemi, M.F.; Luk, K.C.; et al. Cellular milieu imparts distinct pathological alpha-synuclein strains in alpha-synucleinopathies. Nature 2018, 557, 558–563.
- Kovacs, G.G.; Laszlo, L.; Kovacs, J.; Jensen, P.H.; Lindersson, E.; Botond, G.; Molnar, T.; Perczel, A.; Hudecz, F.; Mezo, G.; et al. Natively unfolded tubulin polymerization promoting protein TPPP/p25 is a common marker of alpha-synucleinopathies. Neurobiol. Dis. 2004, 17, 155–162.
- Olah, J.; Lehotzky, A.; Szunyogh, S.; Szenasi, T.; Orosz, F.; Ovadi, J. Microtubule-Associated Proteins with Regulatory Functions by Day and Pathological Potency at Night. Cells 2020, 9, 357.
- Mavroeidi, P.; Arvanitaki, F.; Karakitsou, A.K.; Vetsi, M.; Kloukina, I.; Zweckstetter, M.; Giller, K.; Becker, S.; Sorrentino, Z.A.; Giasson, B.I.; et al. Endogenous oligodendroglial alpha-synuclein and TPPP/p25alpha orchestrate alpha-synuclein pathology in experimental multiple system atrophy models. Acta Neuropathol. 2019, 138, 415–441.
- Kovacs, G.G.; Gelpi, E.; Lehotzky, A.; Hoftberger, R.; Erdei, A.; Budka, H.; Ovadi, J. The brain-specific protein TPPP/p25 in pathological protein deposits of neurodegenerative diseases. Acta Neuropathol. 2007, 113, 153–161.
- Tokesi, N.; Olah, J.; Hlavanda, E.; Szunyogh, S.; Szabo, A.; Babos, F.; Magyar, A.; Lehotzky, A.; Vass, E.; Ovadi, J. Identification of motives mediating alternative functions of the neomorphic moonlighting TPPP/p25. Biochim. Biophys. Acta 2014, 1842, 547–557.
- Lehotzky, A.; Lau, P.; Tokesi, N.; Muja, N.; Hudson, L.D.; Ovadi, J. Tubulin polymerization-promoting protein (TPPP/p25) is critical for oligodendrocyte differentiation. Glia 2010, 58, 157–168.
- Han, S.; Gim, Y.; Jang, E.H.; Hur, E.M. Functions and dysfunctions of oligodendrocytes in neurodegenerative diseases. Front. Cell. Neurosci. 2022, 16, 1083159.
- Ota, K.; Obayashi, M.; Ozaki, K.; Ichinose, S.; Kakita, A.; Tada, M.; Takahashi, H.; Ando, N.; Eishi, Y.; Mizusawa, H.; et al. Relocation of p25alpha/tubulin polymerization promoting protein from the nucleus to the perinuclear cytoplasm in the oligodendroglia of sporadic and COQ2 mutant multiple system atrophy. Acta Neuropathol. Commun. 2014, 2, 136.
- Tokesi, N.; Lehotzky, A.; Horvath, I.; Szabo, B.; Olah, J.; Lau, P.; Ovadi, J. TPPP/p25 promotes tubulin acetylation by inhibiting histone deacetylase 6. J. Biol. Chem. 2010, 285, 17896–17906.
- Szabo, A.; Olah, J.; Szunyogh, S.; Lehotzky, A.; Szenasi, T.; Csaplar, M.; Schiedel, M.; Low, P.; Jung, M.; Ovadi, J. Modulation of Microtubule Acetylation by the Interplay Of TPPP/p25, SIRT2 and New Anticancer Agents with Anti-SIRT2 Potency. Sci. Rep. 2017, 7, 17070.
- Olah, J.; Szenasi, T.; Szunyogh, S.; Szabo, A.; Lehotzky, A.; Ovadi, J. Further evidence for microtubule-independent dimerization of TPPP/p25. Sci. Rep. 2017, 7, 40594.
- Zotter, A.; Bodor, A.; Olah, J.; Hlavanda, E.; Orosz, F.; Perczel, A.; Ovadi, J. Disordered TPPP/p25 binds GTP and displays Mg2+-dependent GTPase activity. FEBS Lett. 2011, 585, 803–808.
- Zotter, A.; Olah, J.; Hlavanda, E.; Bodor, A.; Perczel, A.; Szigeti, K.; Fidy, J.; Ovadi, J. Zn(2)+-induced rearrangement of the disordered TPPP/p25 affects its microtubule assembly and GTPase activity. Biochemistry 2011, 50, 9568–9578.
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. Off. J. Soc. Neurosci. 1988, 8, 2804–2815.
- Bates, C.A.; Zheng, W. Brain disposition of alpha-Synuclein: Roles of brain barrier systems and implications for Parkinson’s disease. Fluids Barriers CNS 2014, 11, 17.
- Takahashi, M.; Tomizawa, K.; Fujita, S.C.; Sato, K.; Uchida, T.; Imahori, K. A brain-specific protein p25 is localized and associated with oligodendrocytes, neuropil, and fiber-like structures of the CA hippocampal region in the rat brain. J. Neurochem. 1993, 60, 228–235.
- Valdinocci, D.; Radford, R.A.; Siow, S.M.; Chung, R.S.; Pountney, D.L. Potential Modes of Intercellular alpha-Synuclein Transmission. Int. J. Mol. Sci. 2017, 18, 469.
- Bras, I.C.; Outeiro, T.F. Alpha-Synuclein: Mechanisms of Release and Pathology Progression in Synucleinopathies. Cells 2021, 10, 375.
- Xia, Y.; Zhang, G.; Han, C.; Ma, K.; Guo, X.; Wan, F.; Kou, L.; Yin, S.; Liu, L.; Huang, J.; et al. Microglia as modulators of exosomal alpha-synuclein transmission. Cell Death Dis. 2019, 10, 174.
- Szenasi, T.; Olah, J.; Szabo, A.; Szunyogh, S.; Lang, A.; Perczel, A.; Lehotzky, A.; Uversky, V.N.; Ovadi, J. Challenging drug target for Parkinson’s disease: Pathological complex of the chameleon TPPP/p25 and alpha-synuclein proteins. Biochim. Biophys. Acta. Mol. Basis Dis. 2017, 1863, 310–323.
- Szunyogh, S.; Olah, J.; Szenasi, T.; Szabo, A.; Ovadi, J. Targeting the interface of the pathological complex of alpha-synuclein and TPPP/p25. Biochim. Biophys. Acta 2015, 1852, 2653–2661.
- Keerthikumar, S.; Gangoda, L.; Liem, M.; Fonseka, P.; Atukorala, I.; Ozcitti, C.; Mechler, A.; Adda, C.G.; Ang, C.S.; Mathivanan, S. Proteogenomic analysis reveals exosomes are more oncogenic than ectosomes. Oncotarget 2015, 6, 15375–15396.
- Kon, T.; Forrest, S.L.; Lee, S.; Martinez-Valbuena, I.; Li, J.; Nassir, N.; Uddin, M.J.; Lang, A.E.; Kovacs, G.G. Neuronal SNCA transcription during Lewy body formation. Acta Neuropathol. Commun. 2023, 11, 185.
- Gould, R.; Brady, S. Identifying mRNAs Residing in Myelinating Oligodendrocyte Processes as a Basis for Understanding Internode Autonomy. Life 2023, 13, 945.
- Cui, H.; Kilpelainen, T.; Zouzoula, L.; Auno, S.; Trontti, K.; Kurvonen, S.; Norrbacka, S.; Hovatta, I.; Jensen, P.H.; Myohanen, T.T. Prolyl oligopeptidase inhibition reduces alpha-synuclein aggregation in a cellular model of multiple system atrophy. J. Cell. Mol. Med. 2021, 25, 9634–9646.
- Kaji, S.; Maki, T.; Kinoshita, H.; Uemura, N.; Ayaki, T.; Kawamoto, Y.; Furuta, T.; Urushitani, M.; Hasegawa, M.; Kinoshita, Y.; et al. Pathological Endogenous alpha-Synuclein Accumulation in Oligodendrocyte Precursor Cells Potentially Induces Inclusions in Multiple System Atrophy. Stem Cell Rep. 2018, 10, 356–365.
- Ferreira, N.; Gram, H.; Sorrentino, Z.A.; Gregersen, E.; Schmidt, S.I.; Reimer, L.; Betzer, C.; Perez-Gozalbo, C.; Beltoja, M.; Nagaraj, M.; et al. Multiple system atrophy-associated oligodendroglial protein p25alpha stimulates formation of novel alpha-synuclein strain with enhanced neurodegenerative potential. Acta Neuropathol. 2021, 142, 87–115.
- Rohan, Z.; Milenkovic, I.; Lutz, M.I.; Matej, R.; Kovacs, G.G. Shared and Distinct Patterns of Oligodendroglial Response in alpha-Synucleinopathies and Tauopathies. J. Neuropathol. Exp. Neurol. 2016, 75, 1100–1109.
- Lehotzky, A.; Olah, J.; Fekete, J.T.; Szenasi, T.; Szabo, E.; Gyorffy, B.; Varady, G.; Ovadi, J. Co-Transmission of Alpha-Synuclein and TPPP/p25 Inhibits Their Proteolytic Degradation in Human Cell Models. Front. Mol. Biosci. 2021, 8, 666026.
- Sanchez-Mirasierra, I.; Ghimire, S.; Hernandez-Diaz, S.; Soukup, S.F. Targeting Macroautophagy as a Therapeutic Opportunity to Treat Parkinson’s Disease. Front. Cell Dev. Biol. 2022, 10, 921314.
- Nilsson, J.; Constantinescu, J.; Nellgard, B.; Jakobsson, P.; Brum, W.S.; Gobom, J.; Forsgren, L.; Dalla, K.; Constantinescu, R.; Zetterberg, H.; et al. Cerebrospinal Fluid Biomarkers of Synaptic Dysfunction are Altered in Parkinson’s Disease and Related Disorders. Mov. Disord. Off. J. Mov. Disord. Soc. 2023, 38, 267–277.
- Vendel, E.; Rottschafer, V.; de Lange, E.C.M. The need for mathematical modelling of spatial drug distribution within the brain. Fluids Barriers CNS 2019, 16, 12.
- Siderowf, A.; Concha-Marambio, L.; Lafontant, D.E.; Farris, C.M.; Ma, Y.; Urenia, P.A.; Nguyen, H.; Alcalay, R.N.; Chahine, L.M.; Foroud, T.; et al. Parkinson’s Progression Markers, I. Assessment of heterogeneity among participants in the Parkinson’s Progression Markers Initiative cohort using alpha-synuclein seed amplification: A cross-sectional study. Lancet. Neurol. 2023, 22, 407–417.
- Kwon, D.H.; Hwang, J.S.; Kim, S.G.; Jang, Y.E.; Shin, T.H.; Lee, G. Cerebrospinal Fluid Metabolome in Parkinson’s Disease and Multiple System Atrophy. Int. J. Mol. Sci. 2022, 23, 1879.
- Schulz, I.; Kruse, N.; Gera, R.G.; Kremer, T.; Cedarbaum, J.; Barbour, R.; Zago, W.; Schade, S.; Otte, B.; Bartl, M.; et al. Systematic Assessment of 10 Biomarker Candidates Focusing on alpha-Synuclein-Related Disorders. Mov. Disord. Off. J. Mov. Disord. Soc. 2021, 36, 2874–2887.
- Mavroudis, I.; Petridis, F.; Chatzikonstantinou, S.; Kazis, D. Alpha-synuclein Levels in the Differential Diagnosis of Lewy Bodies Dementia and Other Neurodegenerative Disorders: A Meta-analysis. Alzheimer Dis. Assoc. Disord. 2020, 34, 220–224.
- Zhou, B.; Wen, M.; Yu, W.F.; Zhang, C.L.; Jiao, L. The Diagnostic and Differential Diagnosis Utility of Cerebrospinal Fluid alpha-Synuclein Levels in Parkinson’s Disease: A Meta-Analysis. Park. Dis. 2015, 2015, 567386.
- Xiang, C.; Cong, S.; Tan, X.; Ma, S.; Liu, Y.; Wang, H.; Cong, S. A meta-analysis of the diagnostic utility of biomarkers in cerebrospinal fluid in Parkinson’s disease. NPJ Park. Dis. 2022, 8, 165.
- Hoftberger, R.; Fink, S.; Aboul-Enein, F.; Botond, G.; Olah, J.; Berki, T.; Ovadi, J.; Lassmann, H.; Budka, H.; Kovacs, G.G. Tubulin polymerization promoting protein (TPPP/p25) as a marker for oligodendroglial changes in multiple sclerosis. Glia 2010, 58, 1847–1857.
- Vincze, O.; Olah, J.; Zadori, D.; Klivenyi, P.; Vecsei, L.; Ovadi, J. A new myelin protein, TPPP/p25, reduced in demyelinated lesions is enriched in cerebrospinal fluid of multiple sclerosis. Biochem. Biophys. Res. Commun. 2011, 409, 137–141.
- Rodger, A.T.; ALNasser, M.; Carter, W.G. Are Therapies That Target alpha-Synuclein Effective at Halting Parkinson’s Disease Progression? A Systematic Review. Int. J. Mol. Sci. 2023, 24, 11022.
- Pujols, J.; Pena-Diaz, S.; Lazaro, D.F.; Peccati, F.; Pinheiro, F.; Gonzalez, D.; Carija, A.; Navarro, S.; Conde-Gimenez, M.; Garcia, J.; et al. Small molecule inhibits alpha-synuclein aggregation, disrupts amyloid fibrils, and prevents degeneration of dopaminergic neurons. Proc. Natl. Acad. Sci. USA 2018, 115, 10481–10486.
- Wagner, J.; Ryazanov, S.; Leonov, A.; Levin, J.; Shi, S.; Schmidt, F.; Prix, C.; Pan-Montojo, F.; Bertsch, U.; Mitteregger-Kretzschmar, G.; et al. Anle138b: A novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta Neuropathol. 2013, 125, 795–813.
- Allen, S.G.; Meade, R.M.; White Stenner, L.L.; Mason, J.M. Peptide-based approaches to directly target alpha-synuclein in Parkinson’s disease. Mol. Neurodegener. 2023, 18, 80.
- Nim, S.; O’Hara, D.M.; Corbi-Verge, C.; Perez-Riba, A.; Fujisawa, K.; Kapadia, M.; Chau, H.; Albanese, F.; Pawar, G.; De Snoo, M.L.; et al. Disrupting the alpha-synuclein-ESCRT interaction with a peptide inhibitor mitigates neurodegeneration in preclinical models of Parkinson’s disease. Nat. Commun. 2023, 14, 2150.
- Simon, C.; Soga, T.; Ahemad, N.; Bhuvanendran, S.; Parhar, I. Kisspeptin-10 Rescues Cholinergic Differentiated SHSY-5Y Cells from alpha-Synuclein-Induced Toxicity In Vitro. Int. J. Mol. Sci. 2022, 23, 5193.
- Kulesskaya, N.; Bhattacharjee, A.; Holmstrom, K.M.; Vuorio, P.; Henriques, A.; Callizot, N.; Huttunen, H.J. HER-096 is a CDNF-derived brain-penetrating peptidomimetic that protects dopaminergic neurons in a mouse synucleinopathy model of Parkinson’s disease. Cell Chem. Biol. 2023.
- Uversky, V.N. A protein-chameleon: Conformational plasticity of alpha-synuclein, a disordered protein involved in neurodegenerative disorders. J. Biomol. Struct. Dyn. 2003, 21, 211–234.
- Sulzer, D.; Edwards, R.H. The physiological role of alpha-synuclein and its relationship to Parkinson’s Disease. J. Neurochem. 2019, 150, 475–486.
- Selkoe, D.; Dettmer, U.; Luth, E.; Kim, N.; Newman, A.; Bartels, T. Defining the native state of alpha-synuclein. Neuro-Degener. Dis. 2014, 13, 114–117.
- Cascella, R.; Bigi, A.; Cremades, N.; Cecchi, C. Effects of oligomer toxicity, fibril toxicity and fibril spreading in synucleinopathies. Cell. Mol. Life Sci. CMLS 2022, 79, 174.
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl. Acad. Sci USA 2011, 108, 4194–4199.

