There are millions of people suffering from Parkinsonism in the world and there is neither a medicine to cure the disease nor an appropriate test to detect the illness at an early stage and to follow the disease/treatment processes at the clinical level [
1,
2]. Alpha-synuclein (SYN) plays a key role in Parkinsonism, which includes Parkinson’s disease (PD), dementia with Lewy bodies (DLB), and multiple system atrophy (MSA) [
3,
4,
5,
6,
7]. Parkinsonism can be characterized by the classical motor symptoms (such as resting tremor, rigidity, bradykinesia, and freezing of gait) as well as non-motor features (including constipation, depression, sleep disorder, cognitive impairment, and dementia) [
6]. In the case of MSA, patients may exhibit either Parkinsonism (MSA-P) or cerebellar ataxia (MSA-C). Despite the many shared symptoms, these diseases differ in the rate of disease progression. Median survival is much shorter for DLB and MSA patients than for PD ones. Current treatments include different therapies (mostly affecting dopamine metabolism, for example, by levodopa), deep brain stimulation, active and passive immunotherapy, and other options for transformative treatment (such as stem cell transplants or gene-targeted treatments) [
1,
8,
9]. Levodopa relieves the motor symptoms through the replacement of lost dopamine; however, MSA-P patients are usually less responsive to levodopa therapy, and it can also worsen neuropsychiatric symptoms [
6].
A major characteristic of these neurological disorders is the atypical protein assembly (proteopathy) leading to cell death, which is one of the key mechanisms of many neurodegenerative diseases. In pathological conditions, the aggregation of SYN results in the formation of insoluble fibrils, which had long been considered the primary structural components of synucleinopathies [
3,
4,
10]. However, there are also striking differences between the different synucleinopathies as there are differences in the brain regions affected and in the cell types with inclusion bodies [
6]. SYN aggregation is typically observed in the substantia nigra pars compacta in PD and in the cerebral cortex and hippocampus in DLB, while the olivopontocerebellar, nigrostriatal, and autonomic systems are affected in MSA. In PD and DLB, SYN-bearing Lewy bodies and Lewy neurites can be observed in neurons, while MSA is characterized by aggregated SYN in glial cytoplasmic inclusions [
3,
4,
5]. The pathological SYN strains from Lewy bodies and glial cytoplasmic inclusions seem to be conformationally and biologically distinct [
11].
In the last decade, however, Tubulin Polymerization Promoting Protein (TPPP) has also emerged as one of the principal actors in the processes underlying neurological disorders [
12,
13,
14]. Pathological interactions of TPPP with SYN can induce SYN assembly that leads to the formation of inclusions as pathological hallmarks characteristic of PD, DLB, and MSA [
12,
15]. TPPP, as a Neomorphic Moonlighting Protein, plays an important role in both physiological and pathological conditions without alterations at the gene level [
16]. TPPP is expressed specifically in oligodendrocytes (OLGs) in the normal brain during differentiation of the progenitor cells, where it is crucial to the formation of projections that results in the ensheathment of the axons [
17]. This ensheathment enables highly efficient signal transmission; moreover, OLGs also provide metabolic and trophic support to the neurons [
18]. TPPP is not present in astroglia or microglia [
17,
19]. In physiological conditions, TPPP modulates the dynamics and stability of the cytoskeletal microtubule system via its bundling and tubulin acetylation-promoting activities [
13]. These physiological functions are mediated by its direct associations with tubulin/microtubules as well as with tubulin deacetylases such as histone deacetylase 6 and sirtuin-2 (SIRT2) [
20,
21]. The bundling and stabilization of microtubules result from the dimerization of the tubulin-attached monomeric TPPP [
22]. The disordered TPPP has a zinc-finger motif and also has a Mg
2+-dependent GTPase activity [
23,
24].
2. Pathological Interaction between TPPP and SYN
An important aspect of Parkinsonism is that the partner proteins in the pathological assembly are expressed in distinct cell types in normal brain: SYN in neurons and TPPP in OLGs, respectively [
17,
31,
32,
33]. This raises the question as to which mechanism can be responsible for their co-localization.
One possibility is the cell-to-cell transmission of SYN from neurons via the extracellular space [
34,
35]. Exosomes, classical exocytosis, and endocytosis as well as direct penetration can be involved in the cell-to-cell transmission of SYN [
36]. The levels of exosomal total and oligomeric SYN increased in plasma in PD patients as compared to healthy controls [
36]. Using immunofluorescence confocal microscopy on its own or coupled with Bifunctional Fluorescence Complementation, the researchers' experiments revealed the co-location/co-enrichment of these two proteins in living human HeLa cells transiently transfected with TPPP and SYN [
37]. The assembly of SYN and TPPP could be observed as well in the cells after their uptake from the medium [
16,
38]. This experimental setup mimics the pathological situation occurring in human brain where these proteins are transmitted via the extracellular space. The mechanisms involved in the cell-to-cell transmission of TPPP are not known. Interestingly, TPPP was detected in exosomes isolated from SH-SY5Y cells by LC-MS/MS-based label-free quantitative proteomics analysis (
http://exocarta.org/gene_summary?gene_id=11076, accessed on 18 December 2023) [
39].
In pathological conditions, another possibility of the inappropriate presence of SYN and TPPP in OLGs and neurons, respectively, might be the translation of their mRNAs. According to the data in the Human Protein Atlas, both SYN mRNA (SNCA: 243.0 nTPM and 51.5 nTPM in excitatory and inhibitory neurons, 89.6 nTPM and 181.8 nTPM in OLG precursor cells and OLGs, respectively) and TPPP mRNA (TPPP: 43.7 nTPM and 31.2 nTPM in excitatory and inhibitory neurons, 6.1 nTPM and 97.5 nTPM in OLG precursor cells and OLGs, respectively) are detected in OLGs and neurons in normal human brains (
https://www.proteinatlas.org/ENSG00000145335-SNCA/single+cell+type,
https://www.proteinatlas.org/ENSG00000171368-TPPP/single+cell+type, accessed on 27 October 2023). Recently, single-nucleus RNA sequencing revealed a high level of SNCA transcripts in inhibitory neurons and OLG progenitor cells but only a low level of these transcripts in excitatory neurons and mature OLGs [
40]. In the case of TPPP, its mRNA is present in neurons but the protein itself is only present in OLGs [
17,
41]. Hence, possible pathological factors include a failure to inhibit translation of the inappropriate mRNA (an inhibition that should occur in physiological conditions) and/or a failure to degrade the newly synthesized protein; these possibilities would be consistent with the absence of the SYN protein in mature OLGs despite the presence of its mRNA. Another possibility would be a failure to put the protein in a place where it would not cause a problem.
The results of recent research are consistent with the above possibilities: the addition of human preformed SYN fibrils to mouse OLGs triggered an increase in endogenous SYN, which was critical for its aggregation with TPPP [
14]; in agreement with this, in rat OLN-AS7 and human MO3.13 OLG models of MSA, TPPP transfection significantly increased the levels of both SYN mRNA and SYN protein [
42]. Moreover, SYN fibrils interfered with the production of proteins associated with neuromodulation and myelination [
43]. Regardless of the exact contribution of these mechanisms, SYN-TPPP assembly is characteristic of PD, DLB, and MSA and is only pathological [
12].
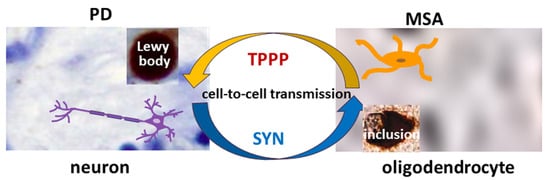
3. The Role of TPPP in the Pathomechanism of Synucleinopathies: Protein Aggregation
TPPP is co-localized with SYN in Lewy bodies in PD and DLB as well as in glial cytoplasmic inclusions in MSA [
12,
15] (
Figure 1). Therefore, TPPP—like SYN—is considered a hallmark of synucleinopathies [
12]. A TPPP-induced SYN strain had different structure and enhanced in vivo prodegenerative properties as compared to the SYN strain; moreover, injection of the preformed fibrils of the SYN-TPPP strain resulted in a shortened lifespan in a mouse model [
44].
Figure 1. The role of SYN and TPPP in the formation of Lewy bodies and glial cytoplasmic inclusions in PD and MSA.
Analysis of the role of TPPP also demonstrated that TPPP is enriched in SYN-bearing Lewy bodies in both PD and DLB [
12,
46]. The oligodendroglial pathology in DLB is generally considered less significant than in MSA. Comparison of DLB and MSA did, however, reveal both shared and distinct patterns. The immunoreactivity of TPPP in the oligodendroglia cytoplasm differed more from the controls for MSA than for DLB; a disintegration of myelin with loss of TPPP nuclear staining was characteristic of MSA, while DLB was more similar to the controls without showing a significant loss of nuclear TPPP.
4. The Role of TPPP in the Dysregulation of Protein Degradation in Parkinsonism
In normal human brain, levels of unwanted proteins as well as aggregates are controlled by the ubiquitin–proteasome system (UPS) and the autophagy–lysosome pathways, which can remove troublesome proteins. One target of these degradative processes is the set of intrinsically disordered proteins (IDPs) involved in the formation of toxic aggregates. Monomeric proteins such as SYN and TPPP are usually degraded by the UPS, while macroautophagy is able to degrade oligomers or aggregates ([
50] and references therein). SYN can also be degraded by chaperone-mediated autophagy and endo-lysosomal degradation. Dysregulation of the cellular degradative pathways has been reported in Parkinsonism, and autophagy modulation has been suggested as a possible strategy for therapeutic intervention [
51].
5. SYN and TPPP as Biomarkers
Synaptic dysfunction and degeneration are central contributors to the pathogenesis and progression of Parkinsonian disorders. Therefore, identification and validation of biomarkers reflecting pathological synaptic alterations are greatly needed [
54]. Human cerebrospinal fluid (CSF) has a composition similar to that of the brain extracellular fluid (ECF), and the two fluids circulate freely together in the brain [
55].
In a recent webinar conference organized by the Michael J. Fox Foundation entitled “Major Research Breakthrough: A New Biomarker for Parkinson’s” (20 April 2023) the significance of the SYN seed amplification assay (SAA) using CSF was highlighted [
56]. This new assay may allow the aggregation of SYN to be used as a biomarker in order to distinguish people with PD from healthy controls even at an early stage of the disease. The cross-sectional analysis included 1123 participants; the assay classified PD patients with high sensitivity and specificity, and could detect low-abundancy SYN. That said, application of this assay for routine laboratory tests may prove challenging. Nevertheless, use of the SYN SAA for biochemical diagnosis of PD and MSA reveals the crucial role it could play in therapeutic development [
56].
In the context of the exploration of candidate biomarkers for MSA, disregarding its close relationship with PD does not seem justifiable. Several recent studies have shown that SYN might be a potential diagnostic biomarker for PD in CSF though the results are inconsistent [
57,
58,
59]. The results of a meta-analysis investigating the diagnostic and differential diagnosis efficacy of CSF SYN in PD have shown that its median concentration is significantly lower in PD compared to controls, but significantly higher in PD as compared to MSA [
60,
61]. However, the median concentration of CSF SYN oligomers was significantly higher in PD than in controls [
60]. This could be due to the fact that the presence of TPPP in the neurons promotes the SYN assembly where these proteins are co-enriched and co-located in the well-separated, individual Lewy bodies.
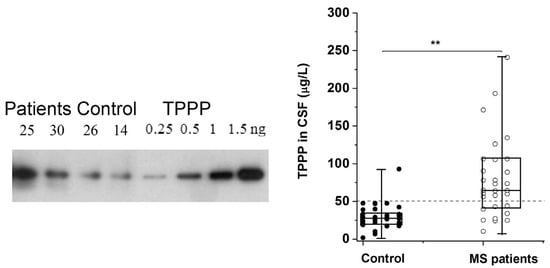
The altered regulation/location of TPPP results in its pathological assembly with SYN in MSA, which leads to demyelination as in the case of Multiple Sclerosis (MS). MS is a chronic, inflammatory, demyelinating disease with a variable extent of remyelination depending on the differentiation of OLGs [
62]. The researchers have developed and validated a sensitive assay based on Western blotting coupled with chemiluminescent detection using human recombinant TPPP and CSF for the quantification of TPPP in the case of different MS patients and controls [
63] (
Figure 2). According to this assay, the median TPPP content of the CSF was 64.7 μg/L for patients, while it was 27.9 μg/L for non-MS patients. The higher level of TPPP in the MS patients was independent of age, gender, and the time between lumbar puncture and relapse [
63]. These results suggest that (i) TPPP-based assay with CSF of the patients may be suitable for the diagnostic testing of MS patients and (ii) this assay could be used to measure TPPP levels in the CSF of MSA and PD patients.
Figure 2. TPPP levels in the CSF of MS and non-MS patients as quantified by Western blot [
63]. A representative Western blot using a specific TPPP antibody is shown for different CSF samples of patients (25 and 30, ○) and the corresponding controls (non-MS patients, ●) (14 and 26). Each box extends from the 25th to the 75th percentile with the middle line representing the median. The vertical bars indicate the full range of TPPP levels. The
p values were determined by Mann–Whitney U tests. **
p < 0.000005. The dashed line corresponds to 50 μg/L [
63].
6. Targeting the Interface of the Pathological SYN-TPPP Complex
Efficient therapies and disease-modifying treatments are under intense research. A search for Parkinsonism produced 100 clinical trials, 57 of which were interventional (
https://clinicaltrials.gov, accessed on 1 December 2023).
A potential strategy to eliminate the excess of these hallmark proteins as well as their toxic aggregates includes the use of small molecules (such as Epigallocatechin gallate, Anle138b, or SynuClean-D), which have the advantages of their usually low cost, high stability, and bioavailability [
64,
65,
66]. However, IDPs such as SYN and TPPP often lack the pocket needed to bind these molecules; an alternative strategy to inhibit protein–protein interactions is to use peptides and peptidomimetics, which is now a rapidly progressing field. Short peptides may have high specificity and low toxicity, although poor bioavailability and limited cellular uptake may pose problems [
67]. Peptides that target different SYN regions have indeed been identified [
67]. For example, Nim and co-workers found PDpep1.3 by a high-throughput, proteome-wide peptide screen; this peptide reduced the accumulation of SYN in a rat model of PD [
68]. A neurohormone peptide and a cerebral dopamine neurotrophic factor (CDNF)-derived peptidomimetic also displayed beneficial effects in PD models [
69,
70].
A therapeutic strategy based on the targeting of either TPPP or SYN is not without risks. One reason for this is that, as IDPs, the structures of these ‘chameleon’ proteins can adopt many conformations, some of which can perturb the drug target [
37,
71]. As demonstrated previously, the SYN protein functions physiologically as a monomer; however, its occurrence as a metastable tetramer in a dynamic equilibrium with the monomers has also been suggested [
72,
73]. Another reason is that they are responsible for essential physiological functions that must be preserved.
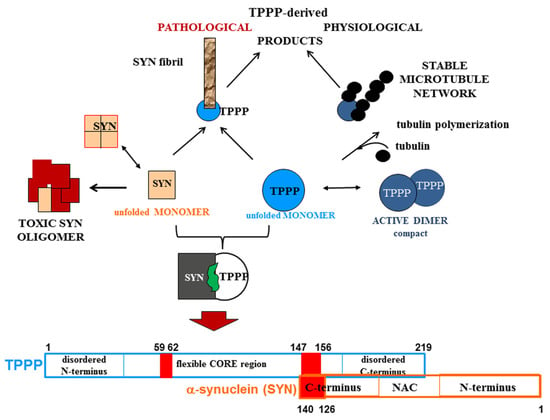
A strategy that the researchers have developed and reported is the drug targeting of the hetero-complexes of SYN and TPPP [
16,
37,
38]; this is because these complexes, which are only pathological, are the initiators of the neurodegenerative processes [
12,
14,
74,
75] (
Figure 3). This strategy is based on targeting the interface where SYN and TPPP are in contact so as to trigger the disassembly of the pathological complexes [
16,
37,
38]. The segments of the interface of both SYN and TPPP that are involved in the formation of their assemblies have been identified by studying the effects of several mutated and truncated forms of the proteins on their association [
16,
37,
38]. The researchers' studies revealed that the truncation of the C-terminal segment of SYN did prevent its hetero-association with TPPP [
38]. This suggests that the SYN126-140 fragment may function as a competitive inhibitor of the association of the two proteins in vitro as well as in a human cell model, where the proteins were added to the medium of CHO cells [
38]. Taken together, these findings substantiate drug-targeting strategies based upon (i) elimination of the toxic species; (ii) proteolytic degradation of the excess and/or pathological partner; and (iii) maintenance/recovery of the physiological function of SYN/TPPP. Moreover, strategies for selecting drugs must also take into account the physiological functions of the validated sites of the hallmark proteins.
Figure 3. Homo- and hetero-associations of TPPP and SYN in physiological and pathological conditions and the interface segment of the SYN-TPPP complex as a potential drug target.
One of the novelties of the strategy is that it is based on the crucial role of TPPP in the formation of SYN assemblies and in the deregulation of their cellular proteolysis; hence, targeting the SYN-TPPP interaction could eliminate the toxic SYN-TPPP assemblies. For this strategy to work, the researchers suggest that autophagy modulation by itself may not be sufficient and that the inhibition/destruction of SYN-TPPP assemblies is also necessary. Such elimination of these assemblies may require adopting and adapting new and emergent techniques.