 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Anne Negre-Salvayre | -- | 4089 | 2024-02-29 09:40:04 | | | |
| 2 | Mona Zou | Meta information modification | 4089 | 2024-03-01 10:03:44 | | |
Video Upload Options
Atherosclerosis is a multifactorial disease of medium and large arteries, characterized by the presence of lipid-rich plaques lining the intima over time. It is the main cause of cardiovascular diseases and death worldwide. Redox imbalance and lipid peroxidation could play key roles in atherosclerosis by promoting a bundle of responses, including endothelial activation, inflammation, and foam cell formation. The oxidation of polyunsaturated fatty acids generates various lipid oxidation products such as reactive carbonyl species (RCS), including 4-hydroxy alkenals, malondialdehyde, and acrolein. RCS covalently bind to nucleophilic groups of nucleic acids, phospholipids, and proteins, modifying their structure and activity and leading to their progressive dysfunction. Protein lipoxidation is the non-enzymatic post-translational modification of proteins by RCS. Low-density lipoprotein (LDL) oxidation and apolipoprotein B (apoB) modification by RCS play a major role in foam cell formation. Moreover, oxidized LDLs are a source of RCS, which form adducts on a huge number of proteins, depending on oxidative stress intensity, the nature of targets, and the availability of detoxifying systems. Many systems are affected by lipoxidation, including extracellular matrix components, membranes, cytoplasmic and cytoskeletal proteins, transcription factors, and other components.
1. Introduction
2. LDL Oxidation and Formation of RCS
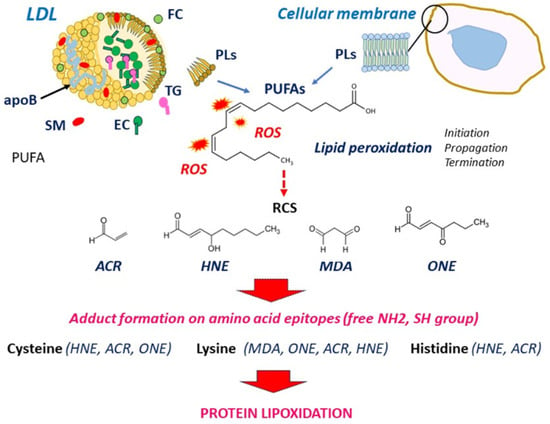
3. Protein Lipoxidation in the Vascular Wall
3.1. Modification of apoB by RCS in LDL: A Main Role in Foam Cell Formation
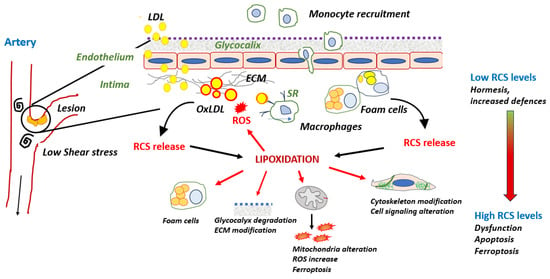
3.2. RCS and Inflammation
Lipoxidation of Transcription Factors and Inflammation
-
NF-κB
-
NLRP3
-
Nrf2
Cyclooxygenase-2 Activation
RCS and ROS
-
Effect of RCS on Mitochondrial ROS Production
-
Effect of RCS on Antioxidant Systems
- -
-
ALDH2
- -
-
Glutathione-S Transferases (GSTs)
- -
-
Thioredoxin 1
- -
-
Peroxiredoxins
-
Effect of RCS on eNOS
3.3. Lipoxidation of Endothelial Barrier Components
Glycocalyx
Extracellular Matrix Proteins
Cytoskeleton Proteins
-
Actin
-
Tubulin
-
Vimentin
-
Integrins and focal adhesions
Cell Signaling Kinases and Growth Factor Receptors
References
- Calabrese, E.J.; Osakabe, N.; Di Paola, R.; Siracusa, R.; Fusco, R.; D’Amico, R.; Impellizzeri, D.; Cuzzocrea, S.; Fritsch, T.; Abdelhameed, A.S.; et al. Hormesis defines the limits of lifespan. Ageing Res. Rev. 2023, 91, 102074.
- Mann, G.E. Nrf2-mediated redox signalling in vascular health and disease. Free Radic. Biol. Med. 2014, 75 (Suppl. S1), S1.
- Nègre-Salvayre, A.; Garoby-Salom, S.; Swiader, A.; Rouahi, M.; Pucelle, M.; Salvayre, R. Proatherogenic effects of 4-hydroxynonenal. Free Radic. Biol. Med. 2017, 111, 127–139.
- Gianazza, E.; Brioschi, M.; Martinez Fernandez, A.; Casalnuovo, F.; Altomare, A.; Aldini, G.; Banfi, C. Lipid Peroxidation in Atherosclerotic Cardiovascular Diseases. Antioxid. Redox Signal 2021, 34, 49–98.
- Frangie, C.; Daher, J. Role of myeloperoxidase in inflammation and atherosclerosis. Biomed. Rep. 2022, 16, 53.
- Negre-Salvayre, A.; Guerby, P.; Gayral, S.; Laffargue, M.; Salvayre, R. Role of reactive oxygen species in atherosclerosis: Lessons from murine genetic models. Free Radic. Biol. Med. 2020, 149, 8–22.
- Jiang, X.; Yang, Z.; Chandrakala, A.N.; Pressley, D.; Parthasarathy, S. Oxidized low density lipoproteins--do we know enough about them? Cardiovasc. Drugs Ther. 2011, 25, 367–377.
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell Longev. 2014, 2014, 360438.
- Reis, A.; Spickett, C.M. Chemistry of phospholipid oxidation. Biochim. Biophys. Acta 2012, 1818, 2374–2387.
- Sousa, B.C.; Pitt, A.R.; Spickett, C.M. Chemistry and analysis of HNE and other prominent carbonyl-containing lipid oxidation compounds. Free Radic. Biol. Med. 2017, 111, 294–308.
- Afonso, C.B.; Spickett, C.M. Lipoproteins as targets and markers of lipoxidation. Redox Biol. 2019, 23, 101066.
- Viedma-Poyatos, Á.; González-Jiménez, P.; Langlois, O.; Company-Marín, I.; Spickett, C.M.; Pérez-Sala, D. Protein Lipoxidation: Basic Concepts and Emerging Roles. Antioxidants 2021, 10, 295.
- Spickett, C.M.; Pitt, A.R. Modification of proteins by reactive lipid oxidation products and biochemical effects of lipoxidation. Essays Biochem. 2020, 64, 19–31.
- Guéraud, F.; Atalay, M.; Bresgen, N.; Cipak, A.; Eckl, P.M.; Huc, L.; Jouanin, I.; Siems, W.; Uchida, K. Chemistry and biochemistry of lipid peroxidation products. Free Radic. Res. 2010, 44, 1098–1124.
- Milkovic, L.; Zarkovic, N.; Marusic, Z.; Zarkovic, K.; Jaganjac, M. The 4-Hydroxynonenal-Protein Adducts and Their Biological Relevance: Are Some Proteins Preferred Targets? Antioxidants 2023, 12, 856.
- Winklhofer-Roob, B.M.; Faustmann, G.; Roob, J.M. Low-density lipoprotein oxidation biomarkers in human health and disease and effects of bioactive compounds. Free Radic. Biol. Med. 2017, 111, 38–86.
- Schaur, R.J.; Siems, W.; Bresgen, N. Eckl PM. 4-Hydroxy-nonenal-A Bioactive Lipid Peroxidation Product. Biomolecules 2015, 5, 2247–2337.
- Gianazza, E.; Brioschi, M.; Fernandez, A.M.; Banfi, C. Lipoxidation in cardiovascular diseases. Redox Biol. 2019, 23, 101119.
- Lee, S.H.; Blair, I.A. Characterization of 4-oxo-2-nonenal as a novel product of lipid peroxidation. Chem. Res. Toxicol. 2000, 13, 698–702.
- Doorn, J.A.; Petersen, D.R. Covalent adduction of nucleophilic amino acids by 4-hydroxynonenal and 4-oxononenal. Chem. Biol. Interact. 2003, 143–144, 93–100.
- Zadeh, M.; Mota-Martorell, N.; Pradas, I.; Martín-Gari, M.; Ayala, V.; Pamplona, R. The Advanced Lipoxidation End-Product Malondialdehyde-Lysine in Aging and Longevity. Antioxidants 2020, 9, 1132.
- Lankin, V.Z.; Tikhaze, A.K.; Melkumyants, A.M. Malondialdehyde as an Important Key Factor of Molecular Mechanisms of Vascular Wall Damage under Heart Diseases Development. Int. J. Mol. Sci. 2022, 24, 128.
- Pirillo, A.; Norata, G.D.; Catapano, A.L. LOX-1, OxLDL, and atherosclerosis. Mediators Inflamm. 2013, 2013, 152786.
- Park, Y.M. CD36, a scavenger receptor implicated in atherosclerosis. Exp. Mol. Med. 2014, 46, e99.
- Tian, K.; Xu, Y.; Sahebkar, A.; Xu, S. CD36 in Atherosclerosis: Pathophysiological Mechanisms and Therapeutic Implications. Curr. Atheroscler. Rep. 2020, 22, 59.
- Levitan, I.; Volkov, S.; Subbaiah, P.V. Oxidized LDL: Diversity, patterns of recognition, and pathophysiology. Antioxid. Redox Signal 2010, 13, 39–75.
- Qin, S. LDL and HDL Oxidative Modification and Atherosclerosis. Adv. Exp. Med. Biol. 2020, 1276, 157–169.
- Goyal, T.; Mitra, S.; Khaidakov, M.; Wang, X.; Singla, S.; Ding, Z.; Liu, S.; Mehta, J.L. Current Concepts of the Role of Oxidized LDL Receptors in Atherosclerosis. Curr. Atheroscler. Rep. 2012, 14, 150–159.
- Hoff, H.F.; O’Neil, J.; Chisolm GM 3rd Cole, T.B.; Quehenberger, O.; Esterbauer, H.; Jürgens, G. Modification of low density lipoprotein with 4-hydroxynonenal induces uptake by macrophages. Arteriosclerosis 1989, 9, 538–549.
- Jürgens, G.; Lang, J.; Esterbauer, H. Modification of human low-density lipoprotein by the lipid peroxidation product 4-hydroxynonenal. Biochim. Biophys. Acta 1986, 875, 103–114.
- Watanabe, K.; Nakazato, Y.; Saiki, R.; Igarashi, K.; Kitada, M.; Ishii, I. Acrolein-conjugated low-density lipoprotein induces macrophage foam cell formation. Atherosclerosis 2013, 227, 51–57.
- Bräsen, J.H.; Häkkinen, T.; Malle, E.; Beisiegel, U.; Ylä-Herttuala, S. Patterns of oxidized epitopes, but not NF-kappa B expression, change during atherogenesis in WHHL rabbits. Atherosclerosis 2003, 166, 13–21.
- Yamada, S.; Funada, T.; Shibata, N.; Kobayashi, M.; Kawai, Y.; Tatsuda, E.; Furuhata, A.; Uchida, K. Protein-bound 4-hydroxy-2-hexenal as a marker of oxidized n-3 polyunsaturated fatty acids. J. Lipid Res. 2004, 45, 626–634.
- Uchida, K.; Toyokuni, S.; Nishikawa, K.; Kawakishi, S.; Oda, H.; Hiai, H.; Stadtman, E.R. Michael addition-type 4-hydroxy-2-nonenal adducts in modified low-density lipoproteins: Markers for atherosclerosis. Biochemistry 1994, 33, 12487–12494.
- Frijhoff, J.; Winyard, P.G.; Zarkovic, N.; Davies, S.S.; Stocker, R.; Cheng, D.; Knight, A.R.; Taylor, E.L.; Oettrich, J.; Ruskovska, T.; et al. Clinical Relevance of Biomarkers of Oxidative Stress. Antioxid. Redox Signal 2015, 23, 1144–1170.
- Libby, P. Inflammation in Atherosclerosis-No Longer a Theory. Clin. Chem. 2021, 67, 131–142.
- Williams, K.J.; Tabas, I. The response-to-retention hypothesis of early atherogenesis. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 551–561.
- de Winther, M.P.; Kanters, E.; Kraal, G.; Hofker, M.H. Nuclear factor kappaB signaling in atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 904–914.
- Mimura, J.; Itoh, K. Role of Nrf2 in the pathogenesis of atherosclerosis. Free Radic. Biol. Med. 2015, 88 Pt B, 221–232.
- Matsumori, A. Nuclear Factor-κB is a Prime Candidate for the Diagnosis and Control of Inflammatory Cardiovascular Disease. Eur. Cardiol. 2023, 18, e40.
- Tall, A.R.; Bornfeldt, K.E. Inflammasomes and Atherosclerosis: A Mixed Picture. Circ. Res. 2023, 132, 1505–1520.
- Ruef, J.; Moser, M.; Bode, C.; Kübler, W.; Runge, M.S. 4-hydroxynonenal induces apoptosis, NF-kappaB-activation and formation of 8-isoprostane in vascular smooth muscle cells. Basic. Res. Cardiol. 2001, 96, 143–150.
- Lee, S.J.; Seo, K.W.; Yun, M.R.; Bae, S.S.; Lee, W.S.; Hong, K.W.; Kim, C.D. 4-Hydroxynonenal enhances MMP-2 production in vascular smooth muscle cells via mitochondrial ROS-mediated activation of the Akt/NF-kappaB signaling pathways. Free Radic. Biol. Med. 2008, 45, 1487–1492.
- Lee, S.J.; Kim, C.E.; Seo, K.W.; Kim, C.D. HNE-induced 5-LO expression is regulated by NF-B/ERK and Sp1/p38 MAPK pathways via EGF receptor in murine macrophages. Cardiovasc. Res. 2010, 88, 352–359.
- Gargiulo, S.; Gamba, P.; Testa, G.; Rossin, D.; Biasi, F.; Poli, G.; Leonarduzzi, G. Relation between TLR4/NF-κB signaling pathway activation by 27-hydroxycholesterol and 4-hydroxynonenal, and atherosclerotic plaque instability. Aging Cell 2015, 14, 569–581.
- Je, J.H.; Lee, J.Y.; Jung, K.J.; Sung, B.; Go, E.K.; Yu, B.P.; Chung, H.Y. NF-kappaB activation mechanism of 4-hydroxyhexenal via NIK/IKK and p38 MAPK pathway. FEBS Lett. 2004, 566, 183–189.
- Page, S.; Fischer, C.; Baumgartner, B.; Haas, M.; Kreusel, U.; Loidl, G.; Hayn, M.; Ziegler-Heitbrock, H.W.; Neumeier, D.; Brand, K. 4-Hydroxynonenal prevents NF-kappaB activation and tumor necrosis factor expression by inhibiting IkappaB phosphorylation and subsequent proteolysis. J. Biol. Chem. 1999, 274, 11611–11618.
- Dou, X.; Li, S.; Wang, Z.; Gu, D.; Shen, C.; Yao, T.; Song, Z. Inhibition of NF-κB activation by 4-hydroxynonenal contributes to liver injury in a mouse model of alcoholic liver disease. Am. J. Pathol. 2012, 181, 1702–1710.
- Byun, M.S.; Choi, J.; Jue, D. Cysteine-179 of IkappaB kinase beta plays a critical role in enzyme activation by promoting phosphorylation of activation loop serines. Exp. Mol. Med. 2006, 38, 546–552.
- Moldogazieva, N.T.; Zavadskiy, S.P.; Astakhov, D.V.; Terentiev, A.A. Lipid peroxidation: Reactive carbonyl species, protein/DNA adducts, and signaling switches in oxidative stress and cancer. Biochem. Biophys. Res. Commun. 2023, 687, 149167.
- Ji, C.; Kozak, K.R.; Marnett, L.J. IkappaB kinase, a molecular target for inhibition by 4-hydroxy-2-nonenal. J. Biol. Chem. 2001, 276, 18223–18228.
- Tirumalai, R.; Rajesh Kumar, T.; Mai, K.H.; Biswal, S. Acrolein causes transcriptional induction of phase II genes by activation of Nrf2 in human lung type II epithelial (A549) cells. Toxicol. Lett. 2002, 132, 27–36.
- Valacchi, G.; Pagnin, E.; Phung, A.; Nardini, M.; Schock, B.C.; Cross, C.E.; van der Vliet, A. Inhibition of NFkappaB activation and IL-8 expression in human bronchial epithelial cells by acrolein. Antioxid. Redox Signal 2005, 7, 25–31.
- Hattori, Y.; Hattori, S.; Kasai, K. 4-hydroxynonenal prevents NO production in vascular smooth muscle cells by inhibiting nuclear factor-kappaB-dependent transcriptional activation of inducible NO synthase. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1179–1183.
- Chapple, S.J.; Cheng, X.; Mann, G.E. Effects of 4-hydroxynonenal on vascular endothelial and smooth muscle cell redox signaling and function in health and disease. Redox Biol. 2013, 1, 319–331.
- Karasawa, T.; Takahashi, M. Role of NLRP3 Inflammasomes in Atherosclerosis. J. Atheroscler. Thromb. 2017, 24, 443–451.
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361, Erratum in Nature 2010, 466, 652.
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687.
- Hsu, C.G.; Chávez, C.L.; Zhang, C.; Sowden, M.; Yan, C.; Berk, B.C. The lipid peroxidation product 4-hydroxynonenal inhibits NLRP3 inflammasome activation and macrophage pyroptosis. Cell Death Differ. 2022, 29, 1790–1803.
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Nunez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357.
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116.
- Ma, Q. Role of Nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426.
- Ma, Q.; He, X. Molecular basis of electrophilic and oxidative defense: Promises and perils of Nrf2. Pharmacol. Rev. 2012, 64, 1055–1081.
- Jakobs, P.; Serbulea, V.; Leitinger, N.; Eckers, A.; Haendeler, J. Nuclear Factor (Erythroid-Derived 2)-Like 2 and Thioredoxin-1 in Atherosclerosis and Ischemia/Reperfusion Injury in the Heart. Antioxid. Redox Signal 2017, 26, 630–644.
- Uruno, A.; Motohashi, H. The Keap1-Nrf2 system as an in vivo sensor for electrophiles. Nitric Oxide 2011, 25, 153–160.
- Pajares, M.; Jiménez-Moreno, N.; García-Yagüe, Á.J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rábano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916.
- Carbone, D.L.; Doorn, J.A.; Kiebler, Z.; Petersen, D.R. Cysteine modification by lipid peroxidation products inhibits protein disulfide isomerase. Chem. Res. Toxicol. 2005, 18, 1324–1331.
- da Costa, R.M.; Rodrigues, D.; Pereira, C.A.; Silva, J.F.; Alves, J.V.; Lobato, N.S.; Tostes, R.C. Nrf2 as a Potential Mediator of Cardiovascular Risk in Metabolic Diseases. Front. Pharmacol. 2019, 10, 382.
- Ishii, T.; Itoh, K.; Ruiz, E.; Leake, D.S.; Unoki, H.; Yamamoto, M.; Mann, G.E. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: Activation by oxidatively modified LDL and 4-hydroxynonenal. Circ. Res. 2004, 94, 609–616.
- Harada, N.; Ito, K.; Hosoya, T.; Mimura, J.; Maruyama, A.; Noguchi, N.; Yagami, K.; Morito, N.; Takahashi, S.; Maher, J.M.; et al. Nrf2 in bone marrow-derived cells positively contributes to the advanced stage of atherosclerotic plaque formation. Free Radic. Biol. Med. 2012, 53, 2256–2262.
- Uchida, K. HNE as an inducer of COX-2. Free Radic. Biol. Med. 2017, 111, 169–172.
- Chacko, B.K.; Wall, S.B.; Kramer, P.A.; Ravi, S.; Mitchell, T.; Johnson, M.S.; Wilson, L.; Barnes, S.; Landar, A.; Darley-Usmar, V.M. Pleiotropic effects of 4-hydroxynonenal on oxidative burst and phagocytosis in neutrophils. Redox Biol. 2016, 9, 57–66.
- Azzu, V.; Parker, N.; Brand, M.D. High membrane potential promotes alkenal-induced mitochondrial uncoupling and influences adenine nucleotide translocase conformation. Biochem. J. 2008, 413, 323–332.
- Raza, H.; John, A. 4-hydroxynonenal induces mitochondrial oxidative stress, apoptosis and expression of glutathione S-transferase A4-4 and cytochrome P450 2E1 in PC12 cells. Toxicol. Appl. Pharmacol. 2006, 216, 309–318.
- Mali, V.R.; Ning, R.; Chen, J.; Yang, X.P.; Xu, J.; Palaniyandi, S.S. Impairment of aldehyde dehydrogenase-2 by 4-hydroxy-2-nonenal adduct formation and cardiomyocyte hypertrophy in mice fed a high-fat diet and injected with low-dose streptozotocin. Exp. Biol. Med. 2014, 239, 610–618.
- Breitzig, M.; Bhimineni, C.; Lockey, R.; Kolliputi, N. 4-Hydroxy-2-nonenal: A critical target in oxidative stress? Am. J. Physiol. Cell Physiol. 2016, 311, C537–C543.
- Picklo, M.J.; Amarnath, V.; McIntyre, J.O.; Graham, D.G.; Montine, T.J. 4-Hydroxy-2(E)-nonenal inhibits CNS mitochondrial respiration at multiple sites. J. Neurochem. 1999, 72, 1617–1624.
- Landar, A.; Zmijewski, J.W.; Dickinson, D.A.; Le Goffe, C.; Johnson, M.S.; Milne, G.L.; Zanoni, G.; Vidari, G.; Morrow, J.D.; Darley-Usmar, V.M. Interaction of electrophilic lipid oxidation products with mitochondria in endothelial cells and formation of reactive oxygen species. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1777–H1787.
- Long, J.; Wang, X.; Gao, H.; Liu, Z.; Liu, C.; Miao, M.; Liu, J. Malonaldehyde acts as a mitochondrial toxin: Inhibitory effects on respiratory function and enzyme activities in isolated rat liver mitochondria. Life Sci. 2006, 79, 1466–1472.
- Sun, L.; Luo, C.; Long, J.; Wei, D.; Liu, J. Acrolein is a mitochondrial toxin: Effects on respiratory function and enzyme activities in isolated rat liver mitochondria. Mitochondrion 2006, 6, 136–142.
- Schneider, C.; Porter, N.A.; Brash, A.R. Autoxidative transformation of chiral omega6 hydroxy linoleic and arachidonic acids to chiral 4-hydroxy-2E-nonenal. Chem. Res. Toxicol. 2004, 17, 937–941.
- Miller, D.M.; Singh, I.N.; Wang, J.A.; Hall, E.D. Administration of the Nrf2–ARE activators sulforaphane and carnosic acid attenuates 4-hydroxy-2-nonenal-induced mitochondrial dysfunction ex vivo. Free Radic. Biol. Medicine 2013, 1, 1–9.
- Sharma, S.; Sharma, P.; Bailey, T.; Bhattarai, S.; Subedi, U.; Miller, C.; Ara, H.; Kidambi, S.; Sun, H.; Panchatcharam, M.; et al. Electrophilic Aldehyde 4-Hydroxy-2-Nonenal Mediated Signaling and Mitochondrial Dysfunction. Biomolecules 2022, 12, 1555.
- Lashin, O.M.; Szweda, P.A.; Szweda, L.I.; Romani, A.M. Decreased complex II respiration and HNE-modified SDH subunit in diabetic heart. Free Radic. Biol. Med. 2006, 40, 886–896.
- Hwang, H.V.; Sandeep, N.; Paige, S.L.; Ranjbarvaziri, S.; Hu, D.Q.; Zhao, M.; Lan, I.S.; Coronado, M.; Kooiker, K.B.; Wu, S.M.; et al. 4HNE Impairs Myocardial Bioenergetics in Congenital Heart Disease-Induced Right Ventricular Failure. Circulation 2020, 142, 1667–1683.
- Lee, J.Y.; Jung, G.Y.; Heo, H.J.; Yun, M.R.; Park, J.Y.; Bae, S.S.; Hong, K.W.; Lee, W.S.; Kim, C.D. 4-Hydroxynonenal induces vascular smooth muscle cell apoptosis through mitochondrial generation of reactive oxygen species. Toxicol. Lett. 2006, 166, 212–221.
- Echtay, K.S.; Pakay, J.L.; Esteves, T.C.; Brand, M.D. Hydroxynonenal and uncoupling proteins: A model for protection against oxidative damage. Biofactors 2005, 24, 119–130.
- Doorn, J.A.; Hurley, T.D.; Petersen, D.R. Inhibition of human mitochondrial aldehyde dehydrogenase by 4-hydroxynon-2-enal and 4-oxonon-2-enal. Chem. Res. Toxicol. 2006, 19, 102–110.
- Balogh, L.M.; Atkins, W.M. Interactions of glutathione transferases with 4-hydroxynonenal. Drug Metab. Rev. 2011, 43, 165–178.
- Sheehan, D.; Meade, G.; Foley, V.M.; Dowd, C.A. Structure, function and evolution of glutathione transferases: Implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem. J. 2001, 360 Pt 1, 1–16.
- Röth, E.; Marczin, N.; Balatonyi, B.; Ghosh, S.; Kovács, V.; Alotti, N.; Borsiczky, B.; Gasz, B. Effect of a glutathione S-transferase inhibitor on oxidative stress and ischemia-reperfusion-induced apoptotic signalling of cultured cardiomyocytes. Exp. Clin. Cardiol. 2011, 16, 92–96.
- Mitchell, A.E.; Morin, D.; Lamé, M.W.; Jones, A.D. Purification, Mass Spectrometric Characterization, and Covalent Modification of Murine Glutathione S-Transferases. Chem. Res. Toxicol. 1995, 8, 1054–1062.
- Go, Y.M.; Halvey, P.J.; Hansen, J.M.; Reed, M.; Pohl, J.; Jones, D.P. Reactive aldehyde modification of thioredoxin-1 activates early steps of inflammation and cell adhesion. Am. J. Pathol. 2007, 171, 1670–1681.
- Perkins, A.; Nelson, K.J.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Peroxiredoxins: Guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 2015, 40, 435–445.
- Roede, J.R.; Carbone, D.L.; Doorn, J.A.; Kirichenko, O.V.; Reigan, P.; Petersen, D.R. In vitro and in silico characterization of peroxiredoxin 6 modified by 4-hydroxynonenal and 4-oxononenal. Chem. Res. Toxicol. 2008, 21, 2289–2299.
- Wong, C.M.; Marcocci, L.; Das, D.; Wang, X.; Luo, H.; Zungu-Edmondson, M.; Suzuki, Y.J. Mechanism of protein decarbonylation. Free Radic. Biol. Med. 2013, 65, 1126–1133.
- Guerby, P.; Tasta, O.; Swiader, A.; Pont, F.; Bujold, E.; Parant, O.; Vayssiere, C.; Salvayre, R.; Negre-Salvayre, A. Role of oxidative stress in the dysfunction of the placental endothelial nitric oxide synthase in preeclampsia. Redox Biol. 2021, 40, 101861.
- Whitsett, J.; Picklo, M.J., Sr.; Vasquez-Vivar, J. 4-Hydroxy-2-nonenal increases superoxide anion radical in endothelial cells via stimulated GTP cyclohydrolase proteasomal degradation. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2340–2347.
- Nakashima, I.; Liu, W.; Akhand, A.A.; Takeda, K.; Kawamoto, Y.; Kato, M.; Suzuki, H. 4-hydroxynonenal triggers multistep signal transduction cascades for suppression of cellular functions. Mol. Aspects Med. 2003, 24, 231–238.
- Shearn, C.T.; Reigan, P.; Petersen, D.R. Inhibition of hydrogen peroxide signaling by 4-hydroxynonenal due to differential regulation of Akt1 and Akt2 contributes to decreases in cell survival and proliferation in hepatocellular carcinoma cells. Free Radic. Biol. Med. 2012, 53, 1–11.
- Weinbaum, S.; Cancel, L.M.; Fu, B.M.; Tarbell, J.M. The Glycocalyx and Its Role in Vascular Physiology and Vascular Related Diseases. Cardiovasc. Eng. Technol. 2021, 12, 37–71.
- Askari, H.; Sadeghinejad, M.; Fancher, I.S. Mechanotransduction and the endothelial glycocalyx: Interactions with membrane and cytoskeletal proteins to transduce force. Curr. Top. Membr. 2023, 91, 43–60.
- Alphonsus, C.S.; Rodseth, R.N. The endothelial glycocalyx: A review of the vascular barrier. Anaesthesia 2014, 69, 777–784.
- Curry, F.E.; Adamson, R.H. Endothelial glycocalyx: Permeability barrier and mechanosensor. Ann. Biomed. Eng. 2012, 40, 828–839.
- Nguyen, T.K.; Paone, S.; Chan, E.; Poon, I.K.H.; Baxter, A.A.; Thomas, S.R.; Hulett, M.D. Heparanase: A Novel Therapeutic Target for the Treatment of Atherosclerosis. Cells 2022, 11, 3198.
- Ko, K.; Suzuki, T.; Ishikawa, R.; Hattori, N.; Ito, R.; Umehara, K.; Furihata, T.; Dohmae, N.; Linhardt, R.J.; Igarashi, K.; et al. Ischemic stroke disrupts the endothelial glycocalyx through activation of proHPSE via acrolein exposure. J. Biol. Chem. 2020, 295, 18614–18624.
- Lin, P.K.; Davis, G.E. Extracellular Matrix Remodeling in Vascular Disease: Defining Its Regulators and Pathological Influence. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 1599–1616.
- Halper, J. Basic Components of Vascular Connective Tissue and Extracellular Matrix. Adv. Pharmacol. 2018, 81, 95–127.
- Xu, J.; Shi, G.P. Vascular wall extracellular matrix proteins and vascular diseases. Biochim. Biophys. Acta 2014, 1842, 2106–2119.
- Greilberger, J.; Schmut, O.; Jürgens, G. In vitro interactions of oxidatively modified LDL with type I, II, III, IV, and V collagen, laminin, fibronectin, and poly-D-lysine. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2721–2728, Erratum in Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1197.
- Dunér, P.; To, F.; Alm, R.; Gonçalves, I.; Fredrikson, G.N.; Hedblad, B.; Berglund, G.; Nilsson, J.; Bengtsson, E. Immune responses against fibronectin modified by lipoprotein oxidation and their association with cardiovascular disease. J. Intern. Med. 2009, 265, 593–603.
- Dunér, P.; To, F.; Berg, K.; Alm, R.; Björkbacka, H.; Engelbertsen, D.; Fredrikson, G.N.; Nilsson, J.; Bengtsson, E. Immune responses against aldehyde-modified laminin accelerate atherosclerosis in Apoe−/− mice. Atherosclerosis 2010, 212, 457–465.
- Dunér, P.; Gonçalves, I.; Grufman, H.; Edsfeldt, A.; To, F.; Nitulescu, M.; Nilsson, J.; Bengtsson, E. Increased aldehyde-modification of collagen type IV in symptomatic plaques--a possible cause of endothelial dysfunction. Atherosclerosis 2015, 240, 26–32.
- Zarkovic, K.; Larroque-Cardoso, P.; Pucelle, M.; Salvayre, R.; Waeg, G.; Nègre-Salvayre, A.; Zarkovic, N. Elastin aging and lipid oxidation products in human aorta. Redox Biol. 2015, 4, 109–117.
- Larroque-Cardoso, P.; Camaré, C.; Nadal-Wollbold, F.; Grazide, M.H.; Pucelle, M.; Garoby-Salom, S.; Bogdanowicz, P.; Josse, G.; Schmitt, A.M.; Uchida, K.; et al. Elastin Modification by 4-Hydroxynonenal in Hairless Mice Exposed to UV-A. Role in Photoaging and Actinic Elastosis. J. Investig. Dermatol. 2015, 135, 1873–1881.
- Larroque-Cardoso, P.; Mucher, E.; Grazide, M.H.; Josse, G.; Schmitt, A.M.; Nadal-Wolbold, F.; Zarkovic, K.; Salvayre, R.; Nègre-Salvayre, A. 4-Hydroxynonenal impairs transforming growth factor-β1-induced elastin synthesis via epidermal growth factor receptor activation in human and murine fibroblasts. Free Radic. Biol. Med. 2014, 71, 427–436.
- Yang, S.; Nugent, M.A.; Panchenko MPEGF antagonizes TGF-beta-induced tropoelastin expression in lung fibroblasts via stabilization of Smad corepressor, T. G.I.F. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, L143–L151.
- Mehta, D.; Malik, A.B. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 2006, 86, 279–367.
- Usatyuk, P.V.; Natarajan, V. Role of mitogen-activated protein kinases in 4-hydroxy-2-nonenal-induced actin remodeling and barrier function in endothelial cells. J. Biol. Chem. 2004, 279, 11789–11797.
- Usatyuk, P.V.; Natarajan, V. Hydroxyalkenals and oxidized phospholipids modulation of endothelial cytoskeleton, focal adhesion and adherens junction proteins in regulating endothelial barrier function. Microvasc. Res. 2012, 83, 45–55.
- Aldini, G.; Dalle-Donne, I.; Vistoli, G.; Maffei Facino, R.; Carini, M. Covalent modification of actin by 4-hydroxy-trans-2-nonenal (HNE): LC-ESI-MS/MS evidence for Cys374 Michael adduction. J. Mass. Spectrom. 2005, 40, 946–954.
- Ozeki, M.; Miyagawa-Hayashino, A.; Akatsuka, S.; Shirase, T.; Lee, W.H.; Uchida, K.; Toyokuni, S. Susceptibility of actin to modification by 4-hydroxy-2-nonenal. J. Chromatogr. B Analyt Technol. Biomed. Life Sci. 2005, 827, 119–126.
- Stewart, B.J.; Doorn, J.A.; Petersen, D.R. Residue-specific adduction of tubulin by 4-hydroxynonenal and 4-oxononenal causes cross-linking and inhibits polymerization. Chem. Res. Toxicol. 2007, 20, 1111–1119.
- Kokubo, J.; Nagatani, N.; Hiroki, K.; Kuroiwa, K.; Watanabe, N.; Arai, T. Mechanism of destruction of microtubule structures by 4-hydroxy-2-nonenal. Cell Struct. Funct. 2008, 33, 51–59.
- Neely, M.D.; Sidell, K.R.; Graham, D.G.; Montine, T.J. The lipid peroxidation product 4-hydroxynonenal inhibits neurite outgrowth, disrupts neuronal microtubules, and modifies cellular tubulin. J. Neurochem. 1999, 72, 2323–2333.
- Neely, M.D.; Boutte, A.; Milatovic, D.; Montine, T.J. Mechanisms of 4-hydroxynonenal-induced neuronal microtubule dysfunction. Brain Res. 2005, 1037, 90–98.
- Shakhov, A.S.; Alieva, I.B. The “Third Violin” in the Cytoskeleton Orchestra-The Role of Intermediate Filaments in the Endothelial Cell’s Life. Biomedicines 2022, 10, 828.
- Mónico, A.; Duarte, S.; Pajares, M.A.; Pérez-Sala, D. Vimentin disruption by lipoxidation and electrophiles: Role of the cysteine residue and filament dynamics. Redox Biol. 2019, 23, 101098.
- Ramos, I.; Stamatakis, K.; Oeste, C.L.; Pérez-Sala, D. Vimentin as a Multifaceted Player and Potential Therapeutic Target in Viral Infections. Int. J. Mol. Sci. 2020, 21, 4675.
- Kim, S.Y.; Jeong, S.J.; Park, J.H.; Cho, W.; Ahn, Y.H.; Choi, Y.H.; Oh, G.T.; Silverstein, R.L.; Park, Y.M. Plasma Membrane Localization of CD36 Requires Vimentin Phosphorylation; A Mechanism by Which Macrophage Vimentin Promotes Atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 792717.
- Björkegren, J.L.M.; Lusis, A.J. Atherosclerosis: Recent developments. Cell 2022, 185, 1630–1645.
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241.
- Falk, E.; Nakano, M.; Bentzon, J.F.; Finn, A.V.; Virmani, R. Update on acute coronary syndromes: The pathologists’ view. Eur. Heart J. 2013, 34, 719–728.
- Attiq, A.; Afzal, S.; Ahmad, W.; Kandeel, M. Hegemony of inflammation in atherosclerosis and coronary artery disease. Eur. J. Pharmacol. 2024, 17, 176338.
- Escargueil-Blanc, I.; Salvayre, R.; Vacaresse, N.; Jürgens, G.; Darblade, B.; Arnal, J.F.; Parthasarathy, S.; Nègre-Salvayre, A. Mildly oxidized LDL induces activation of platelet-derived growth factor beta-receptor pathway. Circulation 2001, 104, 1814–1821.
- Suc, I.; Meilhac, O.; Lajoie-Mazenc, I.; Vandaele, J.; Jürgens, G.; Salvayre, R.; Nègre-Salvayre, A. Activation of EGF receptor by oxidized LDL. FASEB J. 1998, 12, 665–671.
- Gęgotek, A.; Skrzydlewska, E. Biological effect of protein modifications by lipid peroxidation products. Chem. Phys. Lipids 2019, 221, 46–52.
- Liu, W.; Akhand, A.A.; Kato, M.; Yokoyama, I.; Miyata, T.; Kurokawa, K.; Uchida, K.; Nakashima, I. 4-hydroxynonenal triggers an epidermal growth factor receptor-linked signal pathway for growth inhibition. J. Cell Sci. 1999, 112 Pt 14, 2409–2417.
- Auge, N.; Garcia, V.; Maupas-Schwalm, F.; Levade, T.; Salvayre, R.; Negre-Salvayre, A. Oxidized LDL-induced smooth muscle cell proliferation involves the EGF receptor/PI-3 kinase/Akt and the sphingolipid signaling pathways. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1990–1995.
- Zhang, H.; Forman, H.J. 4-Hydroxynonenal activates Src through a non-canonical pathway that involves, E.G.F.R./.P.T.P.1.B. Free Radic. Biol. Med. 2015, 89, 701–707.
- Cantero, A.V.; Portero-Otín, M.; Ayala, V.; Auge, N.; Sanson, M.; Elbaz, M.; Thiers, J.C.; Pamplona, R.; Salvayre, R.; Nègre-Salvayre, A. Methylglyoxal induces advanced glycation end product (AGEs) formation and dysfunction of PDGF receptor-beta: Implications for diabetic atherosclerosis. FASEB J. 2007, 21, 3096–3106.

