1. Introduction
RCS generated during LDL oxidation exhibit biphasic properties, from hormetic and protective effects at low levels to dysfunction and toxicity at higher doses. Hormesis is a defense mechanism based on a dose–response relationship by which low levels of stressors upregulate adaptive and protective responses, whereas higher levels become potentially harmful [
55]. Low RCS concentrations stimulate hormetic responses by activating signaling pathways such as the transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2 (Nrf2)/Kelch-like ECH-associated protein 1 (Keap1) system), resulting in the expression of cytoprotective and antioxidant enzymes and an enhanced expression of anti-inflammatory cellular defenses [
56,
57]. Likewise, RCS modulate NF-κB activation or inhibit the NLRP3 transcription factor and its subsequent inflammatory signaling [
57].
2. LDL Oxidation and Formation of RCS
LDL oxidation in the vascular wall is a complex mechanism that involves several sources of ROS, including NOXs (NADPH oxidases), the mitochondrial electron transport chain, xanthine oxidase, myeloperoxidase, cellular lipoxygenases, uncoupled eNOS, heme, iron, and copper ions [
57,
59,
60,
61,
62]. Lipid peroxidation strongly affects polyunsaturated fatty acids (PUFAs) in three steps (initiation, propagation, and termination), with hydrogen abstraction from a carbon and oxygen insertion [
63,
64]. This peroxidative attack generates a huge variety of lipid peroxidation products, among them lipid peroxyl radicals and lipid hydroperoxides, which undergo structural rearrangements to form RCS [
63,
64,
65,
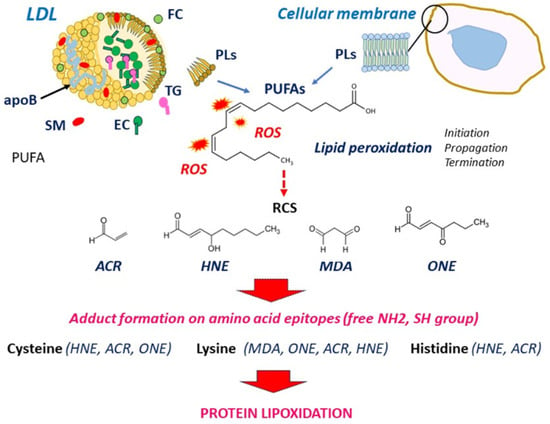
66]. Protein lipoxidation refers to the non-enzymatic post-translational modification of proteins by RCS via their interaction with the nucleophilic side chains of cysteine, histidine, and lysine residues to form Schiff’s bases (addition of the aldehydic group to an amino group of protein), or Michael addition of a nucleophile to α, β-unsaturated aldehydes [
18,
19]. The formation of RCS adducts is very fast, relatively selective, and depends on the protein microenvironment and the specific epitope exposure. The chemistry of adduct formation on proteins, as well as the reversion mechanisms (conjugation with glutathione catalyzed by glutathione S-transferase, oxidation or reduction by aldehyde dehydrogenase or alcohol dehydrogenase), have been largely described and reviewed [
14,
18,
20,
67,
68].
Several electrophilic aldehydes are detected in atherosclerotic lesions [
12,
57]. HNE is one of the most abundant. It is formed by the peroxidation of n-6 PUFAs and could be enzymatically produced by 15-lipoxygenase [
20,
68]. HNE forms Michael adducts with the highest reactivity for cysteine, followed by histidine and lysine, and the lowest reactivity for arginine [
20]. ONE is formed through the oxidation of n-6 PUFAs. It shares structural similarities with HNE, but it is more toxic and reactive on protein nucleophiles, particularly on lysine, on which it forms readily reversible Schiff base that can be oxidized to stable 4-ketoamide [
69,
70].
Acrolein (ACR) is an environmental volatile pollutant present in tobacco smoke and in cooking and exhaust fumes. It is endogenously formed by the peroxidation of PUFAs and through the metabolism of amino acids and polyamines. Acrolein rapidly reacts with cysteine, histidine, and lysine and is detected in oxLDLs and human atherosclerotic lesions [
12]. MDA is a product of PUFA peroxidation, abundantly present in oxLDLs as MDA-lysine adduct of apoB. MDA is highly mutagenic, cytotoxic and carcinogenic. It is largely used as a biomarker of lipid peroxidation to evaluate the extent of oxidative stress [
121,
122]. RCS formation and structure are shown in
Figure 1.
Figure 1. Formation of RCS from PUFA peroxidation in LDLs and cellular membranes. ROS attack PUFAs in LDLs and cellular membranes, which generates lipid oxidation products including RCS. RCS bind and form adducts on free amino groups (lysine, histidine) and thiol groups (cysteine), leading to protein lipoxidation. EC, esterified cholesterol; FC, free cholesterol; PLs, phospholipids; ROS, reactive oxygen species; SM, sphingomyelin; TG, triglycerides.
3. Protein Lipoxidation in the Vascular Wall
3.1. Modification of apoB by RCS in LDL: A Main Role in Foam Cell Formation
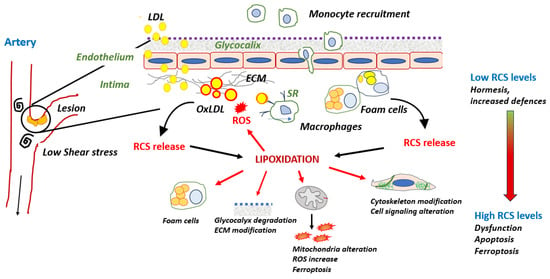
A variety of oxLDLs could be detected in the intima, from minimally/mildly oxLDLs mainly oxidized on their lipid moiety to heavily oxidized LDLs with RCS-modified apoB. Mildly oxLDLs are taken up through scavenger receptors (SR) such as LOX-1, present on endothelial cells or CD36, which is expressed by SMCs, macrophages, and endothelial cells. These mildly oxLDLs are highly inflammatory and contribute to endothelial dysfunction [
123,
124,
125]. Extensively oxidized LDLs contain large amounts of oxidized lipids, with apoB being modified by RCS (MDA, HNE, ACR), which deviates their uptake and metabolism towards the scavenger receptor class A (SR-A) pathway in macrophages [
126,
127]. MDA- or HNE-modified LDLs are a main cause of foam cell and fatty streak formation [
128,
129,
130]. MDA specifically reacts with the terminal ε-amino group of lysine residues involved in the recognition of LDLs by the LDL receptor [
121,
122]. As described by Lankin et al. [
122], MDA-LDLs undergo changes in the molecular conformation of apoB, which promotes the formation of cross-links between LDL particles and changes in electrophoretic patterns pointing out larger LDL formations.
HNE is more effective than MDA for modifying and increasing LDL negative charge and global molecular weight [
68]. HNE-modified LDLs are taken up by macrophages and generate foam cells [
14,
129,
130]. In HNE-modified apoB, Lys residues are the main target of HNE, and the other modified amino acid residues are tyrosine, serine, histidine, and cysteine [
130]. In ACR-modified LDLs, ACR–apoB adducts are mainly formed on Lys residues, which promotes their rapid uptake by macrophages through SR-A1 [
71].
LDL oxidation and some consequences of RCS release on protein lipoxidation possibly occurring in the intima are summarized in Figure 2.
Figure 2. Cellular systems affected by lipoxidation and possibly detected in the intima. RCS-modified apoB promotes the uptake of oxLDLs by macrophages via SR receptors and the formation of foam cells. OxLDLs release RCS in the intimal environment or from macrophagic foam cells. RCS bind various cellular protein systems, thereby promoting their lipoxidation.
The presence of RCS adducts in lesions in human and animal models for atherosclerosis [
131,
132,
133,
134] was demonstrated by immunocytochemistry and immunofluorescence techniques using specific anti-HNE and anti-MDA antibodies. Other adducts could be detected including Michael addition-type 4-hydroxy-2-hexenal (HHE)–histidine adducts [
132], or ACR–lysine adducts (N-alpha-acetyl-N-epsilon-(3-formyl-3,4-dehydropiperidino) lysine [
133]. LDLs and lipid peroxidation within the subendothelial area have multiple consequences—not only the accumulation of foam cells and fatty streaks but also the formation of RCS adducts on cellular and extracellular proteins. RCS adducts are detected on cellular membranes and ECM protein components of atherosclerotic lesions [
12,
126], suggesting their possible implication in atherogenesis.
3.2. RCS and Inflammation
Lipoxidation of Transcription Factors and Inflammation
Inflammation is an early component of plaque [
27]. In the “response to retention” hypothesis [
135], it was proposed that immuno-inflammation in early lesions is a defense mechanism tending to counter the accumulation of modified and oxidized LDLs by recruiting mononuclear cells, which may remove harmful oxLDLs from the environment. In contrast, the chronic inflammatory condition of advanced lesions is a key factor of plaque development and instability. Low RCS concentrations stimulate the activation of the redox-sensitive transcription factor NF-κB and concomitantly activate the anti-inflammatory nuclear factor (erythroid-derived 2)-like 2 (Nrf2) [
136,
137,
138]. High RCS levels elicit an anti-inflammatory response via at least two mechanisms: 1) inhibiting inflammatory transcription factors (NF-κB and the NLRP3 inflammasome pathway) [
139] and 2) stimulating the expression of endogenous antioxidant defenses via Nrf2 [
137].
NF-κB is a key regulator of inflammation and cell survival evoked by proatherogenic stressors [
136,
138]. In unstimulated cells, NF-κB is sequestered in the cytoplasm in an inactive state by its inhibitor, IκBα [
136]. Upon stimulation (by oxidative stress), IκBα is phosphorylated by IκB kinase (IKK), a redox-sensitive regulator of NF-κB activation. This promotes IκBα degradation by the ubiquitin/proteasome pathway and the translocation of NF-κB into the nucleus where it binds specific DNA domains and induces the expression of inflammatory genes [
136,
138]. This includes cytokines, chemokines, macrophage chemotactic factor (MCP)-1, matrix metalloproteinases (MMPs), cyclo-oxygenase (COX)-2, inducible nitric oxide synthase (iNOS), vascular endothelial growth factor (VEGF), adhesion molecules (VCAM-1, ICAM-1, E-selectin) [
138].
At low levels (lower than 10 µM), HNE stimulates the phosphorylation of IκBα and the binding of NF-κB to DNA, which induces MMP2 expression and SMCs proliferation [
140,
141]. In macrophages, HNE activates NF-κB via the EGFR/p38 MAPK pathway, thereby promoting the expression of 5-lipoxygenase and the generation of leukotrienes [
142]. In human U937, HNE and 27-hydroxycholesterol trigger an inflammatory response via Toll-like receptor 4 (TLR4) and NF-κB, leading to cytokine release and MMP-9 upregulation [
143]. Likewise, hydroxyhexenal (HHE)-induced NF-κB activation upregulated p38 MAPK and ERK activities in endothelial cells [
144]. At higher concentrations (10 µM and higher), HNE prevents the activation of NF-κB by LPS in human monocytes by inhibiting the phosphorylation and proteasomal degradation of IκBα [
72]. This inhibitory mechanism could result from the formation of HNE adducts on IκBα, possibly leading to a modification of protein conformation, preventing its phosphorylation by IKK, as reported in hepatocytes in a murine model of alcoholic liver disease [
73]. HNE forms adducts on IKK, particularly on cysteine-179 [
74,
75], which inhibits IKK signaling and NF-κB activation [
76]. Similar observations were reported for ACR, which modifies IKK [
77], thereby inhibiting IκBα phosphorylation and NF-κB activation [
78]. The inhibitory effect of RCS on NF-κB blocks the expression of the inducible NO synthase (iNOS) (a target gene of NF-κB) and NO production evoked by LPS and interferon-γ in SMCs [
145]. In addition, high HNE levels modify c-Jun NH2-terminal kinase (JNK) and upregulate the activating protein-1 transcription factor (AP-1), which promotes apoptosis [
146].
NLRP3 is a major proinflammatory protein complex when associated with the adaptor ASC protein and caspase 1. It plays an important role in atherogenesis [
41,
139,
147]. It is activated by oxLDLs, ROS, cholesterol crystals, and other danger signals, leading to the release of the inflammatory cytokine IL-1β, which aggravates inflammation and promotes cell death by pyroptosis [
148]. Recently, Hsu et al. [
79] reported an inhibitory effect of HNE on NLRP3 activation via a direct binding of HNE to NLRP3 cysteines. This modification alters the interaction between NLRP3 and NEK7, which is an essential partner of inflammasome assembly and activation [
149]. This mechanism could be reversed by N-acetylcysteine and GSH [
149].
Nrf2 is a main regulator of cellular resistance to oxidative stress and electrophiles and a major protective system in atherosclerosis [
56,
150,
151,
152,
153]. This transcription factor controls the expression of antioxidant/detoxifying genes and proteins, which prevents and protects against the onset of oxidative stress outcomes [
56,
150,
151]. In basal conditions, Nrf2 is associated with Keap1 in the cytoplasm. The phosphorylation of Keap1 by GSK-3β promotes its proteasomal degradation after ubiquitination [
150,
151,
152]. Oxidative stress stimulates the release of Nrf2 from the complex with Keap1, allowing its translocation into the nucleus. Nrf2 binds to antioxidant response elements (AREs) on DNA, which initiates the expression of antioxidant and protective genes, including NAD(P)H quinone oxidoreductase 1, glutamate-cysteine ligase, sulfiredoxin 1 and thioredoxin reductase 1, heme oxygenase-1 (HO-1), glutathione S-transferase, the cystine/glutamate amino acid transporter, and other protective systems [
56,
150,
151,
152,
153]. The Nrf2/Keap1 signaling pathway is highly sensitive to electrophiles [
80], which stimulate the expression and nuclear translocation of Nrf2, providing an adaptive response to cellular stress. The mechanisms by which RCS activate Nrf2 involve the presence of several cysteine residues in Keap1, which are highly susceptible to modification by electrophiles [
80], leading to Keap1 degradation and Nrf2 nuclear translocation. Nrf2 is a main effector of the hormetic responses evoked by low RCS levels in vascular cells [
56,
146,
151], via the upregulation of antioxidant, cytoprotective, and antiapoptotic systems, including HO-1 and peroxiredoxin-1. Nrf2 regulates proteasome and autophagy activities [
146,
154] and stimulates GSH synthesis, which prevents the modification of proteins by RCS [
118].
Despite its antioxidant and cytoprotective properties, the role of Nrf2 in atherosclerosis is debated [
155]. Nrf2 activation stimulates the expression of the scavenger receptor CD36 and the formation of foam cells [
156] and could increase the expression of proinflammatory genes in more advanced stages of atherosclerosis [
155]. As reported by Harada et al. [
157], Nrf2 inhibition could be atheroprotective in advanced plaques. All in all, Nrf2 may exhibit pro- or anti-atherogenic properties, depending on the (early or advanced) development of atherosclerotic lesions.
Cyclooxygenase-2 Activation
Cyclooxygenase-2 (COX-2) is a key enzyme involved in the production (from arachidonic acid) of high prostaglandin levels during inflammation and immune responses, particularly in vascular pathophysiology [
158]. COX-2 is rapidly induced in response to ROS, cytokines, growth factors, and HNE, which stimulates COX-2 expression in part via the activation of the p38 MAPK pathway [
158]. HNE-induced COX-2 could result from an accumulation of p53 and sp1 transcription factors (and ubiquitinated proteins) due to a downregulation of proteasome [
158]. Other RCS such as ACR or ONE are unable to stimulate COX-2 expression, which could be HNE-specific. The lipoxidation mechanisms possibly involved in COX-2 expression by HNE are not yet elucidated.
RCS and ROS
GSH depletion by RCS generates ROS and redox imbalance [
75]. Other mechanisms involve the formation of RCS adducts on antioxidant enzymes or alterations of eNOS activity. RCS may also inhibit ROS production, as observed in neutrophils in which HNE could modify and inactivate proteins involved in the respiratory burst (ROS production) and phagocytosis, which reduces both inflammation and antimicrobial defenses [
159].
The mitochondrial electron transport chain is an important source of endogenous cellular ROS [
81], and it is also a main target for RCS [
160,
161,
162]. HNE, MDA or ACR affect respiratory chain activity, decrease the mitochondrial membrane potential, and generate mitochondrial ROS [
82,
83,
84,
85]. HNE exogenously added or endogenously produced in mitochondria via cardiolipin oxidation [
86], decreases the activity of mitochondrial complexes -I and -II [
87,
88]. Several mitochondrial proteins could be modified by HNE or ONE, such as the FAD-containing subunit of succinate dehydrogenase, an essential component of succinate: ubiquinone oxidoreductase (or mitochondrial complex II) [
89]. Hwang et al. [
90] identified several mitochondrial proteins modified by HNE in cardiomyocytes during diabetes, among them NADH dehydrogenase (ubiquinone), iron–sulfur protein 3, aconitate hydratase-1, and heme proteins (myoglobin and cytochrome c1), along with the decreased activity of mitochondrial respiratory chain complex proteins. HNE and HHE form adducts on UCPs and adenine nucleotide translocase (ANT), which contributes to mitochondrial uncoupling by increasing proton leak, regulating membrane potential, and triggering mitochondrial dysfunction [
81]. Increased mitochondrial ROS and dysfunction are involved in HNE-induced vascular SMC apoptosis [
91].
On the other hand, low HNE concentrations may limit ROS production in mitochondria via the activation of the proton transporter function, leading to mild uncoupling that decreases the production of mitochondrial O
2•− [
163]. This mechanism, associated with the modulation of redox-regulating enzymes in mitochondria, could be involved in the activation of the Nrf2/ARE signaling by HNE [
163].
- -
-
ALDH2
Mitochondrial aldehyde dehydrogenase 2 (ALDH-2) is an oxidizing enzyme present in mitochondria and involved in the detoxification of RCS. ALDH-2 could act as a defense mechanism against oxidative stress in cardiovascular diseases. At low levels, HNE and ONE are degraded by ALDH-2, whereas at higher levels, these agents form covalent modifications on this enzyme and inhibit its activity [
93].
- -
-
Glutathione-S Transferases (GSTs)
The conjugation of aldehydes with GSH is a major detoxifying mechanism of reactive electrophiles, which prevents their reaction with cellular nucleophiles and facilitates their elimination. The conjugation with GSH may spontaneously occur, but it is facilitated by enzymes such as the cytosolic glutathione transferases (GSTs) which promote the reduction of hydroperoxides to form oxidized glutathione (GSSG) [
94,
164]. GSTs protect against oxidative injury and regulate GSH homeostasis [
165]. The GSTA4-4 isoform is the most selective for catalyzing the conjugation of GSH with RCS and is a main defense mechanism against oxidative stress [
81,
91,
166]. However, the formation of HNE adducts on the catalytic site of GST inhibits its activity and promotes oxidative stress [
166].
- -
-
Thioredoxin 1
Thioredoxin (Trx-1) is a key antioxidant enzyme involved against oxidative stress through its disulfide reductase activity regulating protein dithiol/disulfide balance. ACR and HNE react with Trx-1 on cysteine-73, inhibiting its enzymatic activity, which potentiates ROS production and promotes monocyte adhesion to endothelial cells [
95].
- -
-
Peroxiredoxins
Peroxiredoxins (PRXs) are ubiquitous peroxide and peroxynitrite-scavenging enzymes [
167]. The modification of PRX1 and PR6 by RCS has been reported [
96,
168]. PRX6 is an important antioxidant protein present in various tissues including cardiac muscle, skin and lung. HNE and ONE promote PRX6 modification and the formation of cross-links, particularly the formation of adducts on cysteine-91-lysine-209, which induces conformational changes and protein inactivation [
96].
The modification of eNOS by HNE and ONE was reported in preeclamptic placentas and in cultured trophoblasts, with subsequent decreased NO generation and trophoblast migration [
97]. Proteomic studies of recombinant eNOS modified by ONE showed the modification of several lysine residues on both oxidase and reductase domains, inhibiting its enzymatic activity [
97]. So far, a direct modification of eNOS in the vascular wall is not known. However, HNE could promote eNOS uncoupling and O
2•−. generation via a depletion in tetrahydrobiopterin (BH4, eNOS co-factor), resulting from the modification by HNE and subsequent proteosomal degradation of the GTP cyclohydrolase I (GTPCH), that is involved in BH4 biosynthesis [
98]. Another mechanism could implicate an inactivation of Akt by HNE [
169,
170], the phosphorylation of eNOS by Akt on serine-1179 being required for its activation and NO production [
169].
3.3. Lipoxidation of Endothelial Barrier Components
In physiological conditions, endothelial cells form a semi-permeable barrier to blood constituents, i.e., cells, macromolecules, albumin, and bioreactive agents. During atherogenesis, alterations of endothelial cell barrier integrity contribute to endothelial dysfunction and increased permeability to LDLs. RCS generated and released during the LDL oxidation process in the intima may play a role in endothelium dysfunction.
Glycocalyx
The glycocalyx is an extracellular matrix component surrounding the endothelium, as an interface between the vascular wall and circulating blood. Endothelial glycocalyx consists of glycoproteins, proteoglycans, glycosaminoglycans, hyaluronic acid, and associated plasma proteins. It is secreted by endothelial cells and located on the luminal side of vessels [
171,
172]. Glycocalyx contributes to mechanotransduction signals in response to stimuli and shear stress and maintains vascular permeability barrier and NO release [
173,
174]. The degradation of glycocalyx components, particularly heparan sulfate, by heparanase (HPSE), contributes to increased endothelial cell permeability, LDL retention, SMC migration, and intimal ECM remodeling [
175]. Exposure of heparan sulfate to MDA or ACR promotes its degradation [
99,
122]. ACR modification on lysine residues of the inactive proform of heparanase (proHPSE) triggers its activation and heparan sulfate degradation, which increases endothelial cell permeability [
99].
Extracellular Matrix Proteins
Endothelial cells are anchored on an underlying basement membrane, the intimal ECM, containing several components including laminin, collagens, fibronectin, heparan sulfate, proteoglycan, or perlecan [
176]. The interaction of endothelial cells with the basement membrane maintains the integrity of the vascular wall [
177]. ECM in the media has a more specialized structure, with elastin/fibrillin/fibulins/microfibril glycoprotein-associated matrices, as well as various components, including type IV collagen, laminins, perlecan, nidogens, or fibronectin. The medial ECM maintains the phenotype and function of contractile SMCs [
176,
177,
178]. Atherogenic inducers and proinflammatory agents, leukocyte and monocyte infiltration, or foam cell accumulation promote ECM remodeling, as well as SMC migration and proliferation in the intima [
176,
177,
178].
RCS released by oxLDLs on the connective tissue in the intima could bind and modify ECM protein components, such as collagen, laminin, or fibronectin. OxLDLs promote in vitro the formation of MDA adducts on fibronectin, laminin, and collagens type I > type V and type III > type IV > type II [
179,
180]. MDA-modified fibronectin is detected in human atherosclerotic lesions, and antibodies specific to this MDA-modified fibronectin could be correlated with the extent of CVD [
180]. Likewise, the presence of MDA-modified laminin was detected in human atherosclerotic lesions and apoE-KO mice, associated with the induction of anti-MDA-modified laminin antibodies, and correlated with a more aggressive development of atherosclerosis [
181]. Duner’s group also identified the presence of MDA-collagen type IV in human endarterectomy lesions. In vitro MDA-modified collagen type IV altered the attachment of endothelial cells and stimulated the expression of VCAM-1 adhesion molecule, suggesting an implication of collagen modification in endothelial dysfunction [
182].
HNE–histidine adducts are age-dependently detected in the three layers of the arterial wall, with a strong expression in the intima associated with atherosclerotic lesions in the media and the adventitia [
183]. These findings confirm that HNE is a main marker of vascular oxidative stress and lipid oxidation and suggest a role for this aldehyde in the development of vasa vasorum and microcapillaries. However, HNE and other RCS do not modify vascular elastin [
183], in contrast to elastin in skins exposed to UV radiations [
184]. In vitro, HNE inhibits the elastogenic activity of TGFβ by forming adducts on EGF receptor, which activates a downstream signal inhibiting TGFβ-induced responses [
185], in agreement with the inhibitory signaling of EGF on tropoelastin expression by TGFβ [
186].
Cytoskeleton Proteins
Cytoskeletal actin, intermediate filaments and microtubules; focal adhesion kinases (FAKs); and adherens junction proteins involved in the regulation of the endothelial barrier [
187] are targeted by RCS.
RCS are potent inducers of actin stress fibers and actin aggregation, via mechanisms implicating ERK1/2, p38 MAPK, JNK, and redox imbalance [
188,
189]. The pretreatment of actin with HNE alters the structure of actin filaments [
100,
101]. LC-ESI-MS/MS studies allowed identifying the site of actin modification by Michael addition of HNE to cysteine-374 [
100,
101].
RCS (HNE and ONE) form adducts on purified bovine brain tubulin, resulting in lysine-dependent protein cross-linking and inhibition of tubulin polymerization, with ONE being more potent than HNE as cross-linker and inhibitor of tubulin assembly [
190]. In addition, LC-MS/MS analysis demonstrated the modifications of several cysteine residues by HNE. In vitro, HNE triggered the destruction of the microtubule network in fibroblasts and in the PC12 cell line (used as model for neuronal differentiation) [
191]. This microtubule disruption by HNE is well described in neurons, possibly via the formation of cysteine adducts on tubulin [
192,
193]. Though not yet reported in the vascular wall, an impairment of tubulin function and cytoskeletal alterations evoked by RCS could be hypothesized in the chronic oxidative stress context of atherosclerosis.
Vimentin is the main protein of endothelial type III intermediate filaments and an important factor of stability and tissue integrity, against mechanical forces exerted by the blood flow [
194]. Links between vimentin, microfilaments, and microtubules coordinate cell polarization and migration as well as endothelial cell function, inflammation, and atherogenesis [
194]. Intermediate filaments (vimentin and lamin) are highly sensitive to oxidative and electrophilic stress, which promote their disruption and fragmentation, leading to the formation of aggresome structures [
102]. HNE forms adducts on nucleophilic (cysteine, histidine, lysine) residues on vimentin, with cysteine-328 being a main target. Cysteine-328 may act as a hub for electrophilic modifications, leading to intermediate filament rearrangements and an extensive reorganization of the vimentin cytoskeletal network, possibly acting either as a mechanism of defense or as a mediator of cell damages [
102,
103]. Vimentin lipoxidation alters the motility and the contractile capacity of fibroblasts [
18]. Recent reports indicated that vimentin in macrophages could play a role in CD36 trafficking and foam cell formation [
195]. From these observations, it could be interesting to evaluate the consequences of vimentin modification by RCS on the accumulation of foam cells in atherosclerotic lesions.
Cell Signaling Kinases and Growth Factor Receptors
In early atherosclerotic lesions, vascular cells and macrophages release various inflammatory cytokines, lipid mediators, and growth factors implicated in the formation of the fibrous cap. SMC phenotypic changes from a contractile to a synthetic state are associated with SMC migration and proliferation in the intima [
4,
196,
197,
198]. Several signaling pathways are modulated by RCS, including MAP kinases, PKC isoforms, cell-cycle regulators, receptor tyrosine kinases, and caspases. The formation of HNE or ACR adducts on receptor tyrosine kinases (RTKs) (PDGFR and EGFR) has been detected either in vitro in vascular cells or in vivo in human carotid endarterectomy plaque, in atheroclerotic lesions of hypercholesterolemic rabbits and apoE
−/− mice [
57,
104,
185]. At low concentrations, these modifications activate the downstream signaling cascade of RTKs, including src, PI3K/Akt, and ERK1/2, leading to cell survival and proliferation [
105,
106,
107,
108,
169,
199,
200]. High HNE levels extensively modify RTKs and trigger their progressive dysfunction. For instance, the accumulation of HNE adducts on PDGFR limits its affinity for PDGF, which decreases PDGF-stimulated cell proliferation and migration [
57]. Likewise, the accumulation of HNE adducts on EGFR inhibits the PI3K/Akt pathway and promotes a switch toward apoptosis [
108].
This entry is adapted from the peer-reviewed paper 10.3390/antiox13020232