Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Zeinab Breijyeh | -- | 4672 | 2024-02-28 20:13:37 | | | |
| 2 | Peter Tang | + 2 word(s) | 4674 | 2024-02-29 06:54:14 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Breijyeh, Z.; Karaman, R. Causes and Risk Factors of Alzheimer’s Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/55688 (accessed on 09 August 2026).
Breijyeh Z, Karaman R. Causes and Risk Factors of Alzheimer’s Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/55688. Accessed August 09, 2026.
Breijyeh, Zeinab, Rafik Karaman. "Causes and Risk Factors of Alzheimer’s Disease" Encyclopedia, https://encyclopedia.pub/entry/55688 (accessed August 09, 2026).
Breijyeh, Z., & Karaman, R. (2024, February 28). Causes and Risk Factors of Alzheimer’s Disease. In Encyclopedia. https://encyclopedia.pub/entry/55688
Breijyeh, Zeinab and Rafik Karaman. "Causes and Risk Factors of Alzheimer’s Disease." Encyclopedia. Web. 28 February, 2024.
Copy Citation
Alzheimer’s disease (AD) is a disorder that causes degeneration of the cells in the brain and it is the main cause of dementia, which is characterized by a decline in thinking and independence in personal daily activities. AD is considered a multifactorial disease: two main hypotheses were proposed as a cause for AD, cholinergic and amyloid hypotheses. Additionally, several risk factors such as increasing age, genetic factors, head injuries, vascular diseases, infections, and environmental factors play a role in the disease.
Alzheimer’s disease
neurodegeneration
β-amyloid peptide
tau protein
risk factors
disease-modifying therapy
chaperons
heat shock proteins
1. Introduction
Alzheimer’s disease (AD) (named after the German psychiatric Alois Alzheimer) is the most common type of dementia and can be defined as a slowly progressive neurodegenerative disease characterized by neuritic plaques and neurofibrillary tangles (Figure 1) as a result of amyloid-beta peptide’s (Aβ) accumulation in the most affected area of the brain, the medial temporal lobe and neocortical structures [1]. Alois Alzheimer noticed a presence of amyloid plaques and a massive loss of neurons while examining the brain of his first patient that suffered from memory loss and change of personality before dying and described the condition as a serious disease of the cerebral cortex. Emil Kraepelin named this medical condition Alzheimer’s disease for the first time in his 8th edition psychiatry handbook [2][3]. Progressive loss of cognitive functions can be caused by cerebral disorder like Alzheimer’s disease (AD) or other factors such as intoxications, infections, abnormality in the pulmonary and circulatory systems, which causes a reduction in the oxygen supply to the brain, nutritional deficiency, vitamin B12 deficiency, tumors, and others [4][5].
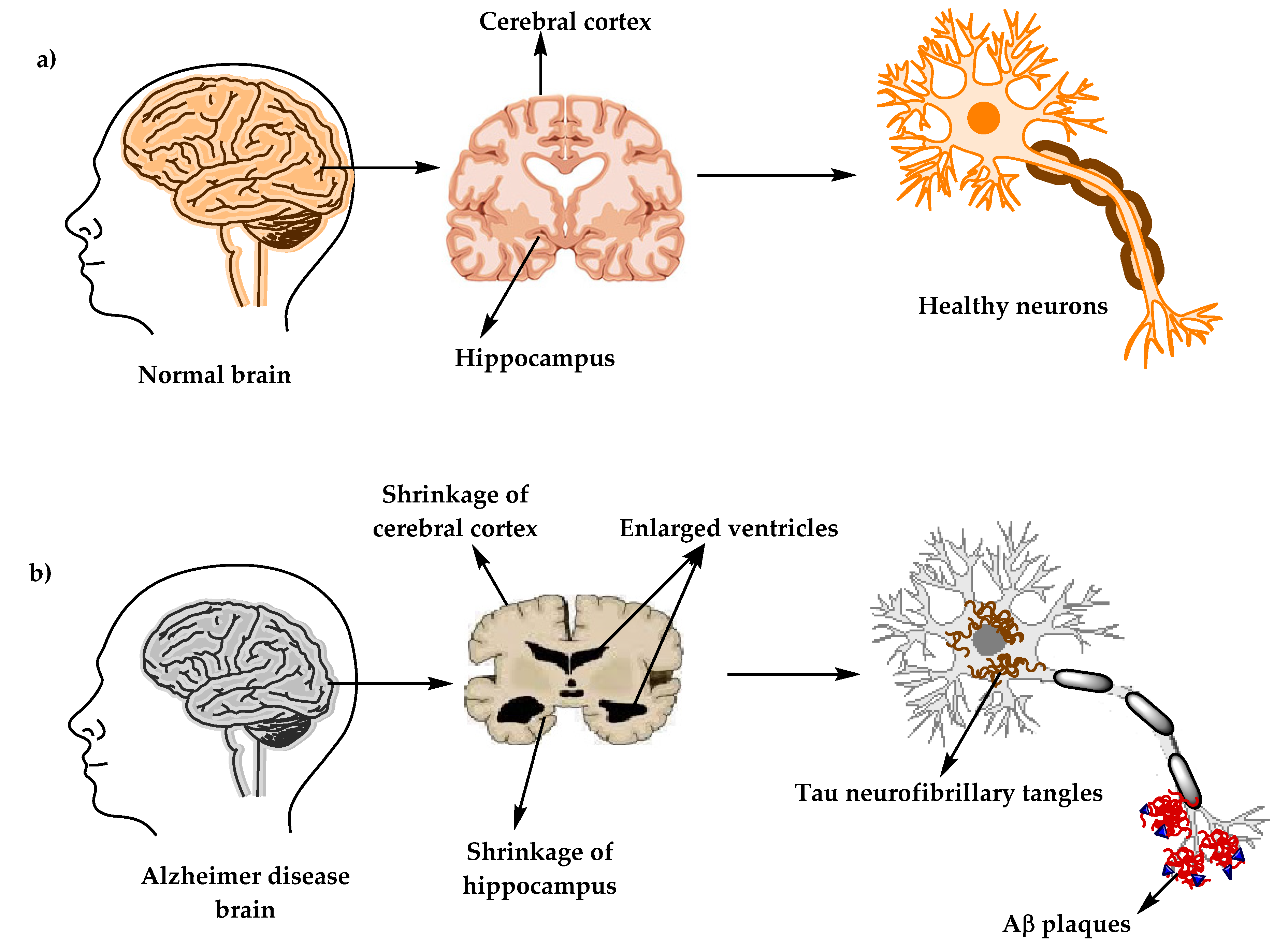
Figure 1. The physiological structure of the brain and neurons in (a) healthy brain and (b) Alzheimer’s disease (AD) brain.
At present, there are around 50 million AD patients worldwide and this number is projected to double every 5 years and will increase to reach 152 million by 2050. AD burden affects individuals, their families, and the economy, with estimated global costs of US$1 trillion annually. At present, there is no cure for Alzheimer’s disease, although there are available treatments that just improve the symptoms [6][7].
2. Alzheimer’s Disease Diagnostic Criteria
A patient suspected to have AD should undergo several tests, including neurological examination, magnetic resonance imaging (MRI) for neurons, laboratory examinations such as vitamin B12, and other tests besides the medical and family history of the patients [8]. Vitamin (vit.) B12 deficiency has been long known for its association with neurologic problems and increasing risks of AD, according to some studies. A special marker of vit. B12 deficiency is elevated homocysteine levels, which can cause brain damage by oxidative stress, increasing calcium influx and apoptosis. Diagnoses of vit. B12 deficiency can be done by measuring serum vit. B12 level alongside complete blood count and serum homocysteine levels tests [9][10].
In 1984, The National Institute of Neurological and Communicative Disorders and Stroke (NINCDS) and the Alzheimer’s Disease and Related Disorders Association (ADRDA) formed a work group (NINCDS-ADRDA) to establish a clinical diagnostic’s criteria for Alzheimer’s disease. This criteria includes: (1) probable Alzheimer’s disease, which can be diagnosed by dementia that is confirmed by neuropsychological tests, progressive memory loss, impaired daily-life activity, and other symptoms like aphasia (impairment of a language), apraxia (a motor skills disorder), and agnosia (a loss of perception). All of these symptoms can start from age 40–90, with the absence of any systemic or brain diseases, (2) possible Alzheimer’s disease can be applied in the absence of neurologic, psychiatric disorders, and the presence of another illness like systemic or brain disorder, but they are not the primary cause of dementia, and (3) definite Alzheimer’s disease, that is confirmed by histopathologic confirmation obtained from a biopsy or autopsy [11][12].
In 2011, The National Institute on Aging—Alzheimer’s Association made several changes and updated the 1984 NINCDS-ADRDA criteria for higher specificity and sensitivity in the diagnosis of Alzheimer’s disease. The newly proposed criteria include probable and possible AD dementia for the use in clinical settings and probable or possible AD dementia with pathophysiological evidence for research purposes, in addition to clinical biomarkers. There are two categories of Alzheimer’s disease biomarkers: (a) markers of brain amyloid such as positron emission tomography (PET) and cerebrospinal fluid (CSF), and (b) markers of neuronal injury like cerebrospinal fluid tau, fluorodeoxyglucose (FDG) for metabolic activity, and magnetic resonance imaging (MRI) for atrophy measurement [13][14][15].
3. Alzheimer’s Disease’s Neuropathology
There are two types of neuropathological changes in AD which provide evidence about disease progress and symptoms and include: (1) positive lesions (due to accumulation), which are characterized by the accumulation of neurofibrillary tangles, amyloid plaques, dystrophic neurites, neuropil threads, and other deposits found in the brains of AD patients. In addition to (2) negative lesions (due to losses), that are characterized by large atrophy due to a neural, neuropil, and synaptic loss. Besides, other factors can cause neurodegeneration such as neuroinflammation, oxidative stress, and injury of cholinergic neurons [16][17][18].
4. The Stages of Alzheimer’s Disease
The clinical phases of Alzheimer’s disease can be classified into (1) pre-clinical or the pre-symptomatic stage, which can last for several years or more. This stage is characterized by mild memory loss and early pathological changes in cortex and hippocampus, with no functional impairment in the daily activities and absence of clinical signs and symptoms of AD [1][19][20]. (2) The mild or early stage of AD, where several symptoms start to appear in patients, such as a trouble in the daily life of the patient with a loss of concentration and memory, disorientation of place and time, a change in the mood, and a development of depression [20][21]. (3) Moderate AD stage, in which the disease spreads to cerebral cortex areas that results in an increased memory loss with trouble recognizing family and friends, a loss of impulse control, and difficulty in reading, writing, and speaking [20]. (4) Severe AD or late-stage, which involves the spread of the disease to the entire cortex area with a severe accumulation of neuritic plaques and neurofibrillary tangles, resulting in a progressive functional and cognitive impairment where the patients cannot recognize their family at all and may become bedridden with difficulties in swallowing and urination, and eventually leading to the patient’s death due to these complications [1][22].
5. Causes and Risk Factors of Alzheimer’s Disease
AD has been considered a multifactorial disease associated with several risk factors (Figure 2) such as increasing age, genetic factors, head injuries, vascular diseases, infections, and environmental factors (heavy metals, trace metals, and others). The underlying cause of pathological changes in Alzheimer’s disease (Aβ, NFTs, and synaptic loss) is still unknown. Several hypotheses were proposed as a cause for AD but two of them are believed to be the main cause: some believe that an impairment in the cholinergic function is a critical risk factor for AD, while others suggest that alteration in amyloid β-protein production and processing is the main initiating factor. However, at present, there is no accepted theory for explaining the AD pathogenesis [23][24].
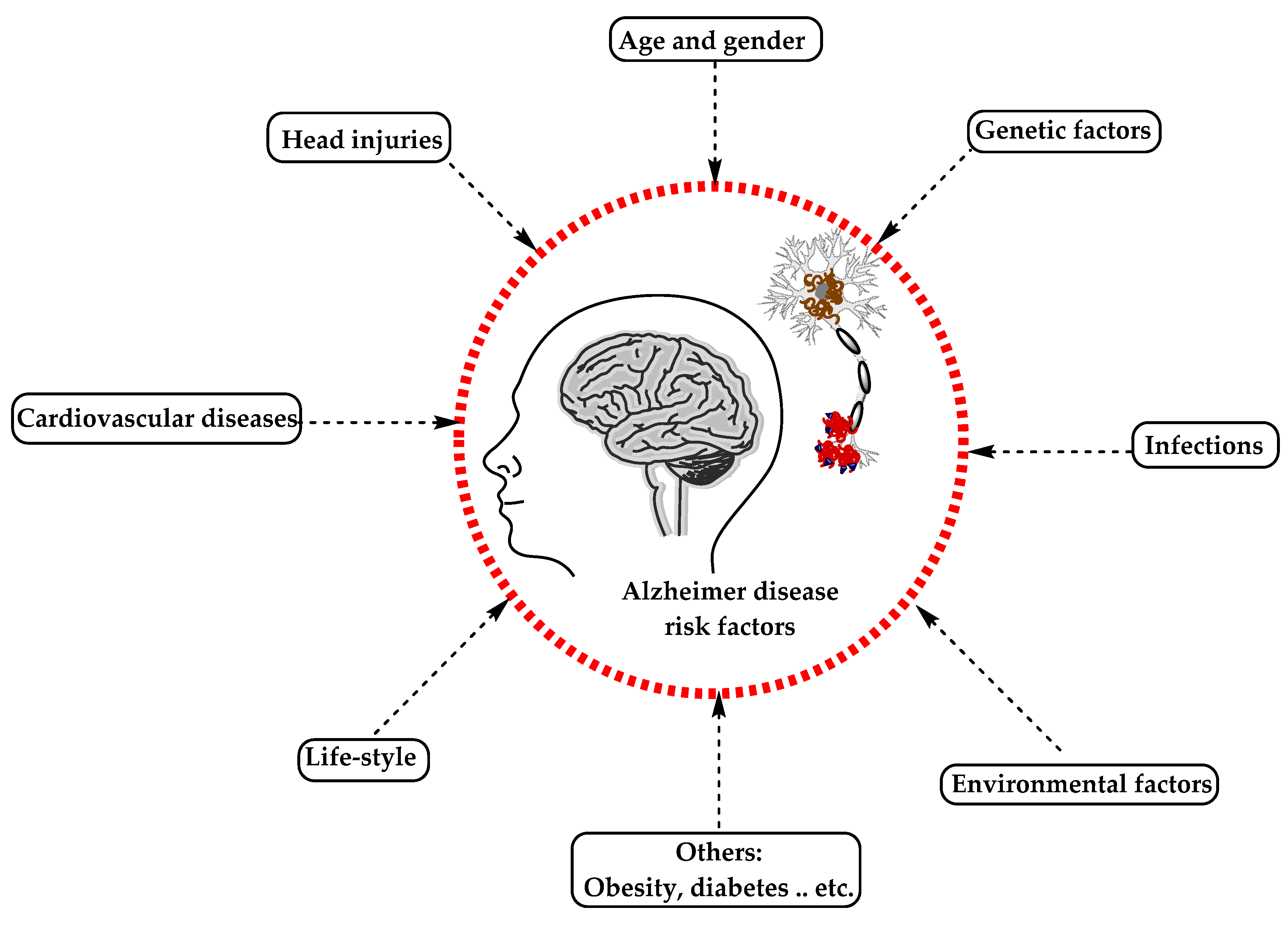
Figure 2. The risk factors for Alzheimer’s disease.
5.1. Alzheimer’s Disease Hypotheses
5.1.1. Cholinergic Hypothesis
In the 1970s, neocortical and presynaptic cholinergic deficits were reported to be related to the enzyme choline acetyltransferase (ChAT), which is responsible for the synthesis of acetylcholine (ACh). Due to the essential role of ACh in cognitive function, a cholinergic hypothesis of AD was proposed. ACh is synthesized in the cytoplasm of cholinergic neurons from choline and acetyl-coenzyme A by the ChAT enzyme and transported to the synaptic vesicles by vesicular acetylcholine transporter (VAChT) (Figure 3). In the brain, ACh is involved in several physiological processes such as memory, attention, sensory information, learning, and other critical functions. Degeneration of the cholinergic neurons was found to take place in AD and to cause alternation in cognitive function and memory loss. Β-amyloid is believed to affect cholinergic neurotransmission and to cause a reduction in the choline uptake and a release of ACh. Studies demonstrated that cholinergic synaptic loss and amyloid fibril formation are related to Aβ oligomers’ neurotoxicity and to interactions between AChE and Aβ peptide. Additional factors also contribute to the progression of AD, such as a reduction in nicotinic and muscarinic (M2) Ach receptors, located on presynaptic cholinergic terminals, and the deficit in excitatory amino acid (EAA) neurotransmission, where glutamate concentration and D-aspartate uptake are significantly reduced in many cortical areas in AD brains. This is in addition to the use of cholinergic receptor antagonists such as scopolamine, which was found to induce amnesia. This effect can be reversed by using compounds that activate acetylcholine formation [25][26][27].
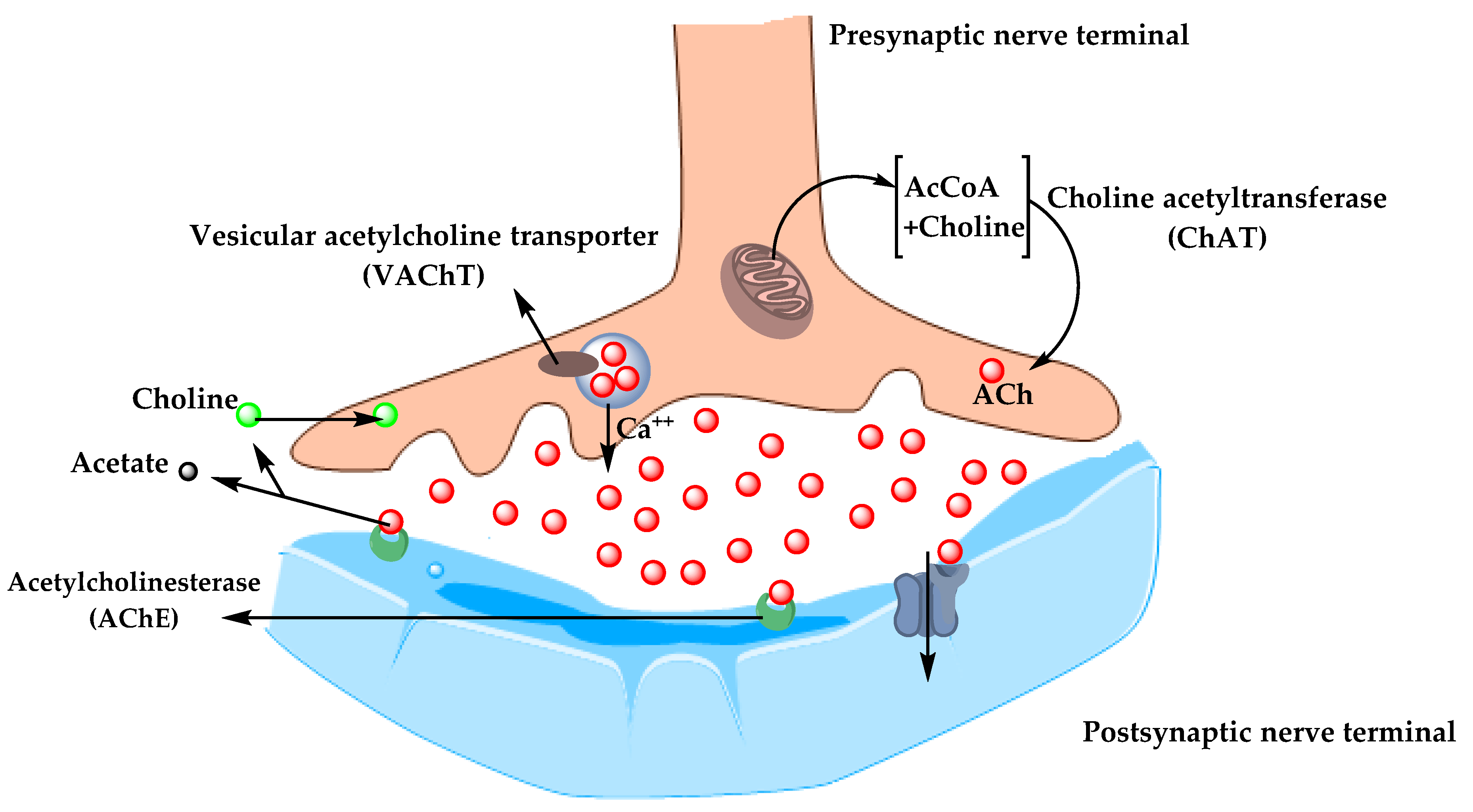
Figure 3. The pathway for the synthesis and transportation of acetylcholine between presynaptic and postsynaptic nerve terminals.
As a result, the cholinergic hypothesis is based on three concepts: reduced presynaptic cholinergic markers in the cerebral cortex, severe neurodegeneration of nucleus basalis of Meynert (NBM) in the basal forebrain, which is the source of cortical cholinergic innervation, and the role of cholinergic antagonists in memory decline compared to the agonists, which have the opposite effect [28].
5.1.2. Amyloid Hypothesis
For decades, it was recognized that abnormal deposition of β-sheets in the central nervous system has a strong correlation with dementia, which led to the concept of the amyloid hypothesis. However, it was found that the amyloid plaques (AP) also deposit in normal healthy brains with aging, which raised the question of whether AP deposition is responsible for AD onset or not? Therefore, in the recent years, alternative hypotheses were proposed for the non-inherited form of AD (NIAD), but at present, the amyloid hypothesis remains the most accepted pathological mechanism for inherited AD (IAD). The amyloid hypothesis suggests that the degradation of Aβ, derived from APP by β- and γ-secretase, is decreased by age or pathological conditions, which leads to the accumulation of Aβ peptides (Aβ40 and Aβ42). Increasing the ratio of Aβ42/Aβ40 induces Aβ amyloid fibril formation, resulting in neurotoxicity and tau pathology induction, and consequently, leading to neuronal cell death and neurodegeneration. AD risk factors and mutations of several genes like APP, PSEN1, and PSEN2 were found to affect Aβ catabolism and anabolism, which rapidly cause an accumulation of Aβ and fast progression of neurodegeneration [29][30][31].
5.2. Alzheimer’s Disease Risk Factors
5.2.1. Aging
The most important risk factor in AD is aging. Younger individuals rarely have this disease, and most AD cases have a late onset that starts after 65 years of age [32]. Aging is a complex and irreversible process that occurs through multiple organs and cell systems with a reduction in the brain volume and weight, a loss of synapses, and ventricles’ enlargement in specific areas accompanied by SP deposition and NFT. Moreover, several conditions might emerge during aging such as glucose hypometabolism, cholesterol dyshomeostasis, mitochondria dysfunction, depression, and cognitive decline. These changes also appear in normal aging, which makes it difficult to distinguish the cases in early AD [33][34]. AD can be divided based on age of onset into early-onset AD (EOAD), the rare form with around 1–6% of cases, in which most of them are familial AD characterized by having more than one member in more than one generation with AD, and ranges from 30–60 or 65 years. The second type is the late-onset AD (LOAD), which is more common with age of onset above 65 years. Both types may occur in people who have a family with a positive history of AD and families with a late-onset disease [35].
5.2.2. Genetics
Genetic factors were discovered over the years and were found to play a major role in the development of AD. 70% of the AD cases were related to genetic factors: most cases of EOAD are inherited in an autosomal dominant pattern and mutations in the dominant genes such as Amyloid precursor protein (APP), Presenilin-1 (PSEN-1), Presenilin-2 (PSEN-2), and apolipoprotein E (ApoE) are associated with AD [36][37].
Herein, the researchers discuss the strong genetic risk factors in AD.
-
Amyloid Precursor Protein (APP)
APP is a type I transmembrane protein cleaved by α-, β-, and γ-secretase to release Aβ and other proteins and is encoded by the APP gene on chromosome 21. Thirty mutations have been found in the APP gene in which twenty-five of them are related to AD and cause an accumulation of Aβ with elevated amounts. Meanwhile, there is one protective mutation, A673T, which protects against AD by decreasing Aβ, Aβ40, and Aβ42 secretion [38][39]. All mutations surround the secretase cleavage site, for example, the KM670/671NL mutation in mouse models has shown an increasing level of amyloid plaques in the hippocampus and cortex with no NFTs. A673V, D678H, D678N, E682K, and K687N mutations have shown cortical atrophy, whereas E682K has shown hippocampal atrophy. Neuropathological reports for the A673V mutation demonstrated a presence of NFTs and Aβ, activation of microglia and astrocytes, and neuronal loss, compared to the rest of the mentioned mutations, which show no change in the intracellular Aβ according to neuropathological reports [38][40]. Other mutations such as T714I, V715A, V715M, V717I, V717L, L723P, K724N, and I716V affect the γ-secretase cleavage site and cause an increase in the Aβ42/Aβ40 ratio, while E693G, E693K, D694N, and A692G mutations affect the α-secretase cleavage site and cause polymorphic aggregates with the ability to disrupt bilayer integrity. Also, the E693delta is a deletion mutation that enhances the formation of synaptotoxic Aβ [41][42].
-
Presenilin-1 (PSEN-1) and Presenilin-2 (PSEN-2)
PSEN1 and PSEN2 genes are also the autosomal dominant form of EOAD located on chromosomes 14 and 1, respectively. PSEN-2 and PSEN-1 are homologous, with 67% similarity, with a difference in the N-terminus and the hydrophilic region. Mutation in PSEN1 gene is more common, with more than 200 mutations, while a rare form with less than 40 mutations was identified in the PSEN2 gene [43][44].
PSEN1 is a core protein that activates the γ-secretase complex and plays an important role in the production of Aβ from APP. Knockout studies of PSEN1 showed synaptic dysfunction and memory impairment in mice, which indicate its essential role in maintaining memory and neurons [41]. PSEN1 mutations are simple ones which include single amino acid substitution, and severe mutation can result from the substitutions of two amino acids [45]. Mutations in the PSEN1 gene increase the ratio of Aβ42/Aβ40 by decreasing Aβ40 levels. The results obtained by Sun et al. study demonstrated that C410Y or L435F mutations in PSEN1 knock-in mice increased the Aβ42/Aβ40 ratio due to a greater reduction in Aβ40 [46].
In contrast, PSEN-2 mutations are rare and play a minor role in Aβ production. Any mutation in PSEN-2 might have a severe effect on the Aβ 42/40 ratio, causing familial AD in the presence of normal PSEN-1 alleles. Some of the PSEN-2 mutations cause a significant increase in γ-secretase activity with an elevation in the Aβ-42 and Aβ 42/40 ratio level, such as N141I, T122P, M239V, and M239I, while others are rare polymorphisms and have no effect on Aβ-42, -40, and Aβ 42/40 ratio levels and are not considered as pathogenic mutations [43][47].
-
Apolipoprotein E (ApoE)
ApoE protein is a glycoprotein expressed highly in the liver and brain astrocytes and some microglia and serves as a receptor-mediated endocytosis ligand for lipoprotein particles like cholesterol, which is essential for myelin production and normal brain function. The ApoE gene located on chromosome 19 has three isoforms, ApoE2, ApoE3, and ApoE4, due to single-nucleotide polymorphisms (SNPs) which cause changes in the coding sequence. The ApoEε4 allele is a strong risk factor for both EOAD and LOAD compared to ApoEε2 and ApoEε3 alleles that are associated with a lower risk and protective effect, respectively [48]. ApoEε4 plays an important role in Aβ deposition as a senile plaque and causes cerebral amyloid angiopathy (CAA), which is known as a marker for AD [49]. ApoEε4 was also shown to be associated with vascular damage in the brain, which leads to AD pathogenesis [50].
-
ATP Binding Cassette Transporter A1 (ABCA1)
Adenosine triphosphate (ATP)-binding cassette transporter A1 (ABCA1) is part of a large ABC transporters family that regulate cholesterol efflux in the circulation, like apolipoproteins-AI (ApoAI), and into the brain, like ApoE. In addition, ABCA1 maintains the stability of ApoE lipidation and serves as a mediator for high-density lipoprotein (HDL) generation, which reflects its role in atherosclerosis and cardiovascular diseases. Studies on the AD mice model showed that ABCA1 deficiency increases amyloid plaques and eliminates the lipidation of ApoE [51]. In humans, a mutation in ABCA1 results in Tangier disease, which is characterized by low levels of high-density lipoprotein (HDL) and ApoAI in plasma, accumulation of cholesterol in tissues, and AD pathogenesis [52].
-
Clusterin Gene (CLU) and Bridging Integrator 1 (BIN1)
In contrast to PSEN1, PSEN2, and APP mutations, which result in familial or EOAD, clusterin (CLU) and Bridging Integrator 1 (BIN1) genes are novel risk factors for LOAD. In 2009, Genome-Wide Association Studies (GWAS) identified the CLU gene located on chromosome 8, which is upregulated in the cortex and hippocampus of AD brains, in addition to AD cerebrospinal fluid (CSF) and plasma, which make the CLU a promising biomarker for AD. The CLU may play a protective role by interacting with Aβ and promoting its clearance, or a neurotoxic role by reducing Aβ clearance. The Aβ ratio values determine whether the CLU role is neuroprotective or neurotoxic [53].
BIN1 is a Bin-Amphiphysin-Rvs (BAR) adaptor protein that is involved in the production of membrane curvature and other endocytosis cellular functions. BIN1 has several isoforms: some are found in the brain, where they interact with different proteins such as clathrin, synaptojanin, and amphiphysin 1, and others in which they regulate synaptic vesicle endocytosis. Recently, BIN1 was recognized as the second most important risk factor for LOAD after ApoE, where it plays a role in Aβ production and as a tau and NFT pathology modulator [54][55].
-
Evolutionarily Conserved Signaling Intermediate in Toll pathway (ECSIT)
A significant accumulation of Aβ in AD brains increases protein oxidation, which reflects the critical role of mitochondria in Aβ cytotoxicity and AD pathogenesis. Evolutionarily conserved signaling intermediate in Toll pathway (ECSIT) gene is located on chromosome 19 and is associated with increasing the risk of AD. ECSIT encodes the adapting protein that functions as a cytoplasmic and signaling protein and is responsible for stabilizing the mitochondrial respiratory complex. Moreover, the adaptor protein is involved in the activation of nuclear factor (NF)-κB, interferon regulatory factors (IRFs), and activating protein-1. Also, it is involved in coupling immune toll-like receptor (TLR), homeostatic bone morphogenetic pathway (BMP), and transforming growth factor-beta (TGF-b) pathways [56][57].
ECSIT interacts with mitochondrial proteins such as Lon protease homolog (LONP1) and glutaryl-CoA dehydrogenase (GCDH), which are involved in intra-mitochondrial proteolysis and redox signaling respectively, followed by interactions with AD seed nitric oxide synthase (NOS3). Moreover, studies have shown certain interactions of ECSIT with the AD genes ApoE, PSEN-1, and PSEN-2. These interactions support the role of ECSIT as a molecular link in oxidative stress, inflammation, and mitochondrial dysfunction in AD [56][58].
-
Estrogen Receptor Gene (ESR)
AD affects both women and men, but nearly two-thirds of AD cases are women. Several studies have shown that women with AD experience worse mental deterioration than men. Additionally, on the genetic level, some genes’ variation, like the ApoE4 allele, significantly increases AD risk in women compared to men. Other studies documented that AD risk in women is associated with the loss of ovarian hormones during menopause due to the fact that estrogen regulates several activities in the brain, such as neurotransmission, neural development, survival, protection against oxidative stress, reduction of Aβ peptide levels, and attenuation of tau hyperphosphorylation. The estrogen activity is mediated through estrogen receptors (ERs) (intracellular, transmembrane, and membrane-bound ERs). The two major subtypes of these receptors are ERα and Erβ, which are encoded by two distinct genes and are located on chromosome 6 and 14, respectively. ERα receptor is found in the hypothalamus and amygdala, whereas ERβ receptors are in the hippocampus and cortex. Single nucleotide polymorphisms (SNPs) in ERβ and ERα genes may affect exogenous estrogen in older women and influence cognitive aging. PvuII (rs9340799) and Xbal (rs223493) are examples of SNPs found in ERα and are associated with AD and cognitive impairment. Also, several SNPs in ERβ have been proven to increase the risk of AD in women [59][60][61][62].
-
Other Genes
Other genes’ polymorphism associated with increasing the risk of AD include vitamin D receptor (VDR) gene polymorphism, which affects the affinity of vitamin D to its receptor and may cause neurodegenerative diseases and neuronal damage [63]. Moreover, epigenetic factors like DNA methylation, histone, and chromatin modifications were demonstrated to be involved in AD [23][64].
5.2.3. Environmental Factors
Aging and genetic risk factors cannot explain all cases of AD. Environmental risk factors including air pollution, diet, metals, infections, and many others may induce oxidative stress and inflammation and increase the risk for developing AD. Herein, the researchers report the most important environmental factors and their relationships with AD [65][66].
-
Air Pollution
The air pollution is characterized by modifying the nature of the atmosphere through the introduction of chemical, physical, or biological pollutants. It is associated with respiratory and cardiovascular diseases and recently, its association with AD was documented. Six air pollutants have been defined by National Ambient Air Quality Standards (NAAQSs) in the USA as a threat to human health, including ozone (O3), nitrogen oxides (NOx), carbon monoxide (CO), particulate matter (PM), sulfur dioxide (SO2), and lead. Studies on animals and cellular models have shown that an exposure to high levels of air pollution can result in a damage to the olfactory mucosa and bulb, in addition to the frontal cortex region, similar to that observed in AD. In individuals exposed to air pollutants, there is a link between oxidative stress, neuroinflammation, and neurodegeneration, with the presence of hyper-phosphorylated tau and Aβ plaques in the frontal cortex. The air pollution can cause an increase in Aβ42 formation, accumulation, and impaired cognitive function [67][68].
-
Diet
In recent years, the number of studies on the role of nutrition in AD have been increased. Several dietary supplements such as antioxidants, vitamins, polyphenols, and fish were reported to decrease the risk of AD, whereas saturated fatty acids and high-calorie intake were associated with increasing the risk of AD [69]. The food processing causes degradation of heat-sensitive micronutrients (e.g., vitamin C and folates), loss of large amounts of water, and formation of toxic secondary products (advanced glycation end products, AGEs) from non-enzymatic glycation of free amino groups in proteins, lipids, and nucleic acids. The toxic effect of AGEs is referred to as their ability to induce oxidative stress and inflammation by modifying the structure and function of the cell surface receptors and body proteins. Different studies demonstrated that elevated AGEs serum level is associated with cognitive decline and progression of AD. The AGE receptor (RAGE) is located in different places within the body, including microglia and astrocytes, and was established to be overexpressed in the brain of AD patients and serve as a transporter and a cell surface receptor for Aβ [70]. Malnutrition is another risk factor for AD. Deficiency in nutrients such as folate, vitamin B12, and vitamin D may cause a decrease in cognitive function, in addition to the fact that patients with AD suffer from problems associated with eating and swallowing, which may increase the risk of malnutrition [71].
-
Metals
Metals are found in nature and biological systems and can be divided into bio-metals that have a physiological function in living organisms (e.g., copper, zinc, and iron), and toxicological metals which do not possess any biological function (e.g., aluminum and lead) [72]. Aluminum is used significantly in the industries such as processed foods, cosmetics, medical preparations, medicines, and others. In the body, aluminum is bound to plasma transferrin and to citrate molecules that can mediate the transfer of aluminum to the brain. Studies demonstrated that Al accumulates in the cortex, hippocampus, and cerebellum areas, where it interacts with proteins and causes misfolding, aggregation, and phosphorylation of highly phosphorylated proteins like tau protein, characteristic of AD [73]. Lead competes with the binding site of bio-metals like calcium and can cross the blood–brain barrier (BBB) rapidly, where it can modify neural differentiation and synaptogenesis and cause severe damage. Studies revealed that an acute exposure to lead was associated with AD and caused an increase of β-secretase expression and Aβ accumulation. Cadmium is a carcinogenic water-soluble metal that can cross the BBB and cause neurological diseases like AD. Results have demonstrated that Cadmium ions are involved in the aggregation of Aβ plaques and the self-aggregation of tau in the AD brain. The data accumulated on metals support the notion that they are among the risk factors involved in the development of AD [74].
-
Infections
Chronic infections to the central nervous system (CNS) can cause an accumulation of Aβ plaques and NFT, therefore, they are included among the risk factors in AD. Studies by Dr. Itzhaki showed that the DNA of herpes simplex virus (HSV-1) was found in patients with ApoE-ε4 allele carriers, which explains the high risk for developing AD. HSV-1 can replicate in the brain, which can result in the activation of the inflammatory response and an increase in Aβ deposition, resulting in damage to neurons and gradual development of AD. On the other hand, the study results by Miklossy and Balin’s have revealed the role of chronic bacterial infections in AD. For example, syphilitic dementia caused by spirochete bacteria (Treponema pallidum), which are accumulated in the cerebral cortex, produced lesions similar to neurofibrillary tangles, which led to devastating neurodegenerative disorders. Besides, Chlamydia pneumonia bacterium can trigger late-onset AD by activation of astrocyte and cytotoxic microglia, disrupt calcium regulation and apoptosis, resulting in deterioration of cognitive function, and increase the risk of AD [75][76][77].
5.2.4. Medical Factors
Several risk factors are related to the development of Alzheimer’s disease. Adding to this list, older people with AD usually have medical conditions such as cardiovascular disease (CVD), obesity, diabetes, and others. All of these conditions are associated with increased risk of AD [78][79].
-
Cardiovascular Disease (CVDs)
CVDs are recognized as an important risk factor for AD, such as the stroke that is associated with increased risk of dementia due to a neural tissue loss, which enhances degenerative effect and influences amyloid and tau pathology. Atrial fibrillation also causes embolisms which leads to stroke and a decrease in memory and cognitive functions. Moreover, heart failure affects the pumping function of the heart and results in insufficient blood supply to the body and hypo-perfusion of the brain that leads to hypoxia and neural damage. The coronary heart disease’s hypothesis indicates that atherosclerosis, peripheral artery disease, hypo-perfusion, and emboli are all related to increased risk of AD. Hypertension is associated with thickening of vessel walls and narrowing of the lumen which reduce the cerebral blood flow, and in chronic cases, it may cause cerebral edema, which all participate as risk factors for AD and CVD. The CVD is a modifiable risk factor and by focusing on its relationship with AD, a pathway to prevent and delay the disease can be obtained [79][80].
-
Obesity and Diabetes
Obesity is a term used for too much body fat in individuals due to consuming more calories than they burn and can be calculated by using the body mass index (BMI). Increasing the body fat is associated with a decreased brain blood supply which promotes brain ischemia, memory loss, and vascular dementia. The obesity, unhealthy diet, and other factors can cause impaired glucose tolerance (IGT) or diabetes, which is characterized by hyperglycemia that affects peripheral tissues and blood vessels. Chronic hyperglycemia can induce cognitive impairment as a result of increasing amyloid-beta accumulation, oxidative stress, mitochondrial dysfunction, and neuroinflammation. Obesity is characterized by increasing pro-inflammatory cytokines secretions from adipose tissue, which stimulate macrophages and lymphocytes and eventually lead to local and systemic inflammation. This inflammation promotes insulin resistance, hyperinsulinemia, and as a consequence, hyperglycemia. Obesity is a well-known risk factor for type 2 diabetes, CVDs, and cancer, which are identified as risk factors for dementia and AD. The brain inflammation causes an increase in microglia and results in reduced synaptic plasticity and impaired neurogenesis. Microglia can affect insulin receptor substrate 1 (IRS-1) and block intracellular insulin signaling, which has an important role in neural health. Therefore, alteration in insulin action can result in Aβ accumulation and reduce the tau protein degradation associated with AD [81][82][83][84].
References
- De-Paula, V.J.; Radanovic, M.; Diniz, B.S.; Forlenza, O.V. Alzheimer’s disease. Sub-Cell. Biochem. 2012, 65, 329–352.
- Cipriani, G.; Dolciotti, C.; Picchi, L.; Bonuccelli, U. Alzheimer and his disease: A brief history. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2011, 32, 275–279.
- Blass, J.P. Alzheimer’s disease. Dis. A Mon. Dm 1985, 31, 1–69.
- Terry, R.D.; Davies, P. Dementia of the Alzheimer type. Annu. Rev. Neurosci. 1980, 3, 77–95.
- Rathmann, K.L.; Conner, C.S. Alzheimer’s disease: Clinical features, pathogenesis, and treatment. Drug Intell. Clin. Pharm. 1984, 18, 684–691.
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments in alzheimer disease: An update. J. Cent. Nerv. Syst. Dis. 2020, 12.
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020, 396, 413–446.
- Schachter, A.S.; Davis, K.L. Alzheimer’s disease. Dialogues Clin. Neurosci. 2000, 2, 91–100.
- Jatoi, S.; Hafeez, A.; Riaz, S.U.; Ali, A.; Ghauri, M.I.; Zehra, M. Low Vitamin B12 levels: An underestimated cause of minimal cognitive impairment and dementia. Cureus 2020, 12, e6976.
- Cho, H.S.; Huang, L.K.; Lee, Y.T.; Chan, L.; Hong, C.T. Suboptimal baseline serum Vitamin B12 is associated with cognitive decline in people with Alzheimer’s disease undergoing cholinesterase inhibitor treatment. Front. Neurol. 2018, 9, 325.
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944.
- Neugroschl, J.; Wang, S. Alzheimer’s disease: Diagnosis and treatment across the spectrum of disease severity. Mt. Sinai J. Med. N. Y. 2011, 78, 596–612.
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2011, 7, 263–269.
- Mayeux, R.; Stern, Y. Epidemiology of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006239.
- Yaari, R.; Fleisher, A.S.; Tariot, P.N. Updates to diagnostic guidelines for Alzheimer’s disease. Prim. Care Companion Cns Disord. 2011, 13, 11f01262.
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189.
- Spires-Jones, T.L.; Hyman, B.T. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 2014, 82, 756–771.
- Singh, S.K.; Srivastav, S.; Yadav, A.K.; Srikrishna, S.; Perry, G. Overview of Alzheimer’s disease and some therapeutic approaches targeting abeta by using several synthetic and herbal compounds. Oxidative Med. Cell. Longev. 2016, 2016, 7361613.
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2016, 12, 292–323.
- Kumar, A.; Sidhu, J.; Goyal, A. Alzheimer Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020; Available online: https://www.ncbi.nlm.nih.gov/books/NBK499922/ (accessed on 8 December 2020).
- Wattmo, C.; Minthon, L.; Wallin, A.K. Mild versus moderate stages of Alzheimer’s disease: Three-year outcomes in a routine clinical setting of cholinesterase inhibitor therapy. Alzheimer’s Res. Ther. 2016, 8, 7.
- Apostolova, L.G. Alzheimer disease. Continuum 2016, 22, 419–434.
- Armstrong, R.A. Risk factors for Alzheimer’s disease. Folia Neuropathol. 2019, 57, 87–105.
- Anand, P.; Singh, B. A review on cholinesterase inhibitors for Alzheimer’s disease. Arch. Pharmacal Res. 2013, 36, 375–399.
- Babic, T. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. Psychiatry 1999, 67, 558.
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115.
- Monczor, M. Diagnosis and treatment of Alzheimer’s disease. Curr. Med. Chem. Cent. Nerv. Syst. Agents 2005, 5, 5–13.
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain A J. Neurol. 2018, 141, 1917–1933.
- Paroni, G.; Bisceglia, P.; Seripa, D. Understanding the amyloid hypothesis in Alzheimer’s disease. J. Alzheimer’s Dis. Jad 2019, 68, 493–510.
- Kametani, F.; Hasegawa, M. Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer’s disease. Front. Neurosci. 2018, 12, 25.
- Ricciarelli, R.; Fedele, E. The amyloid cascade hypothesis in Alzheimer’s disease: It’s time to change our mind. Curr. Neuropharmacol. 2017, 15, 926–935.
- Guerreiro, R.; Bras, J. The age factor in Alzheimer’s disease. Genome Med. 2015, 7, 106.
- Riedel, B.C.; Thompson, P.M.; Brinton, R.D. Age, APOE and sex: Triad of risk of Alzheimer’s disease. J. Steroid Biochem. Mol. Biol. 2016, 160, 134–147.
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581.
- Bekris, L.M.; Yu, C.E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227.
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. Off. J. Am. Coll. Med Genet. 2016, 18, 421–430.
- Khanahmadi, M.; Farhud, D.D.; Malmir, M. Genetic of Alzheimer’s disease: A narrative review article. Iran. J. Public Health 2015, 44, 892–901.
- Li, N.M.; Liu, K.F.; Qiu, Y.J.; Zhang, H.H.; Nakanishi, H.; Qing, H. Mutations of beta-amyloid precursor protein alter the consequence of Alzheimer’s disease pathogenesis. Neural Regen. Res. 2019, 14, 658–665.
- Tcw, J.; Goate, A.M. Genetics of beta-Amyloid precursor protein in Alzheimer’s disease. Cold Spring Harb. Perspect. Med. 2017, 7, a024539.
- Bi, C.; Bi, S.; Li, B. Processing of mutant beta-amyloid precursor protein and the clinicopathological features of familial Alzheimer’s disease. Aging Dis. 2019, 10, 383–403.
- Dai, M.H.; Zheng, H.; Zeng, L.D.; Zhang, Y. The genes associated with early-onset Alzheimer’s disease. Oncotarget 2018, 9, 15132–15143.
- Zhao, J.; Liu, X.; Xia, W.; Zhang, Y.; Wang, C. Targeting amyloidogenic processing of APP in Alzheimer’s disease. Front. Mol. Neurosci. 2020, 13, 137.
- Cai, Y.; An, S.S.; Kim, S. Mutations in presenilin 2 and its implications in Alzheimer’s disease and other dementia-associated disorders. Clin. Interv. Aging 2015, 10, 1163–1172.
- Lanoiselee, H.M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017, 14, e1002270.
- De Strooper, B. Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. Embo Rep. 2007, 8, 141–146.
- Kelleher, R.J., 3rd; Shen, J. Presenilin-1 mutations and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 629–631.
- Walker, E.S.; Martinez, M.; Brunkan, A.L.; Goate, A. Presenilin 2 familial Alzheimer’s disease mutations result in partial loss of function and dramatic changes in Abeta 42/40 ratios. J. Neurochem. 2005, 92, 294–301.
- Kim, J.; Basak, J.M.; Holtzman, D.M. The role of apolipoprotein E in Alzheimer’s disease. Neuron 2009, 63, 287–303.
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118.
- Giau, V.V.; Bagyinszky, E.; An, S.S.; Kim, S.Y. Role of apolipoprotein E in neurodegenerative diseases. Neuropsychiatr. Dis. Treat. 2015, 11, 1723–1737.
- Koldamova, R.; Fitz, N.F.; Lefterov, I. ATP-binding cassette transporter A1: From metabolism to neurodegeneration. Neurobiol. Dis. 2014, 72 Pt A, 13–21.
- Nordestgaard, L.T.; Tybjaerg-Hansen, A.; Nordestgaard, B.G.; Frikke-Schmidt, R. Loss-of-function mutation in ABCA1 and risk of Alzheimer’s disease and cerebrovascular disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2015, 11, 1430–1438.
- Foster, E.M.; Dangla-Valls, A.; Lovestone, S.; Ribe, E.M.; Buckley, N.J. Clusterin in Alzheimer’s disease: Mechanisms, genetics, and lessons from other pathologies. Front. Neurosci. 2019, 13, 164.
- Holler, C.J.; Davis, P.R.; Beckett, T.L.; Platt, T.L.; Webb, R.L.; Head, E.; Murphy, M.P. Bridging integrator 1 (BIN1) protein expression increases in the Alzheimer’s disease brain and correlates with neurofibrillary tangle pathology. J. Alzheimer’s Dis. Jad 2014, 42, 1221–1227.
- Andrew, R.J.; De Rossi, P.; Nguyen, P.; Kowalski, H.R.; Recupero, A.J.; Guerbette, T.; Krause, S.V.; Rice, R.C.; Laury-Kleintop, L.; Wagner, S.L.; et al. Reduction of the expression of the late-onset Alzheimer’s disease (AD) risk-factor BIN1 does not affect amyloid pathology in an AD mouse model. J. Biol. Chem. 2019, 294, 4477–4487.
- Soler-Lopez, M.; Badiola, N.; Zanzoni, A.; Aloy, P. Towards Alzheimer’s root cause: ECSIT as an integrating hub between oxidative stress, inflammation and mitochondrial dysfunction. Hypothetical role of the adapter protein ECSIT in familial and sporadic Alzheimer’s disease pathogenesis. Bioessays News Rev. Mol. Cell. Dev. Biol. 2012, 34, 532–541.
- Mi Wi, S.; Park, J.; Shim, J.H.; Chun, E.; Lee, K.Y. Ubiquitination of ECSIT is crucial for the activation of p65/p50 NF-kappaBs in Toll-like receptor 4 signaling. Mol. Biol. Cell 2015, 26, 151–160.
- Soler-Lopez, M.; Zanzoni, A.; Lluis, R.; Stelzl, U.; Aloy, P. Interactome mapping suggests new mechanistic details underlying Alzheimer’s disease. Genome Res. 2011, 21, 364–376.
- Zhao, L.; Woody, S.K.; Chhibber, A. Estrogen receptor beta in Alzheimer’s disease: From mechanisms to therapeutics. Ageing Res. Rev. 2015, 24, 178–190.
- Sundermann, E.E.; Maki, P.M.; Bishop, J.R. A review of estrogen receptor alpha gene (ESR1) polymorphisms, mood, and cognition. Menopause 2010, 17, 874–886.
- Yaffe, K.; Lindquist, K.; Sen, S.; Cauley, J.; Ferrell, R.; Penninx, B.; Harris, T.; Li, R.; Cummings, S.R. Estrogen receptor genotype and risk of cognitive impairment in elders: Findings from the Health ABC study. Neurobiol. Aging 2009, 30, 607–614.
- Goumidi, L.; Dahlman-Wright, K.; Tapia-Paez, I.; Matsson, H.; Pasquier, F.; Amouyel, P.; Kere, J.; Lambert, J.C.; Meirhaeghe, A. Study of estrogen receptor-alpha and receptor-beta gene polymorphisms on Alzheimer’s disease. J. Alzheimer’s Dis. Jad 2011, 26, 431–439.
- Khorram Khorshid, H.R.; Gozalpour, E.; Saliminejad, K.; Karimloo, M.; Ohadi, M.; Kamali, K. Vitamin D Receptor (VDR) polymorphisms and late-onset Alzheimer’s disease: An association study. Iran. J. Public Health 2013, 42, 1253–1258.
- Liu, X.; Jiao, B.; Shen, L. The epigenetics of Alzheimer’s Disease: Factors and therapeutic implications. Front. Genet. 2018, 9, 579.
- Wainaina, M.N.; Chen, Z.; Zhong, C. Environmental factors in the development and progression of late-onset Alzheimer’s disease. Neurosci. Bull. 2014, 30, 253–270.
- Grant, W.B.; Campbell, A.; Itzhaki, R.F.; Savory, J. The significance of environmental factors in the etiology of Alzheimer’s disease. J. Alzheimer’s Dis. Jad 2002, 4, 179–189.
- Moulton, P.V.; Yang, W. Air pollution, oxidative stress, and Alzheimer’s disease. J. Environ. Public Health 2012, 2012, 472751.
- Croze, M.L.; Zimmer, L. Ozone atmospheric pollution and Alzheimer’s disease: From epidemiological facts to molecular mechanisms. J. Alzheimer’s Dis. Jad 2018, 62, 503–522.
- Hu, N.; Yu, J.T.; Tan, L.; Wang, Y.L.; Sun, L.; Tan, L. Nutrition and the risk of Alzheimer’s disease. Biomed. Res. Int. 2013, 2013, 524820.
- Abate, G.; Marziano, M.; Rungratanawanich, W.; Memo, M.; Uberti, D. Nutrition and AGE-ing: Focusing on Alzheimer’s disease. Oxidative Med. Cell. Longev. 2017, 2017, 7039816.
- Koyama, A.; Hashimoto, M.; Tanaka, H.; Fujise, N.; Matsushita, M.; Miyagawa, Y.; Hatada, Y.; Fukuhara, R.; Hasegawa, N.; Todani, S.; et al. Malnutrition in Alzheimer’s disease, dementia with lewy bodies, and frontotemporal lobar degeneration: Comparison using serum albumin, total protein, and hemoglobin level. PLoS ONE 2016, 11, e0157053.
- Adlard, P.A.; Bush, A.I. Metals and Alzheimer’s disease. J. Alzheimer’s Dis. Jad 2006, 10, 145–163.
- Colomina, M.T.; Peris-Sampedro, F. Aluminum and Alzheimer’s disease. Adv. Neurobiol. 2017, 18, 183–197.
- Huat, T.J.; Camats-Perna, J.; Newcombe, E.A.; Valmas, N.; Kitazawa, M.; Medeiros, R. Metal toxicity links to Alzheimer’s disease and neuroinflammation. J. Mol. Biol. 2019, 431, 1843–1868.
- Sochocka, M.; Zwolinska, K.; Leszek, J. The infectious etiology of Alzheimer’s disease. Curr. Neuropharmacol. 2017, 15, 996–1009.
- Fulop, T.; Itzhaki, R.F.; Balin, B.J.; Miklossy, J.; Barron, A.E. Role of microbes in the development of Alzheimer’s disease: State of the art—An international symposium presented at the 2017 IAGG congress in San Francisco. Front. Genet. 2018, 9, 362.
- Muzambi, R.; Bhaskaran, K.; Brayne, C.; Smeeth, L.; Warren-Gash, C. Common bacterial infections and risk of incident cognitive decline or dementia: A systematic review protocol. BMJ Open 2019, 9, e030874.
- Stampfer, M.J. Cardiovascular disease and Alzheimer’s disease: Common links. J. Intern. Med. 2006, 260, 211–223.
- Santos, C.Y.; Snyder, P.J.; Wu, W.C.; Zhang, M.; Echeverria, A.; Alber, J. Pathophysiologic relationship between Alzheimer’s disease, cerebrovascular disease, and cardiovascular risk: A review and synthesis. Alzheimer’s Dement. 2017, 7, 69–87.
- De Bruijn, R.F.; Ikram, M.A. Cardiovascular risk factors and future risk of Alzheimer’s disease. BMC Med. 2014, 12, 130.
- Alford, S.; Patel, D.; Perakakis, N.; Mantzoros, C.S. Obesity as a risk factor for Alzheimer’s disease: Weighing the evidence. Obes. Rev. Off. J. Int. Assoc. Study Obes. 2018, 19, 269–280.
- Pegueroles, J.; Jimenez, A.; Vilaplana, E.; Montal, V.; Carmona-Iragui, M.; Pane, A.; Alcolea, D.; Videla, L.; Casajoana, A.; Clarimon, J.; et al. Obesity and Alzheimer’s disease, does the obesity paradox really exist? A magnetic resonance imaging study. Oncotarget 2018, 9, 34691–34698.
- Anjum, I.; Fayyaz, M.; Wajid, A.; Sohail, W.; Ali, A. Does obesity increase the risk of dementia: A literature review. Cureus 2018, 10, e2660.
- Lee, H.J.; Seo, H.I.; Cha, H.Y.; Yang, Y.J.; Kwon, S.H.; Yang, S.J. Diabetes and Alzheimer’s disease: Mechanisms and nutritional aspects. Clin. Nutr. Res. 2018, 7, 229–240.
More
Information
Subjects:
Pharmacology & Pharmacy
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
4.4K
Revisions:
2 times
(View History)
Update Date:
29 Feb 2024
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No