Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Zeinab Breijyeh | -- | 2165 | 2024-02-28 20:12:27 | | | |
| 2 | Peter Tang | + 1 word(s) | 2166 | 2024-02-29 06:03:49 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Jubeh, B.; Breijyeh, Z.; Karaman, R. Antibacterial Prodrugs to Overcome Bacterial Resistance. Encyclopedia. Available online: https://encyclopedia.pub/entry/55687 (accessed on 10 August 2026).
Jubeh B, Breijyeh Z, Karaman R. Antibacterial Prodrugs to Overcome Bacterial Resistance. Encyclopedia. Available at: https://encyclopedia.pub/entry/55687. Accessed August 10, 2026.
Jubeh, Buthaina, Zeinab Breijyeh, Rafik Karaman. "Antibacterial Prodrugs to Overcome Bacterial Resistance" Encyclopedia, https://encyclopedia.pub/entry/55687 (accessed August 10, 2026).
Jubeh, B., Breijyeh, Z., & Karaman, R. (2024, February 28). Antibacterial Prodrugs to Overcome Bacterial Resistance. In Encyclopedia. https://encyclopedia.pub/entry/55687
Jubeh, Buthaina, et al. "Antibacterial Prodrugs to Overcome Bacterial Resistance." Encyclopedia. Web. 28 February, 2024.
Copy Citation
Prodrugs are pharmacologically inactive entities of active drugs that undergo biotransformation before eliciting their pharmacological effects. A prodrug strategy can be used to revive drugs discarded due to a lack of appropriate pharmacokinetic and drug-like properties, or high host toxicity. A special advantage of the use of the prodrug approach in the era of bacterial resistance is targeting resistant bacteria by developing prodrugs that require bacterium-specific enzymes to release the active drug.
prodrugs
biotransformation
targeting
β-lactam antibiotics
β-lactamases
pathogens
resistance
1. Introduction
Nowadays, the issue of pathogens resistant to drugs and the urgent need for new compounds that are capable of eradicating these pathogens are well known and understood. Most of the resistance mechanisms by bacteria have been discovered and described, such as enzymatic degradation, target modification, overexpression of efflux pumps and decreased uptake. Multidrug-resistant bacteria show resistance to three or more antibiotic classes, and are a major cause of mortality as indicated by the World Health Organization (WHO) [1].
Penicillin was discovered in the 1940s, and shortly after, resistance was developed. By the 1950s, many β-lactam antibiotics were discovered; however, resistance was identified in the 1960s. Later on, fluoroquinolones were introduced to treat Gram-negative bacteria in the 1980s, but resistance was quickly developed due to chromosomal mutations. Antibiotic discovery continued over decades, and millions of drugs were introduced to the market; this has led to less expensive antibiotics and the irresponsible dispensing of antibiotics without prescriptions, with the absence of regulatory guidelines in many countries. This has all contributed to the development of resistant strains [2][3].
2. Prodrugs
Prodrugs are pharmacologically inactive compounds that inter- or intra-convert inside the body into active forms and non-toxic moieties by enzymatic or chemical reactions. In prodrug design, the addition or removal of certain parts of the parent drug can alter its bioavailability, absorption, and permeability without affecting its pharmacological activity. Prodrugs are classified into three types:(1) carrier-linked prodrugs in which the active drug is linked to a promoiety, which is removed by an enzymatic or chemical reaction to release the active drug (Figure 1),(2) bioprecursor prodrugs in which a molecular modification is made to the active drug and that can undergo molecular modification by oxidation or reduction reactions to release the active drug, and finally (3) double prodrugs that are attached to each other by two linkers that can be cleaved by different mechanisms, such as a co-drug, in which two biologically active drugs are linked in a single molecule.
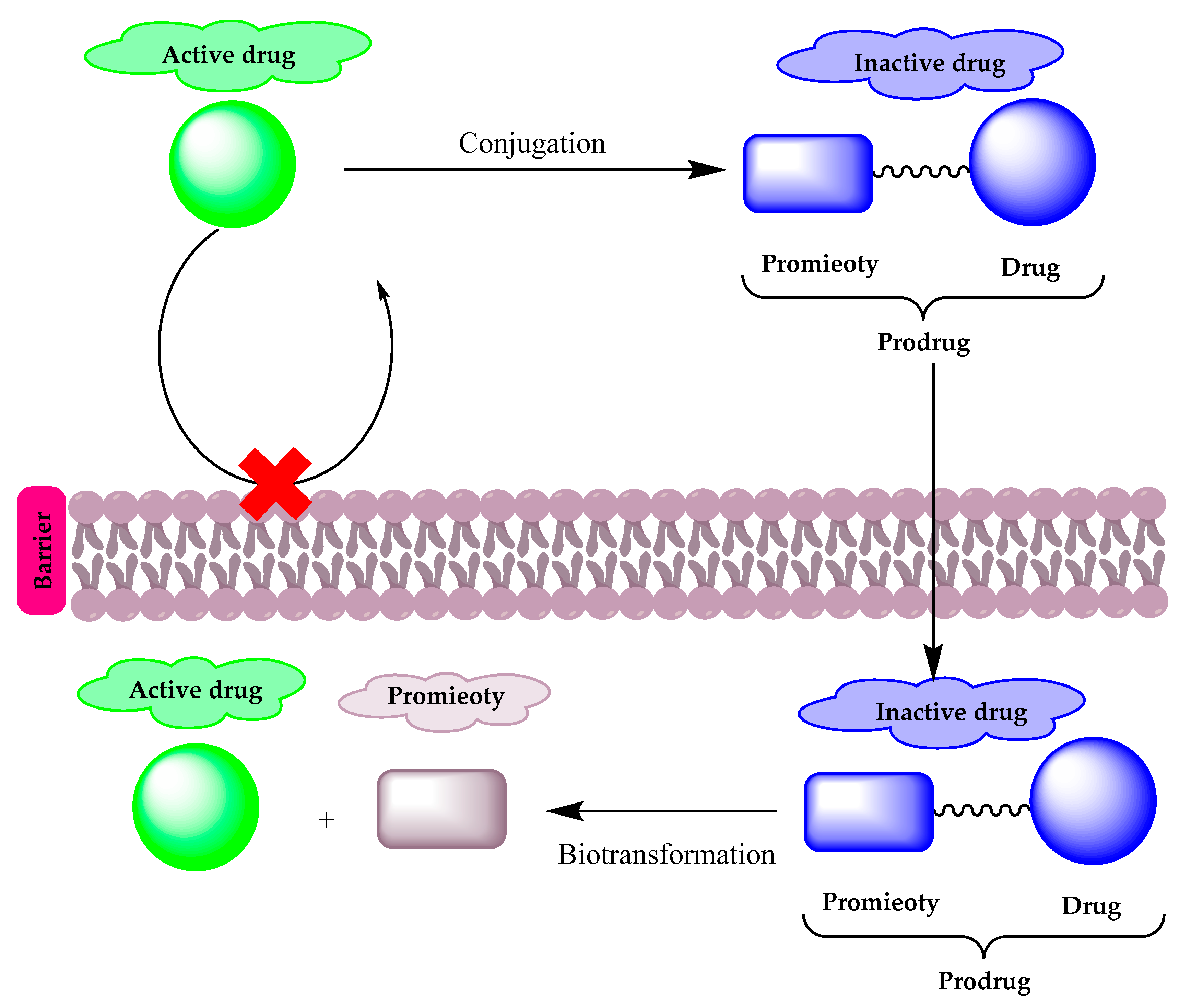
Figure 1. A diagram illustrating the prodrug concept.
Most of the β-lactam antibiotics are poorly absorbed, and the prodrug approach was used to improve their oral bioavailability.Pivampicillin (1) (Figure 2) is an ampicillin derivative that is considered one of the first prodrugs to be developed, which is enzymatically hydrolyzed by esterases to give the active drug ampicillin, with inactive pivalic acid. Moreover, talampicillin (2), bacampicillin (3) andhetacillin (4) (Figure 2) are all ester prodrugs that were developed to improve ampicillin bioavailability. Another form of prodrug is made by attaching a linker to the active drug to combine it with other antibiotics or different drugs such as sultamicillin (5) (Figure 2), which is a prodrug that links ampicillin and sulbactam by a methylene group [4][5][6][7]. Other prodrugs were developed to improve the activity of existing antibiotics including ethionamide (6), isoniazid (7) and pyrazinamide (8) (Figure 2), used to treat Mycobacterium tuberculosis. In addition, metronidazole (9) (Figure 2) acts as a prodrug, which is used for the treatment of Helicobacter pylori and anaerobic infections. Metronidazole requires reduction by specific enzymes to be activated; the redox intermediate could be responsible for killing the microorganisms by targeting intracellular components such asthe bacterial cell membrane, RNA, DNA and proteins.
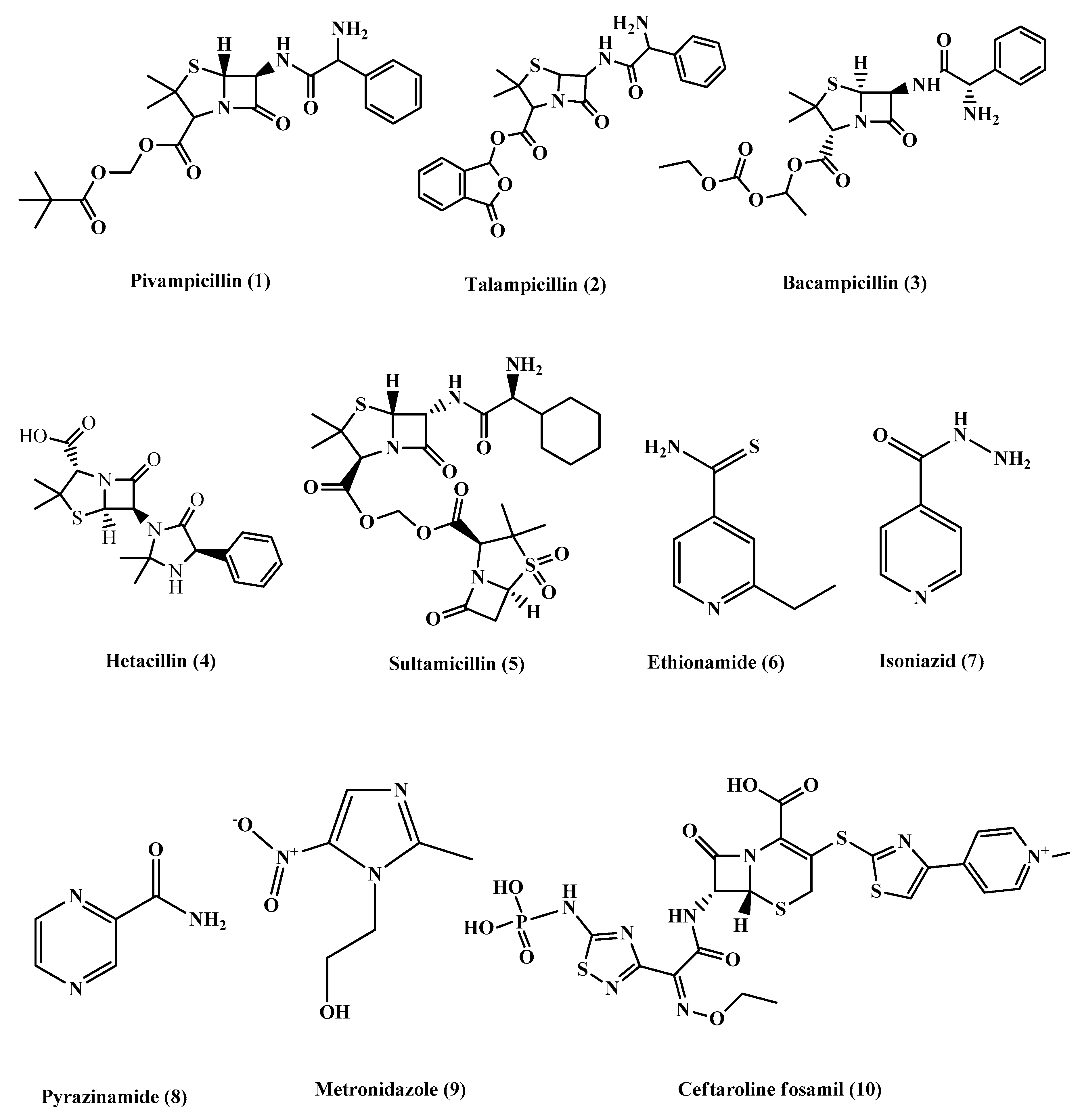
Figure 2. Chemical structures of pivampicillin, talampicillin, bacampicillin, hetacillin, Sultamicillin, ethionamide, isoniazid, pyrazinamide, metronidazole and ceftaroline fosamil.
Ceftaroline fosamil (10) (Figure 2) is an example of cephalosporin prodrugs that upon activation give the active metabolite ceftaroline, which acts by binding to penicillin-binding proteins that inhibit bacterial cell wall synthesis (Figure 3).Ceftaroline fosamil has potent activity against the bacteria causing community-acquired bacterial pneumonia (CABP), such as multidrug-resistant Streptococcus pneumoniae(MDRSP), penicillin-resistant Streptococcus pneumonia (PRSP) and methicillin-resistant Staphylococcus aureus (MRSA) [8].
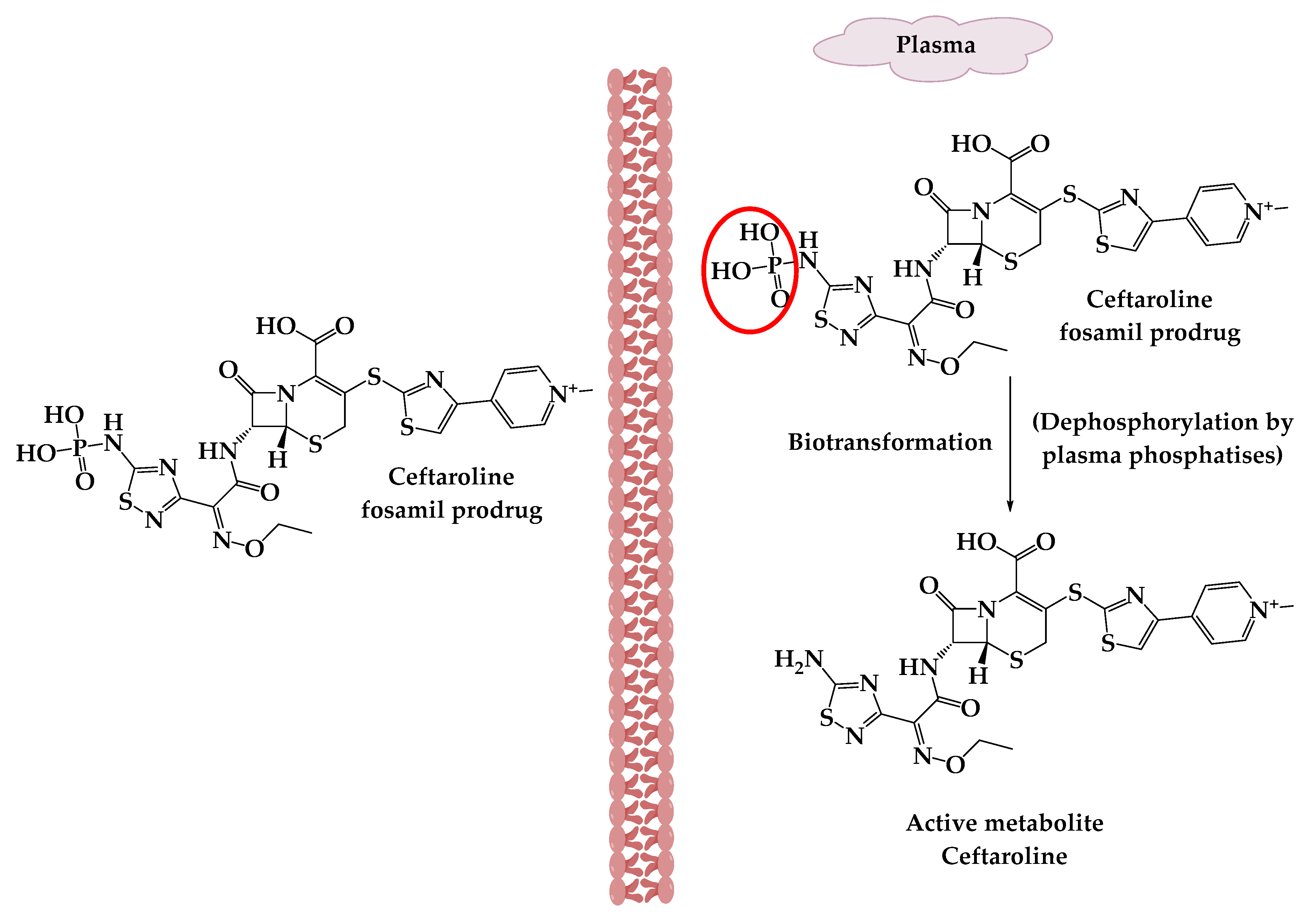
Figure 3. The activation of ceftaroline fosamil prodrug.
3. Prodrug Applications against Resistant Bacterial Pathogens
In combating antibiotic resistance, prodrugs have been applied either to revive antibiotics that possess activity against resistant pathogens—but that cannot be used clinically because of their suboptimal pharmacokinetics—or as a targeting drug tool to overcome the resistance barriers and minimize host toxicity. Herein, the researchers describe the recently reported antibiotic prodrugs for which the prodrug approach was the key to fight resistance.
3.1. A β-Lactamase-Activated Ciprofloxacin Prodrug
The most important and prevalent determinant of antibiotic resistance is the expression of β-lactamase enzymes, which hydrolyze the β-lactam antibiotics, preventing their interaction with the penicillin-binding proteins, their therapeutic targets. Extended-spectrum β-lactamases that have the ability to cleave a wide range of β-lactam antibiotics, such as the CTX-M class (extended-spectrum β-lactamases active on CefoTaXime, first isolated in Munich), are of particular concern [9][10][11]. Broad-spectrum antibiotics, like ciprofloxacin, are increasingly used to treat infections caused by β-lactam-resistant bacterial infections, especially Escherichia coli-caused urinary tract infections [12]. The problem is that the use of broad-spectrum antibiotics disrupts the microbiota, the beneficial gastrointestinal bacteria, which normally lead to secondary infections caused by other antibiotic-resistant bacteria [13][14].
3.2. Cephalosporin-3-Diazeniumdiolate
Cephalosporin-3′-diazeniumdiolates (C3Ds) are a new class of nitric oxide (NO) donor prodrugs. These prodrugs have a β-lactam ring in their structures and are designed to selectively deliver NO to bacterial infection sites after the reaction with β-lactamases and the cleavage of β-lactam by transpeptidase [15][16]. Currently, there are some C3Ds that are under development, including PYRRO-C3D (pyrro-cephalosporin-3′-diazeniumdiolate) (11), and DEA-C3D(diethylamin-cephalosporin-3′-diazeniumdiolate) (12) (Figure 4). DEA-C3D, the prototypical example of a C3D, contains the phenacetyl side chain of cefaloram, a first-generation cephalosporin, and the diazeniumdiolate NO donor. In vitro studies have shown that DEA-C3D was able to disperse biofilms formed by multiple clinical isolates of Pseudomonas aeruginosa, and that when combined with colistin, it caused the near-complete eradication of P. aeruginosa biofilms [17].
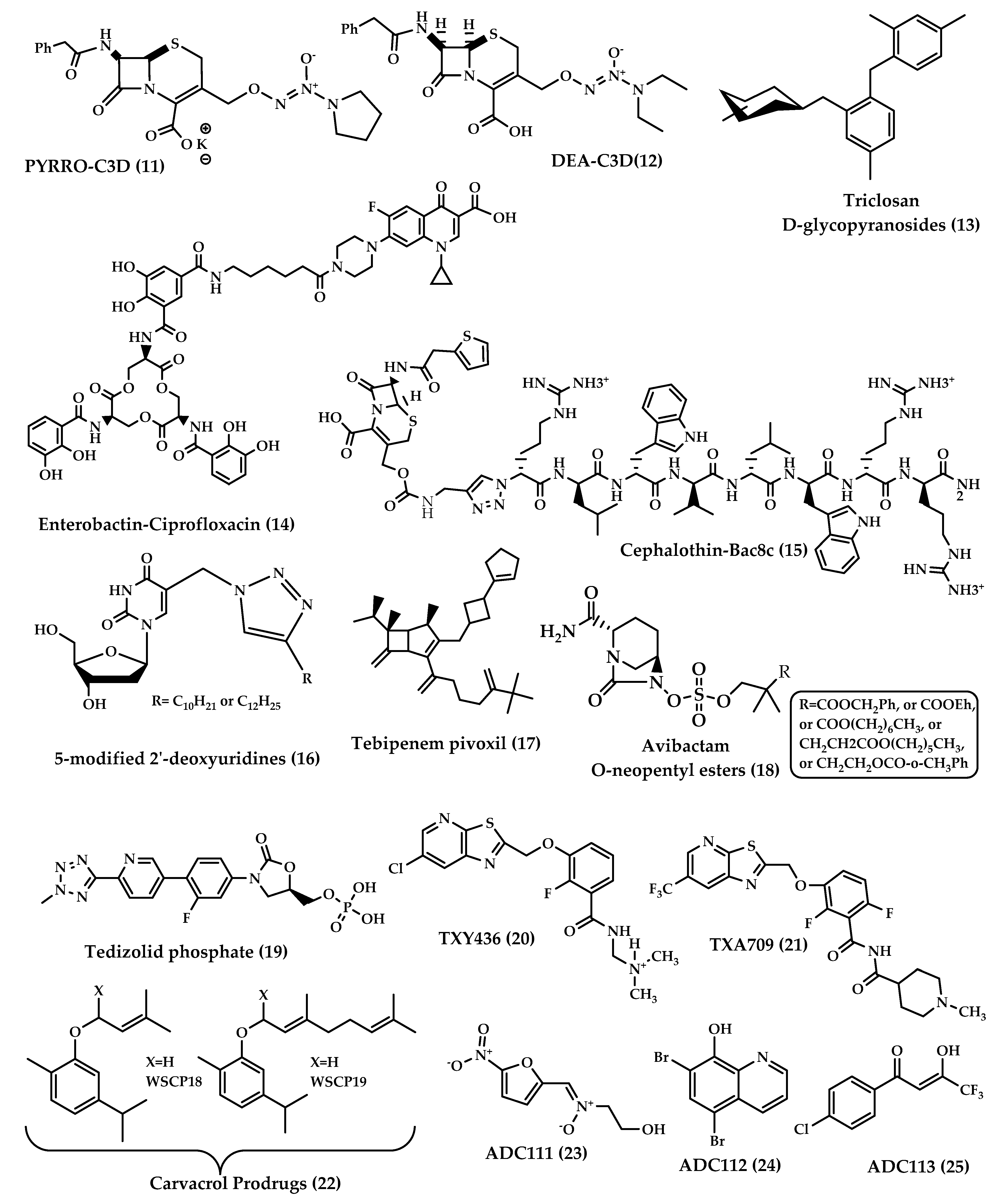
Figure 4. Chemical structures of 11–25.
3.3. Triclosan Glycoside Prodrugs
The idea of using glycoside derivatives of antibacterials as bacterium-targeting prodrugs came about as a result of the discovery of glycosidase enzyme expression in bacteria [18][19]. Triclosan is an antibacterial and antifungal agent that has been used as a disinfectant. Triclosan acts by inhibiting fatty acid synthesis, and in high concentrations, it disrupts the cell wall [19]. This agent is used only topically due to its low solubility at physiological pH. It was expected that glycoside derivatives on the hydroxyl group of triclosan would enhance bacterial uptake by active transport. Glycoside derivatives of triclosan (α-D-glycopyranosides and β-D-glycopyranosides) (13) (Figure 4) have the ability to inhibit the growth of Gram-positive and Gram-negative bacteria and showed potent, selective antibacterial activity and increased aqueous solubility compared to triclosan. This earned them the advantage to be used orally for the treatment of systemic infections [20].
3.4. Enterobactin-Antibiotic Conjugates
The prodrug approach has been used to attain drug targeting with certain antibiotics. Zheng and Nolan [21] presented a strategy to achieve intracellular antibiotic targeting, as well as pathogen-specific activity, by making siderophore-antibiotic conjugates. This strategy is based on linking antibiotics with enterobactin—a siderophore that has receptors on bacterial surfaces and is responsible for the uptake of iron—generating inactive prodrugs. This linkage enables the uptake of the siderophore along with the antibiotic linked to it, and then the hydrolysis of the conjugate and the activation of the antibiotic take place in the bacterial cytoplasm, involving specific cytoplasmic enzymes [22]. Enterobactin (Ent) is a tricatecholatesiderophore (iron carrier) naturally produced by enteric Gram-negative bacteria like E. coli, and is secreted in the host vertebrate to acquire iron. Recently, enterobactin-ciprofloxacin conjugate (14) (Figure 4) discovery was reported by Neumann et al. [21]. Ent–Ciprofloxacin is a conjugate that has ciprofloxacin attached to an alkyl linker at one of the catechols of Ent; the result is an inactive prodrug of ciprofloxacin that is guided into the cytoplasm by Ent uptake machinery. Intracellularly, the prodrug is activated by the cytoplasmic esteraseIroD, an enzyme that is only expressed by E. coli that express the iroA gene cluster—a pathogen-associated cluster—which makes the prodrug selective for E. coli that have the iroA gene cluster, while having no activity against nonpathogenic clusters [21].
3.5. Antimicrobial Peptide Prodrugs
Antimicrobial peptides (AMPs), also known as host defense peptides, are peptidic molecular mediators of innate immunity found in multicellular organisms that have antimicrobial activity [23][24]. AMPs exert direct microbicidal activity by having an amphipathic and cationic nature that enables them to be inserted in microbial cytoplasmic membranes, increasing permeation and causing cell lysis [24][25][26]. Interests in the development of AMP antibiotics, especially against resistant bacteria, are increasing [27][28][29][30][31], but unwanted cell toxicity forms a major limitation for their improvement. The prodrug strategy is a promising solution to solve the toxicity problem and fulfill bacterial selectivity, by making AMP prodrugs that permit the cationic feature to be transiently reduced by the reversible conjugation of an anionic promoiety and that can be activated by specific bacterial enzymes [32].
3.6. Prodrugs of 5-Modified 2ʹ-Deoxyuridines
Pyrimidine nucleoside derivatives with a lengthy substituent at the C-5 position of the nucleobase were shown to have in vitro antimicrobial activity [33][34]. The mechanism of action is unclear, but it was shown that some derivatives selectively inhibit the microorganismic enzyme flavin-dependent thymidylate synthase (ThyX), an enzyme that is absent in mammals. Other derivatives demonstrated destruction of the mycobacterial cell wall [35].
3.7. The Tebipenempivoxil Prodrug
Tebipenem (SPR859) is a β-lactam antibiotic belonging to the carbapenem family and is active against Gram-negative and Gram-positive pathogens, but its high hydrophilicity limits its oral absorption. Tebipenempivoxil (17) (Figure 4) is an orally-administrated pivaloyloxymethyl ester prodrug of tebipenemwith better absorption and high bioavailability, which is currently approved only in Japan as a granule formulation for the treatment of ear, nose, throat and respiratory infections in pediatrics. Tebipenempivoxil HBr salt is currently under development for the treatment of complicated urinary tract infections in adults. Pharmacokinetics and pharmacodynamics activity showed that tebipenempivoxil HBr is a good alternative oral treatment for resistant Gram-negative pathogens and may serve as a new antibacterial agent [36].
3.8. Avibactam Prodrugs
β-lactamase inhibitors restored the effectiveness of β-lactam antibiotics. None of the β-lactamase inhibitors other than clavulanic acid are orally available. This is the case of avibactam, which is a potent diazabicyclooctane inhibitor of a wide spectrum of β-lactamases but lacks the proper oral bioavailability. Gordon et al. [37] have reported the synthesis and testing of avibactam O-neopentyl ester prodrugs (18) (Figure 4) designed to mask the charged sulfate moiety in avibactam’s structure, a moiety that causes a major obstacle for oral absorption. Coupled with anappropriate antibiotic, avibactam has the potential to treat serious Gram-negative infections without the need for intravenous injections [37].
3.9. Tedizolid Phosphate (TR701)
Tedizolid phosphate (19) (Figure 4) is an orally absorbed phosphate prodrug of tedizolid (TR700): an antibiotic of the new class oxazolidinones. Among this class, linezolid is the only marketed oxazolidinone. Oxazolidinone antibiotics are protein synthesis inhibitors that bind to the 50S ribosome and prevent the formation of the 70S complex [38]. Oxazolidinones are unlikely to have cross-sensitivity with other antibiotics because of a unique site of action that is the ribosomal peptidyltransferase center [39].
3.10. FtsZ-Targeting Benzamide Prodrugs
Fts-Z (Filamenting temperature-sensitive mutant Z) is a prokaryote-specific protein involved in bacterial cell division; this protein represents a new antibiotic target. The compound PC190723 was among the first FtsZ-Targeting Benzamides to be proved effective against methicillin-sensitive and resistant Staphylococcus aureus (MSSA and MRSA). The poor pharmacokinetics and drug-like properties of PC190723 hindered its clinical development, but the development of the N-Mannich base prodrug TXY436 (20) (Figure 4) followed, which exhibited oral bioavailability and efficacy, as well as enhanced intravenous efficacy [40].
3.11. Carvacrol Prodrugs
Carvacrol is a natural monoterpene; it is a component of phenolic essential oils particularly abundant in plants belonging to the Lamiaceae family. Carvacrol is a compound with emerging potential for application in microbial infection management because of its antimicrobial activity. Biofilm formation is one mechanism by which bacteria have developed resistance to drugs. Carvacrol can inhibit the growth of bacterial biofilms and interfere with biofilm formation, hence carvacrol has recently attracted much attention [41].
Carvacrol has an amphipathic structure that allows it to spread through the polar matrix of bacterial biofilms and to disrupt bacterial membranes; it increases the fluidity, permeability, and perturbation of the cytoplasmatic membranes. Carvacrol acts on biofilms produced by Gram-positive bacteria, especially those produced by S. aureus and Staphylococcus epidermidis. It disintegrates the outer membrane of Gram-negative bacteria as well [42].
3.12. ADC111, ADC112 and ADC113
The need for broad-spectrum antibiotics can be met by the development of prodrugs that are activated by enzymes that are bacterium-specific, to give reactive compounds that could kill persisters and accumulate over time. Hence, a screen of 55,000 compounds of prodrugs was done by Fleck et al. [43]. The screen was directed to prodrugs that have been discarded in conventional high-throughput screening campaigns due to the lack of specificity of the mechanism of action. The screen was based on identifying compounds that nonspecifically inhibit the reduction of alamarBlue, a viability dye, and then testing for cytotoxicity and eliminating generally-toxic compounds. Twenty hit compounds were active against E. coli. Out of the 20 hits, three prodrugs were further developed: ADC111 (23), ADC112 (24) and ADC113 (25).
References
- Gajdacs, M. The Concept of an Ideal Antibiotic: Implications for Drug Design. Molecules 2019, 24, 892.
- Ventola, C.L. The antibiotic resistance crisis: Part 1: Causes and threats. Pharm. Ther. 2015, 40, 277–283.
- Zaman, S.B.; Hussain, M.A.; Nye, R.; Mehta, V.; Mamun, K.T.; Hossain, N. A Review on Antibiotic Resistance: Alarm Bells are Ringing. Cureus 2017, 9, e1403.
- Monserrat-Martinez, A.; Gambin, Y.; Sierecki, E. Thinking Outside the Bug: Molecular Targets and Strategies to Overcome Antibiotic Resistance. Int. J. Mol. Sci. 2019, 20, 1255.
- Smyth, R.D.; Pfeffer, M.; Van Harken, D.R.; Cohen, A.; Hottendorf, G.H. Human pharmacokinetics and disposition of sarmoxicillin, a lipophilic amoxicillin prodrug. Antimicrob. Agents Chemother. 1981, 19, 1004–1012.
- Jusko, W.J.; Lewis, G.P.; Schmitt, G.W. Ampicillin and hetacillin pharmacokinetics in normal and anephric subjects. Clin. Pharmacol. Ther. 1973, 14, 90–99.
- Jones, K.H. Bioavailability of talampicillin. Br. Med. J. 1977, 2, 232–233.
- Shirley, D.A.; Heil, E.L.; Johnson, J.K. Ceftaroline fosamil: A brief clinical review. Infect Dis. Ther. 2013, 2, 95–110.
- Kong, K.F.; Schneper, L.; Mathee, K. Beta-lactam antibiotics: From antibiosis to resistance and bacteriology. Apmis 2010, 118, 1–36.
- Bush, K. Proliferation and significance of clinically relevant β-lactamases. Ann. N. Y. Acad. Sci. 2013, 1277, 84–90.
- Cantón, R.; González-Alba, J.M.; Galán, J.C. CTX-M enzymes: Origin and diffusion. Front. Microbiol. 2012, 3, 110.
- Scheld, W.M. Maintaining fluoroquinolone class efficacy: Review of influencing factors. Emerg. Infect. Dis. 2003, 9, 1.
- Becattini, S.; Taur, Y.; Pamer, E.G. Antibiotic-induced changes in the intestinal microbiota and disease. Trends Mol. Med. 2016, 22, 458–478.
- Stewardson, A.J.; Gaïa, N.; Francois, P.; Malhotra-Kumar, S.; Delemont, C.; de Tejada, B.M.; Schrenzel, J.; Harbarth, S.; Lazarevic, V.; Wp, S. Collateral damage from oral ciprofloxacin versus nitrofurantoin in outpatients with urinary tract infections: A culture-free analysis of gut microbiota. Clin. Microbiol. Infect. 2015, 21, 344.
- Allan, R.N.; Kelso, M.J.; Rineh, A.; Yepuri, N.R.; Feelisch, M.; Soren, O.; Brito-Mutunayagam, S.; Salib, R.J.; Stoodley, P.; Clarke, S.C. Cephalosporin-NO-donor prodrug PYRRO-C3D shows β-lactam-mediated activity against Streptococcus pneumoniae biofilms. Nitric Oxide 2017, 65, 43–49.
- Barraud, N.; Kardak, B.G.; Yepuri, N.R.; Howlin, R.P.; Webb, J.S.; Faust, S.N.; Kjelleberg, S.; Rice, S.A.; Kelso, M.J. Cephalosporin-3′-diazeniumdiolates: Targeted NO-Donor Prodrugs for Dispersing Bacterial Biofilms. Angew. Chem. Int. Ed. 2012, 51, 9057–9060.
- Soren, O.; Rineh, A.; Silva, D.G.; Cai, Y.; Howlin, R.P.; Allan, R.N.; Feelisch, M.; Davies, J.C.; Connett, G.J.; Faust, S.N. Cephalosporin nitric oxide-donor prodrug DEA-C3D disperses biofilms formed by clinical cystic fibrosis isolates of Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2019, 75, 117–125.
- Kämpfer, P.; Rauhoff, O.; Dott, W. Glycosidase profiles of members of the family Enterobacteriaceae. J. Clin. Microbiol. 1991, 29, 2877–2879.
- Levy, C.W.; Roujeinikova, A.; Sedelnikova, S.; Baker, P.J.; Stuitje, A.R.; Slabas, A.R.; Slabas, A.R.; Rice, D.W.; Rafferty, J.B. Molecular basis of triclosan activity. Nature 1999, 398, 383–384.
- Howse, G.L.; Bovill, R.A.; Stephens, P.J.; Osborn, H.M. Synthesis and antibacterial profiles of targeted triclosan derivatives. Eur. J. Med. Chem. 2019, 162, 51–58.
- Neumann, W.; Sassone-Corsi, M.; Raffatellu, M.; Nolan, E.M. Esterase-catalyzed siderophore hydrolysis activates an enterobactin–ciprofloxacin conjugate and confers targeted antibacterial activity. J. Am. Chem. Soc. 2018, 140, 5193–5201.
- Zheng, T.; Nolan, E.M. Enterobactin-mediated delivery of β-lactam antibiotics enhances antibacterial activity against pathogenic Escherichia coli. J. Am. Chem. Soc. 2014, 136, 9677–9691.
- Lai, Y.; Gallo, R.L. AMPed up immunity: How antimicrobial peptides have multiple roles in immune defense. Trends Immunol. 2009, 30, 131–141.
- Hancock, R.E.; Haney, E.F.; Gill, E.E. The immunology of host defence peptides: Beyond antimicrobial activity. Nat. Rev. Immunol. 2016, 16, 321.
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250.
- Peschel, A.; Sahl, H.-G. The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat. Rev. Microbiol. 2006, 4, 529–536.
- Chung, P.Y.; Khanum, R. Antimicrobial peptides as potential anti-biofilm agents against multidrug-resistant bacteria. J. Microbiol. Immunol. Infect. 2017, 50, 405–410.
- Zhou, Y.; Peng, Y. Synergistic effect of clinically used antibiotics and peptide antibiotics against Gram-positive and Gram-negative bacteria. Exp. Ther. Med. 2013, 6, 1000–1004.
- Samy, R.P.; Stiles, B.G.; Franco, O.L.; Sethi, G.; Lim, L.H. Animal venoms as antimicrobial agents. Biochem. Pharmacol. 2017, 134, 127–138.
- Wang, K.; Yan, J.; Chen, R.; Dang, W.; Zhang, B.; Zhang, W.; Song, J.; Wang, R. Membrane-active action mode of polybia-CP, a novel antimicrobial peptide isolated from the venom of Polybia paulista. Antimicrob. Agents Chemother. 2012, 56, 3318–3323.
- das Neves, R.C.; Mortari, M.R.; Schwartz, E.F.; Kipnis, A.; Junqueira-Kipnis, A.P. Antimicrobial and antibiofilm effects of peptides from venom of social Wasp and scorpion on multidrug-resistant Acinetobacter baumannii. Toxins 2019, 11, 216.
- Forde, É.; Shafiy, G.; Fitzgerald-Hughes, D.; Strömstedt, A.A.; Devocelle, M. Action of antimicrobial peptides and their prodrugs on model and biological membranes. J. Pept. Sci. 2018, 24, e3086.
- Ferrari, V.; Serpi, M. Nucleoside analogs and tuberculosis: New weapons against an old enemy. Future Med. Chem. 2015, 7, 291–314.
- Serpi, M.; Ferrari, V.; Pertusati, F. Nucleoside derived antibiotics to fight microbial drug resistance: New utilities for an established class of drugs? J. Med. Chem. 2016, 59, 10343–10382.
- Khandazhinskaya, A.L.; Alexandrova, L.A.; Matyugina, E.S.; Solyev, P.N.; Efremenkova, O.V.; Buckheit, K.W.; Wilkinson, M.; Buckheit, R.W.; Chernousova, L.N.; Smirnova, T.G. Novel 5′-Norcarbocyclic Pyrimidine Derivatives as Antibacterial Agents. Molecules 2018, 23, 3069.
- McEntee, L.; Johnson, A.; Farrington, N.; Unsworth, J.; Dane, A.; Jain, A.; Cotroneo, N.; Critchley, I.; Melnick, D.; Parr, T. Pharmacodynamics of Tebipenem: New Options for Oral Treatment of Multidrug-Resistant Gram-Negative Infections. Antimicrob. Agents Chemother. 2019, 63, e00603.
- Gordon, E.M.; Duncton, M.A.; Gallop, M.A. Orally absorbed derivatives of the β-lactamase inhibitor avibactam. Design of novel prodrugs of sulfate containing drugs. J. Med. Chem. 2018, 61, 10340–10344.
- Moellering, R., Jr. Linezolid: The first oxazolidinone antimicrobial. Ann. Intern. Med. 2003, 138, 135.
- Kanafani, Z.A.; Corey, G.R. Tedizolid (TR-701): A new oxazolidinone with enhanced potency. Expert Opin. Investig. Drugs 2012, 21, 515–522.
- Kaul, M.; Mark, L.; Zhang, Y.; Parhi, A.K.; LaVoie, E.J.; Pilch, D.S. An FtsZ-targeting prodrug with oral antistaphylococcal efficacy in vivo. Antimicrob. Agents Chemother. 2013, 57, 5860–5869.
- Marinelli, L.; Di Stefano, A.; Cacciatore, I. Carvacrol and its derivatives as antibacterial agents. Phytochem. Rev. 2018, 17, 903–921.
- Marinelli, L.; Fornasari, E.; Eusepi, P.; Ciulla, M.; Genovese, S.; Epifano, F.; Fiorito, S.; Turkez, H.; Rtc, S.; Mingoia, M. Carvacrol prodrugs as novel antimicrobial agents. Eur. J. Med. Chem. 2019, 178, 515–529.
- Fleck, L.E.; North, E.J.; Lee, R.E.; Mulcahy, L.R.; Casadei, G.; Lewis, K. A screen for and validation of prodrug antimicrobials. Antimicrob. Agents Chemother. 2014, 58, 1410–1419.
More
Information
Subjects:
Pharmacology & Pharmacy
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Revisions:
2 times
(View History)
Update Date:
29 Feb 2024
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No