Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Argen Mamazhakypov | -- | 4175 | 2024-02-27 10:14:03 | | | |
| 2 | Lindsay Dong | Meta information modification | 4175 | 2024-02-28 04:21:34 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Mamazhakypov, A.; Maripov, A.; Sarybaev, A.S.; Schermuly, R.T.; Sydykov, A. Mast Cells in Cardiac Remodeling. Encyclopedia. Available online: https://encyclopedia.pub/entry/55517 (accessed on 25 July 2026).
Mamazhakypov A, Maripov A, Sarybaev AS, Schermuly RT, Sydykov A. Mast Cells in Cardiac Remodeling. Encyclopedia. Available at: https://encyclopedia.pub/entry/55517. Accessed July 25, 2026.
Mamazhakypov, Argen, Abdirashit Maripov, Akpay S. Sarybaev, Ralph Theo Schermuly, Akylbek Sydykov. "Mast Cells in Cardiac Remodeling" Encyclopedia, https://encyclopedia.pub/entry/55517 (accessed July 25, 2026).
Mamazhakypov, A., Maripov, A., Sarybaev, A.S., Schermuly, R.T., & Sydykov, A. (2024, February 27). Mast Cells in Cardiac Remodeling. In Encyclopedia. https://encyclopedia.pub/entry/55517
Mamazhakypov, Argen, et al. "Mast Cells in Cardiac Remodeling." Encyclopedia. Web. 27 February, 2024.
Copy Citation
In response to various stressors, cardiac chambers undergo structural remodeling. Long-term exposure of the right ventricle (RV) to pressure or volume overload leads to its maladaptive remodeling, associated with RV failure and increased mortality. While left ventricular adverse remodeling is well understood and therapeutic options are available or emerging, RV remodeling remains underexplored, and no specific therapies are currently available.
mast cells
right ventricle
cardiac remodeling
1. Introduction
Cardiac remodeling refers to changes in size, mass, geometry, and function of heart, that develop in the course of various cardiovascular pathologies. It initially represents an adaptive process of the heart in response to mechanical, neurohumoral, or other stressors, with the primary aim of preserving cardiac function [1]. However, sustained exposure to pathological factors triggers the development of maladaptive cardiac remodeling. Cardiac remodeling can affect either the left or right ventricle (RV) or both ventricles and is associated with unfavorable outcomes and treatment responses [1][2]. It is now recognized as an important aspect of cardiovascular disease progression and is emerging as a therapeutic target in heart failure therapy [1].
Currently approved therapies have been efficient in reducing mortality and morbidity of left-heart failure patients [3][4][5]. Both pharmacological and non-pharmacological therapies beneficially impacted prognosis in heart failure patients by modulating the cardiac remodeling process [5][6][7]. Modulation of the renin-angiotensin-aldosterone (RAAS) and sympathetic nervous system is the cornerstone of pharmacotherapy of left heart failure [8]. Increased activity of the sympathetic nervous and RAAS has also been implicated in the pathophysiology for pressure overload-related RV failure [9][10][11]. Consequently, angiotensin-converting enzyme inhibitors, angiotensin II receptor antagonists, and β-blockers have demonstrated efficacy in reversing RV, remodeling in animal models [9][10]. However, currently, they are not recommended for therapy of RV failure associated with pulmonary hypertension due to inconsistent results in clinical trials [12]. It is important to note that most of the previous trials were small, underpowered, enrolled patients with systemic RV or predominantly focused on the pulmonary vasculature and did not assess RV remodeling [13][14]. In addition, the differences between the RV and left ventricle might account for the dissimilar responses of the failing ventricles to inhibitors of neurohormonal activity [15][16]. However, despite advances in treatment, the burden of heart failure remains high, emphasizing the ongoing need for further research and the development of novel management strategies [3].
Cardiac remodeling develops in response to stressors such as volume or pressure overload. Such unfavorable hemodynamic conditions occur in a variety of diseases, including hypertension, valvular pathologies, chronic pulmonary diseases, obesity, and metabolic disorders. To counteract the sustained rise in wall stress caused by excessive pressure and/or volume load, the myocardium undergoes phenotypic and functional transformations, which encompass a sequence of molecular, cellular, and interstitial alterations. Cardiomyocytes become enlarged due to new contractile protein synthesis, contributing to the increased size and mass of the affected cardiac chamber [17][18]. In addition to cardiomyocyte changes, cardiac remodeling involves alterations in other cell types and extracellular matrix organization. Fibroblast activation and proliferation result in the amplified synthesis of extracellular matrix proteins [19]. Changes in coronary microvascular endothelial cells lead to alterations in the coronary microvasculature and blood supply to the heart [20]. One of the important features of cardiac remodeling is the recruitment and accumulation of diverse inflammatory and immune cells within the myocardium [21][22], which can mediate both protective and deleterious effects [21].
2. Right Ventricular Remodeling
Recent studies have provided strong evidence to recognize the pivotal role of the RV in cardiovascular pathologies [23]. Functional and structural changes of the RV define the prognosis in patients with various cardiovascular diseases, including congenital heart disease [24], pulmonary arterial hypertension [25], myocardial infarction [26], advanced left heart failure [27] and stable coronary artery disease [28]. The recognition of the RV as a critical player in the progression of cardiovascular and respiratory conditions has sparked increased research attention towards the RV in recent years [29][30][31].
Pressure overload is a key pathogenetic factor for RV remodeling and dysfunction, which is associated with the release and subsequent accumulation of a myriad of bioactive molecules in the circulation and within the cardiac tissue [32][33]. These bioactive molecules have the potential to directly impose deleterious effects on the RV and modulate its response to pressure or volume overload [22].
Similar to the remodeling of the left ventricle, RV remodeling in response to pressure or volume overload is intricately associated with alterations in the function of cardiomyocytes, fibroblasts, endothelial cells, and various immune and inflammatory cells [34][35][36]. These cells are critically involved in cardiac remodeling, and their dysfunction can be caused by a multitude of factors and mediators synthesized and released by various cells within the RV myocardium [36]. Interactions between different cell types play a vital role in determining the overall outcome.
Another hallmark of RV remodeling is the increased production and deposition of extracellular matrix proteins. Ultimately, this alters the microenvironment of the myocardial cells and leads to a rise in tissue stiffness and expansion of the intercellular space within the heart [37].
The pathogenesis of the deleterious events during the course of RV remodeling involves a complex interplay of various processes and signaling pathways, including inflammation [22], extracellular matrix synthesis [38], calcium homeostasis [39], endothelial cell dysfunction [40], nitric oxide (NO) synthesis [41], endothelial-mesenchymal transition [42], matricellular protein synthesis [43] and growth factor signaling pathways such as transforming growth factor-β (TGF-β) [44] and apelin [45]. These interactions collectively dictate the fate of the RV under pressure or volume overload, and a better understanding of these intricate processes is essential to developing effective therapeutic strategies for the prevention or management of RV dysfunction and failure.
3. Mast Cell Biology
The pivotal role of mast cells in allergic responses has long been recognized [46]. However, recent studies have suggested their involvement in various pathological processes associated with different non-allergic diseases, such as tissue remodeling, repair, fibrosis, and angiogenesis [46][47]. Mast cells represent immune cells residing within the peripheral tissues and originate from bone marrow-derived precursor cells, regulated by key growth factors such as c-KIT ligand stem cell factor [48].
Mast cells express numerous cell surface antigens, including the cytokine receptor KIT (CD117) and the high-affinity receptor for the Fc region of immunoglobulin E (FcεRI). They are often identified as c-kit+FcεRI+ [49][50][51]. Mast cells also express the pan leukocyte antigen (CD45); however, they do not express surface antigens specific for other hematopoietic cells like CD2, CD3, CD4, CD11a, CD11b, CD11c, CD14, CD15, CD19, etc. [52][53]. Evaluation of the expression of these antigens allows distinguishing between mast cells and other CD45+ cells [54].
The most commonly used method for histochemical identification of mast cells in tissue sections is toluidine blue staining [55]. Morphologically, mast cells are identified by the presence of multiple large metachromatic granules in their cytoplasm, which store a variety of mediators, cytokines, and proteases [56].
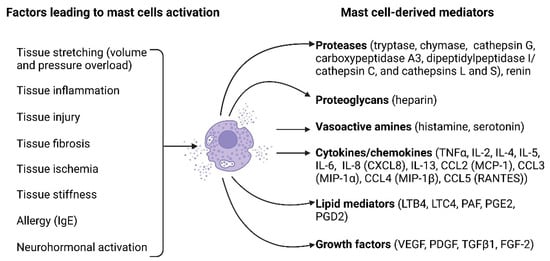
Mast cell activation can be mediated by the binding of antigens and antibodies to their membrane [57]. Additionally, activation may occur in response to diverse stimuli, including neuropeptides (substance P, vasoactive intestinal peptide), calcitonin gene-related peptide, and neurotensin), basic compounds (compound 48/80), inflammatory mediators, and certain drugs [57]. Figure 1 illustrates the key factors causing mast cell activation and the biologically active substances released by mast cells upon their activation.
Figure 1. Mast cell activation factors and mast cell-derived mediators. A multitude of factors can induce mast cell activation and encompass tissue stretching due to pressure or volume overload, tissue inflammation, tissue injury, tissue fibrosis, tissue ischemia, tissue stiffness, allergies, and neurohormonal activation. Upon activation, mast cells release a diverse array of biologically active substances, including proteases (such as tryptase, chymase, cathepsin G, carboxypeptidase A3, dipeptidyl peptidase I (DPPI, cathepsin C), cathepsins L and S, and renin), proteoglycans (heparin), vasoactive amines (histamine and serotonin), cytokines/chemokines (such as TNFα, IL-2, IL-4, IL-5, IL-6, IL-8 [CXCL8], IL-13, CCL2 [MCP-1], CCL3 [MIP-1α], CCL4 [MIP-1β], and CCL5 [RANTES]), lipid mediators (LTB4, LTC4, PAF, PGE2, PGD2), and growth factors (VEGF, PDGF, TGFβ1, FGF-2). The figure was created using BioRender.com, accessed on 25 November 2023.
Upon mast cell activation, their degranulation follows [58]. Degranulated mast cells in tissues can be identified using toluidine blue staining by a reduced granule content in their cytoplasm and by extracellular metachromatic granules in their immediate vicinity [59][60]. For the quantification of mast cell activation, the number of degranulated cells is expressed as a percent of the total number of mast cells [61]. Expression of some surface antigens, including CD63, CD200R1 and CD203c, was reported to significantly increase because of mast cell degranulation and was proposed as a surrogate molecular marker for mast cell activation [62].
Mast cells exert their effects through the release of various mediators, which are pre-stored in the granules or synthesized upon mast cell activation. The biologically active substances released by mast cells are then involved in various processes. Many factors released by mast cells, including histamine, heparin, proteoglycans, serotonin, and proteases, are preformed and stored in the granules [63].
4. Mast Cell-Deficient Lines to Study Cardiac Physiology and Remodeling
Mast cell-deficient lines have significantly deepened our knowledge of mast cell biology and their role in diseases. The most frequently used in biomedical research mast cell-deficient mouse lines carry naturally occurring mutations in the white spotting (W) locus on mouse chromosome 5, which encodes the c-KIT proto-oncogene [64]. All these mutations lead to reduced receptor tyrosine kinase KIT-dependent signaling and severe mast cell deficiency, as the development of mast cells is critically dependent on the binding of the growth factor stem cell factor to its receptor KIT [65].
KitW/W-v mice are double heterozygotes, which carry two mutant alleles, W and W-v [66]. The W mutant allele is characterized by a point mutation at a splicing site in the transcript, resulting in a truncated KIT protein lacking kinase activity, whereas the Wv mutant allele represents a point mutation in the kinase domain of the c-KIT coding sequence, resulting in decreased tyrosine kinase activity [66]. KitW/W homozygotes mice display severe anemia and neutropenia; however, they die around the first week of life [67]. KitW-v homozygotes also exhibit severe anemia, and many of them survive to maturity (hence “v” for “viable”) [67]. KitW/W-v mice, which combine the severe KitW with the milder KitW-v mutation, have widely been used in biomedical research for their favorable survival and vigor [68].
KitW-sh/W-sh mice bear a spontaneous inversion mutation W-sash (W-sh) in the transcriptional regulatory elements upstream of the c-KIT transcription start site [69]. The defects caused by the W-sh mutations are confined to melanogenesis and mast cells. Heterozygotes are black with a broad white belt at the midline (hence the name and symbol sash) [70]. KitW-sh/W-sh mice are fertile and not anemic, which makes them more preferable than KitW/W-v mice in certain disease models [71]. However, the inversion in KitW-sh/W-sh mice also disrupts the gene encoding the pro-atrial natriuretic peptide (ANP) convertase corin, which is responsible for proteolytic cleavage of pro-ANP to produce ANP [72].
5. Mast Cells in Healthy Hearts
Besides cardiomyocytes, endothelial cells and fibroblasts, heart tissue contains a number of other cell types, including various immune and inflammatory cells [73][74]. The immune cells in the healthy myocardium comprise mast cells, macrophages, T lymphocytes (CD4+ and CD8+), natural killer cells, eosinophils, B cells, dendritic cells, neutrophils, monocytes, and plasma cells. These immune cells are involved in the maintenance of tissue homeostasis, regulation of inflammation, tissue repair, and heart protection against potential pathogens [74].
Cardiac mast cells are generally located in the interstitial space between cardiomyocytes and are closely associated with coronary vessels [75] and nerves [76]. They constitute only a small part of the immune cells present within the healthy myocardium. According to a recent single-cell RNA sequencing analysis, mast cells constituted less than 1% of all CD45+ immune cells in the healthy mouse myocardium [54]. In this study, the authors identified mast cells in cluster 19 of the single cell sequencing results, which expressed the mast cell marker Mcpt8 and the mast cell-associated cytokines IL4 and IL13 [54].
Mast cell distribution exhibits significant variation within the wall of mouse hearts; they are most prevalent in the epicardium (50%) and myocardium (45%), with fewer numbers in the endocardium (5%) [77]. In contrast, in healthy rats, mast cell numbers appear to be similar in both the subepicardial and subendocardial layers of the left ventricle [78].
Mast cells are known for their heterogeneity, so the phenotypes of cardiac mast cells differ from those found in other tissues. In contrast to mast cells found in human skin and lungs, mast cells in human hearts demonstrate slightly different patterns of mediator release and synthesis [79]. Upon cross-linking of IgE receptors on human heart mast cells, histamine, tryptase, leukotriene C4, and prostaglandin D2 are released, although they differ quantitatively and qualitatively compared to skin and lung mast cells [79]. Mast cell heterogeneity is also evident from the patterns of mediator release. Human heart mast cells respond to C5a and protamine by releasing histamine in a similar way as skin mast cells; however, they differ from lung mast cells in this aspect [79].
6. Mast Cells in Right Ventricular Physiology and Remodeling
Various immune and inflammatory cells are crucially involved in cardiac remodeling [22]. Notably, the content of immune cells in the RV differs from that in the left ventricle in physiological as well as pathological conditions [80]. Specifically, the number of CD45+/CD11b/c+ cells was on average four times higher in the RV than in the left ventricle in rats exposed to either normoxia or hypoxia, suggesting a higher content of immune cells in the RV. A substantial body of evidence has accumulated supporting the involvement of mast cells in left heart hypertrophy and failure. However, the role of mast cells in RV hypertrophy and failure remains insufficiently explored.
In pathological conditions associated with left ventricular remodeling, mast cell numbers in the left ventricle were increased and exhibited more dynamic changes compared to the RV in animals as well as in humans.
Activation of mast cells in response to pressure overload occurs early after exposure to the stressor, as evidenced by increased mast cell degranulation in the RV of mice 3 days following pulmonary artery banding (PAB) surgery [81].
The source of increased mast cell numbers in RV remodeling remains to be elucidated. In left ventricular remodeling induced by angiotensin II infusion, the increase in mast cell density was shown to result from the maturation of pre-existing immature mast cells [82].
SHR commonly serves as a model of systemic arterial hypertension and left ventricular hypertrophy. Enhanced myocardial fibrosis, increased collagen volume fraction, and elevated hydroxyproline levels in the left ventricle in SHR were closely linked to an increased accumulation of mast cells in the left ventricle [83].
The evidence for the pathological role of mast cells in adverse RV remodeling was further provided in experiments exploiting mast cell-deficient mice and pharmacological inhibition of mast cell activation [84]. Treatment of wild-type mice subjected to pressure overload by PAB with the mast cell stabilizer cromolyn significantly improved RV remodeling and function [84].
Accumulating evidence suggests that mast cells may also play a protective role in myocardial remodeling [51][85]. Moreover, mast cells and their granules exert protective effects in the acute phase after myocardial infarction in rats via improving cardiomyocyte survival and vascularization [86]. Mast cell population heterogeneity could account for the diversity of mast cell effects [87].
7. Mechanisms of Mast Cell-Mediated Effects on the Heart
7.1. Effects of Mast Cells on Cardiomyocytes
Mediators released by activated mast cells may exert direct effects on cardiomyocytes and impact their survival, apoptosis, contraction, and arrhythmogenesis. Experiments with adoptive transfer of bone marrow-derived mast cells from wild-type mice or corresponding knockout mice to mast cell-deficient KitW-sh/W-sh mice with diabetic cardiomyopathy demonstrated a role of mast cell-derived IL6 and TNF-α in promoting cardiomyocyte death [88]. The mast cell-derived mediator histamine, which is present in the human heart at high concentrations, was shown to reduce cell viability and induce cardiomyocyte apoptosis [89]. In line with these observations, pharmacological inhibition, or genetic disruption of the histamine H2 receptor slowed heart failure progression in mice subjected to pressure overload through a reduction of myocardial apoptosis [89].
In cardiac volume overload, chymase uptake by cardiomyocytes resulted in myosin degradation and cardiomyocyte dysfunction [90]. In contrast, mast cell deficiency led to reduced contractility and myofilament Ca2+ sensitization after myocardial infarction in mice, suggesting an important role of cardiac mast cells in preserving postischemic cardiac function [51].
7.2. Mast Cells and Extracellular Matrix Modulation
Myocardial fibrosis plays a pivotal role in the pathogenesis and progression of various cardiac diseases [38]. Elucidating the precise mechanisms underlying myocardial fibrosis development is crucial for understanding disease pathogenesis and identifying novel therapeutic targets [91]. Myocardial fibrosis involves an interplay of versatile processes, including the synthesis and turnover of extracellular matrix proteins, the activation and proliferation of cardiac fibroblasts, as well as the synthesis of diverse inflammatory mediators and growth factors with potent pro-fibrotic properties [92]. These intricate events are meticulously orchestrated by various cell types residing within the myocardium, including various immune cells [92][93][94].
Recent studies have implicated mast cells as significant contributors to myocardial fibrosis [95]. In patients with idiopathic dilated cardiomyopathy, a notable association was demonstrated between mast cell density and the severity of myocardial fibrosis [96].
Experimental studies provided further evidence to support the link between mast cell activation and myocardial fibrosis development. In Dahl salt-sensitive rats, degranulated mast cells were found to localize near fibrotic regions in the myocardium [97].
Chymase inhibition with chymostatin reduced myocardial active TGF-β1 levels in rats subjected to pressure overload by transverse aortic constriction (TAC), suggesting that activation of latent TGF-β1 is one of the pathways by which cardiac mast cell-derived chymase contributes to myocardial fibrosis [98]. Besides TGF-β1 activation, chymase induced TGF-β1 expression in a dose-dependent fashion in rat cardiac fibroblasts [99].
Another pathway by which mast cells can contribute to myocardial fibrosis is the activation of the RAAS in the heart. Mast cells are recognized as a significant source of renin in the myocardium, which consequently converts angiotensinogen to angiotensin I [76][100]. Next, cathepsin G [101] and chymase [102][103] derived from activated mast cells convert angiotensin I into angiotensin II. Angiotensin II subsequently exerts pro-fibrotic effects on the myocardium [104] through angiotensin II receptors, which are present on cardiac fibroblasts and mediate multiple pro-fibrotic effects [104][105].
Activated mast cells release a number of other factors, including histamine [89] and tryptase [106][107] that might account for their pro-fibrotic activities on cardiac fibroblasts. Treatment of isolated cardiac fibroblasts with tryptase induced increased collagen synthesis and fibroblast proliferation [106][107]. Collagen production was enhanced via activation of the protease-activated receptor-2 and ERK1/2 signaling [107]. The relevance of in vitro findings was further corroborated by the reduction of myocardial fibrosis in spontaneously hypertensive rats treated with a tryptase inhibitor [107].
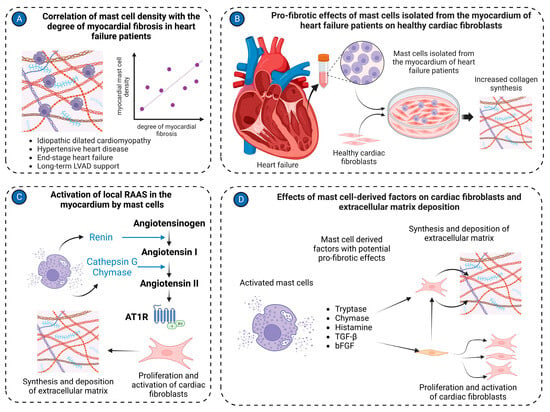
MMP/tissue inhibitors of the metalloproteinases system play a key role in the modulation of the extracellular matrix in pathological conditions. Several lines of evidence implicate mast cells in the regulation of this system in volume overload-induced cardiac remodeling (Figure 2).
Figure 2. Mast cell in cardiac fibrosis. (A) Mast cell density positively correlates with the degree of myocardial fibrosis in the left ventricular tissues isolated from patients with various cardiac pathologies, including idiopathic dilated cardiomyopathy, hypertensive heart disease, end-stage heart failure, and long-term LVAD support; (B) Mast cells isolated from cardiac tissues of heart failure patients stimulate activation of healthy cardiac fibroblasts, causing them to synthesize and release extracellular matrix molecules in co-culture experiments; (C) Mast cells activate the local RAAS in the myocardium. Renin produced by mast cells can convert angiotensinogen to angiotensin I. Cathepsin G and chymase derived from mast cells transform angiotensin I to angiotensin II. Angiotensin II stimulates cardiac fibroblasts to synthesize extracellular matrix molecules; (D) Several factors derived from mast cells, such as tryptase, chymase, histamine, TGF-β, and bFGF, activate cardiac fibroblasts to myofibroblasts and stimulate production of extracellular matrix molecules. The figure was created using BioRender.com, accessed on 25 November 2023.
7.3. Mast Cells and Myocardial Vascularization
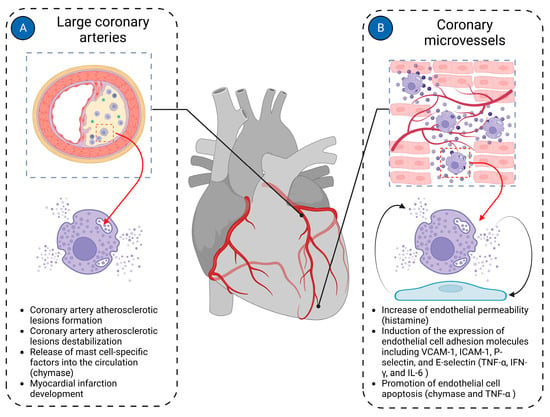
Most cardiac pathologies are associated with changes in myocardial vascularization [40][108]. Mast cells are present in the walls of both microvessels and larger coronary arteries, suggesting their potential involvement in both micro- and macrovascular pathologies of the coronary arteries (Figure 3).
Figure 3. Mast cells in cardiac vasculature. Mast cells in the cardiac vasculature have been implicated in the pathologies of both large coronary arteries and coronary microvasculature. (A) In large coronary arteries, mast cells are mainly involved in the pathogenesis of coronary atherosclerotic plaque formation and contribute to its instability, ultimately leading to the development of myocardial infarction; (B) In coronary microvessels, mast cells primarily interact with endothelial cells, modulating their functions. Histamine derived from mast cells increases the permeability of endothelial cells. Mast cell-derived TNF-α, IFN-γ, and IL-6 induce expression of endothelial cell adhesion molecules such as VCAM-1, ICAM-1, P-selectin, and E-selectin. Chymase and TNF-α derived from mast cells promote endothelial cell apoptosis. The figure was created using BioRender.com, accessed on 25 November 2023.
Mast cells have been shown to induce left ventricular diastolic dysfunction in various animal models of heart failure [109][110]. Abnormal mast cell activation was identified as a cause of cardiac microvessel disease in Leprdb/db female mice, another experimental model of diastolic dysfunction associated with heart failure with preserved ejection fraction [61]. The cardiac microvessel disease was characterized by enhanced vessel permeability due to disruption of endothelial adherens junctions by mast cell-derived histamine [61]. Histamine released by mast cells may interact with both H1 and H2 receptors to exert its effects on the cardiac endothelial cells and contribute to heart failure development [111].
Increased serum tryptase levels were reported in patients with stable coronary artery disease, suggesting that chronic low-grade inflammation in atherosclerotic plaques triggers mast cell activation [112]. In vitro experiments demonstrated that activated mast cells might contribute to plaque erosion by inducing endothelial cell apoptosis [113][114].
7.4. Mast Cells and Myocardial Inflammation
Myocardial inflammation in various cardiac pathologies is recognized as a significant contributor to adverse myocardial remodeling in both ventricles [22]. The importance of mast cells in this process is emphasized by the fact that they possess a variety of inflammatory mediators in their granules and are able to rapidly generate more inflammatory agents upon activation [115]. Activated mast cells release a number of inflammatory factors, including interleukins (IL-1, IL-3, IL-4, IL-5, IL-6, IL-8, IL-10, IL-13, and IL-17), TNF-α, and chemokines (CXCL8/IL-8, CCL2/MCP-1, CCL3/MIP-1α, CCL4/MIP-1β, and CCL5/RANTES) [56].
Inflammatory mediators released by mast cells can enhance myocardial inflammation by activating further inflammatory processes. In canine experimental cardiac ischemia-reperfusion, mast cells primarily released TNF-α, which then exacerbated myocardial inflammation and cardiac injury by upregulating IL-6 in infiltrating leukocytes and initiating the cytokine cascade [116]. In line with this report, attenuated myocardial injury in mast cell-deficient KitW/KitW-v mice following myocardial ischemia-reperfusion was associated with lower serum IL-6 compared to their wild-type counterparts [117].
Markedly elevated myocardial TNF-α levels in response to cardiac volume overload were observed in wild-type rats at 5 days post-fistula; conversely, TNF-α was almost undetectable in the hearts of mast cell-deficient rats [118].
8. Mast Cells as a Therapeutic Target
Preclinical studies have provided evidence that mast cells may represent an attractive target in the prevention and treatment of heart diseases. Treatment approaches in the context of heart failure may involve targeting different stages of mast cell development and activation, including blocking the processes that activate mast cells, targeting specific mast cell-derived mediators and their receptors on cardiac cells, limiting mast cell proliferation, or promoting mast cell apoptosis [119].
Prevention of mast cell activation by stabilization of their membranes has demonstrated efficacy in improving myocardial remodeling in various experimental models of heart failure. In SHR, mast cell stabilization with nedocromil prevented left ventricular fibrosis, inflammatory cell recruitment, and cytokine overexpression in the myocardium without affecting blood pressure or left ventricular hypertrophy [106]. Mast cell stabilization with cromolyn sodium attenuated left ventricular remodeling and left ventricular diastolic dysfunction in ovariectomized Fischer rats [109], ameliorated left ventricular diastolic dysfunction in Leprdb/db female mice [61], and attenuated RV dilatation and improved RV function in mice subjected to PAB [84]. The class effects of these drugs were further supported by the prevention of the transition from compensated hypertrophy to heart failure in animals subjected to abdominal aortic banding using another mast cell stabilizing agent, tranilast [120].
Many currently approved pulmonary arterial hypertension therapies represent repurposed drugs, and drug repurposing continues to be an attractive approach to developing novel PAH therapies [121]. One of the advantages of already approved drugs is that they have a well-established safety profile [122]. This helps mitigate the costs and time associated with novel drug development [122]. Cromolyn sodium is safe and has been approved for treatment of mastocytosis and allergic diseases, such as bronchial asthma, conjunctivitis, rhinitis, etc. Therefore, cromolyn represents a promising candidate for clinical development for the treatment of RV failure in humans.
Mast cell-derived chymase has been identified as one of the key mast cell-specific targets in various cardiac pathologies [123]. Chymase inhibition improved cardiac remodeling in various animal models, including rapid ventricular pacing-induced heart failure in dogs [103], LAD-occlusion-induced myocardial infarction in rats [124] and hamsters [125], ischemia-reperfusion cardiac remodeling in pigs [126], and pressure overload-induced left ventricular remodeling in rats [98].
Blocking receptors for mast cell-derived mediators such as histamine H2 receptor improved cardiac function in TAC mice [89] and in dogs with pacemaker-driven tachycardia [127]. Interestingly, a large prospective observational cohort study of participants without cardiovascular disease at baseline showed that baseline use of H2 receptor antagonists was associated with a 62% lower risk of incident heart failure [128]. The beneficial effects of H2 receptor antagonists on all-cause mortality in patients with different clinical forms of pulmonary hypertension suggest that these drugs exert direct effects on the right ventricle [129]. In line with these observations, H2 receptor antagonist use in the general population was associated with lower RV mass and smaller RV end-diastolic volume [130].
References
- Cohn, J.N.; Ferrari, R.; Sharpe, N. Cardiac remodeling—Concepts and clinical implications: A consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J. Am. Coll. Cardiol. 2000, 35, 569–582.
- Azevedo, P.S.; Polegato, B.F.; Minicucci, M.F.; Paiva, S.A.; Zornoff, L.A. Cardiac Remodeling: Concepts, Clinical Impact, Pathophysiological Mechanisms and Pharmacologic Treatment. Arq. Bras. Cardiol. 2016, 106, 62–69.
- Pazos-López, P.; Peteiro-Vázquez, J.; Carcía-Campos, A.; García-Bueno, L.; de Torres, J.P.; Castro-Beiras, A. The causes, consequences, and treatment of left or right heart failure. Vasc Health Risk Manag 2011, 7, 237–254.
- Shah, A.; Gandhi, D.; Srivastava, S.; Shah, K.J.; Mansukhani, R. Heart Failure: A Class Review of Pharmacotherapy. P&T 2017, 42, 464–472.
- Iacoviello, M.; Palazzuoli, A.; Gronda, E. Recent advances in pharmacological treatment of heart failure. Eur. J. Clin. Investig. 2021, 51, e13624.
- Ishii, H.; Amano, T.; Matsubara, T.; Murohara, T. Pharmacological intervention for prevention of left ventricular remodeling and improving prognosis in myocardial infarction. Circulation 2008, 118, 2710–2718.
- Kim, G.H.; Uriel, N.; Burkhoff, D. Reverse remodelling and myocardial recovery in heart failure. Nat. Rev. Cardiol. 2018, 15, 83–96.
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). With the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. J. Heart Fail. 2022, 24, 4–131.
- Ameri, P.; Bertero, E.; Meliota, G.; Cheli, M.; Canepa, M.; Brunelli, C.; Balbi, M. Neurohormonal activation and pharmacological inhibition in pulmonary arterial hypertension and related right ventricular failure. Heart Fail. Rev. 2016, 21, 539–547.
- Klinke, A.; Schubert, T.; Müller, M.; Legchenko, E.; Zelt, J.G.E.; Shimauchi, T.; Napp, L.C.; Rothman, A.M.K.; Bonnet, S.; Stewart, D.J.; et al. Emerging therapies for right ventricular dysfunction and failure. Cardiovasc Diagn Ther 2020, 10, 1735–1767.
- Handoko, M.L.; de Man, F.S.; Allaart, C.P.; Paulus, W.J.; Westerhof, N.; Vonk-Noordegraaf, A. Perspectives on novel therapeutic strategies for right heart failure in pulmonary arterial hypertension: Lessons from the left heart. Eur. Respir. Rev. 2010, 19, 72–82.
- Konstam, M.A.; Kiernan, M.S.; Bernstein, D.; Bozkurt, B.; Jacob, M.; Kapur, N.K.; Kociol, R.D.; Lewis, E.F.; Mehra, M.R.; Pagani, F.D.; et al. Evaluation and Management of Right-Sided Heart Failure: A Scientific Statement From the American Heart Association. Circulation 2018, 137, e578–e622.
- Amsallem, M.; Mercier, O.; Kobayashi, Y.; Moneghetti, K.; Haddad, F. Forgotten No More: A Focused Update on the Right Ventricle in Cardiovascular Disease. JACC Heart Fail 2018, 6, 891–903.
- Rubin, L.J. The adrenergic nervous system as a therapeutic target in pulmonary arterial hypertension: A cautionary tale. Eur. Respir. J. 2016, 48, 617–618.
- Taverne, Y.; Sadeghi, A.; Bartelds, B.; Bogers, A.; Merkus, D. Right ventricular phenotype, function, and failure: A journey from evolution to clinics. Heart Fail. Rev. 2021, 26, 1447–1466.
- Reddy, S.; Bernstein, D. Molecular Mechanisms of Right Ventricular Failure. Circulation 2015, 132, 1734–1742.
- Haque, Z.K.; Wang, D.Z. How cardiomyocytes sense pathophysiological stresses for cardiac remodeling. Cell. Mol. Life Sci. 2017, 74, 983–1000.
- Scarborough, E.A.; Uchida, K.; Vogel, M.; Erlitzki, N.; Iyer, M.; Phyo, S.A.; Bogush, A.; Kehat, I.; Prosser, B.L. Microtubules orchestrate local translation to enable cardiac growth. Nat. Commun. 2021, 12, 1547.
- Frangogiannis, N.G. The extracellular matrix in myocardial injury, repair, and remodeling. J. Clin. Investig. 2017, 127, 1600–1612.
- Segers, V.F.M.; Brutsaert, D.L.; De Keulenaer, G.W. Cardiac Remodeling: Endothelial Cells Have More to Say Than Just NO. Front. Physiol. 2018, 9, 382.
- Kologrivova, I.; Shtatolkina, M.; Suslova, T.; Ryabov, V. Cells of the Immune System in Cardiac Remodeling: Main Players in Resolution of Inflammation and Repair After Myocardial Infarction. Front. Immunol. 2021, 12, 664457.
- Sydykov, A.; Mamazhakypov, A.; Petrovic, A.; Kosanovic, D.; Sarybaev, A.S.; Weissmann, N.; Ghofrani, H.A.; Schermuly, R.T. Inflammatory Mediators Drive Adverse Right Ventricular Remodeling and Dysfunction and Serve as Potential Biomarkers. Front. Physiol. 2018, 9, 609.
- Tello, K.; Naeije, R.; de Man, F.; Guazzi, M. Pathophysiology of the right ventricle in health and disease: An update. Cardiovasc. Res. 2023, 119, 1891–1904.
- Cho, Y.K.; Ma, J.S. Right ventricular failure in congenital heart disease. Korean J. Pediatr. 2013, 56, 101–106.
- Prisco, S.Z.; Thenappan, T.; Prins, K.W. Treatment Targets for Right Ventricular Dysfunction in Pulmonary Arterial Hypertension. JACC Basic Transl. Sci. 2020, 5, 1244–1260.
- Shahar, K.; Darawsha, W.; Yalonetsky, S.; Lessick, J.; Kapeliovich, M.; Dragu, R.; Mutlak, D.; Reisner, S.; Agmon, Y.; Aronson, D. Time Dependence of the Effect of Right Ventricular Dysfunction on Clinical Outcomes After Myocardial Infarction: Role of Pulmonary Hypertension. J. Am. Heart Assoc. 2016, 5.
- Dini, F.L.; Pugliese, N.R.; Ameri, P.; Attanasio, U.; Badagliacca, R.; Correale, M.; Mercurio, V.; Tocchetti, C.G.; Agostoni, P.; Palazzuoli, A. Right ventricular failure in left heart disease: From pathophysiology to clinical manifestations and prognosis. Heart Fail. Rev. 2023, 28, 757–766.
- Sumin, A.N.; Korok, E.V.; Sergeeva, T.Y. Impaired right ventricular filling in patients with a chronic coronary syndrome. Med. Ultrason. 2021, 23, 311–318.
- Edward, J.; Banchs, J.; Parker, H.; Cornwell, W. Right ventricular function across the spectrum of health and disease. Heart 2023, 109, 349–355.
- Mandoli, G.E.; De Carli, G.; Pastore, M.C.; Cameli, P.; Contorni, F.; D’Alessandro, M.; Bargagli, E.; Mondillo, S.; Cameli, M. Right cardiac involvement in lung diseases: A multimodality approach from diagnosis to prognostication. J. Intern. Med. 2021, 289, 440–449.
- Mamazhakypov, A.; Sommer, N.; Assmus, B.; Tello, K.; Schermuly, R.T.; Kosanovic, D.; Sarybaev, A.S.; Weissmann, N.; Pak, O. Novel Therapeutic Targets for the Treatment of Right Ventricular Remodeling: Insights from the Pulmonary Artery Banding Model. Int. J. Environ. Res. Public Health 2021, 18, 8927.
- Jabagi, H.; Mielniczuk, L.M.; Liu, P.P.; Ruel, M.; Sun, L.Y. Biomarkers in the Diagnosis, Management, and Prognostication of Perioperative Right Ventricular Failure in Cardiac Surgery—Are We There Yet? J. Clin. Med. 2019, 8, 559.
- Pradhan, N.M.; Mullin, C.; Poor, H.D. Biomarkers and Right Ventricular Dysfunction. Crit. Care Clin. 2020, 36, 141–153.
- Kret, M.; Arora, R. Pathophysiological basis of right ventricular remodeling. J. Cardiovasc. Pharmacol. Ther. 2007, 12, 5–14.
- Avazmohammadi, R.; Mendiola, E.A.; Li, D.S.; Vanderslice, P.; Dixon, R.A.F.; Sacks, M.S. Interactions Between Structural Remodeling and Hypertrophy in the Right Ventricle in Response to Pulmonary Arterial Hypertension. J. Biomech. Eng. 2019, 141, 0910161–09101613.
- Lother, A.; Kohl, P. The heterocellular heart: Identities, interactions, and implications for cardiology. Basic Res. Cardiol. 2023, 118, 30.
- Sharifi Kia, D.; Kim, K.; Simon, M.A. Current Understanding of the Right Ventricle Structure and Function in Pulmonary Arterial Hypertension. Front. Physiol. 2021, 12, 641310.
- Egemnazarov, B.; Crnkovic, S.; Nagy, B.M.; Olschewski, H.; Kwapiszewska, G. Right ventricular fibrosis and dysfunction: Actual concepts and common misconceptions. Matrix Biol. 2018.
- Roe, A.T.; Frisk, M.; Louch, W.E. Targeting cardiomyocyte Ca2+ homeostasis in heart failure. Curr. Pharm. Des. 2015, 21, 431–448.
- Frump, A.L.; Bonnet, S.; de Jesus Perez, V.A.; Lahm, T. Emerging role of angiogenesis in adaptive and maladaptive right ventricular remodeling in pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L443–L460.
- Dupont, M.; Tang, W.H. Right ventricular afterload and the role of nitric oxide metabolism in left-sided heart failure. J. Card. Fail. 2013, 19, 712–721.
- Park, J.F.; Clark, V.R.; Banerjee, S.; Hong, J.; Razee, A.; Williams, T.; Fishbein, G.; Saddic, L.; Umar, S. Transcriptomic Analysis of Right Ventricular Remodeling in Two Rat Models of Pulmonary Hypertension: Identification and Validation of Epithelial-to-Mesenchymal Transition in Human Right Ventricular Failure. Circ. Heart Fail. 2021, 14, e007058.
- Imoto, K.; Okada, M.; Yamawaki, H. Expression profile of matricellular proteins in hypertrophied right ventricle of monocrotaline-induced pulmonary hypertensive rats. J. Vet. Med. Sci. 2017, 79, 1096–1102.
- Frangogiannis, N.G. Transforming growth factor-β in myocardial disease. Nat. Rev. Cardiol. 2022, 19, 435–455.
- Falcão-Pires, I.; Gonçalves, N.; Henriques-Coelho, T.; Moreira-Gonçalves, D.; Roncon-Albuquerque, R., Jr.; Leite-Moreira, A.F. Apelin decreases myocardial injury and improves right ventricular function in monocrotaline-induced pulmonary hypertension. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H2007–H2014.
- Galli, S.J.; Tsai, M. Mast cells: Versatile regulators of inflammation, tissue remodeling, host defense and homeostasis. J. Dermatol. Sci. 2008, 49, 7–19.
- Bruno, K.A.; Mathews, J.E.; Yang, A.L.; Frisancho, J.A.; Scott, A.J.; Greyner, H.D.; Molina, F.A.; Greenaway, M.S.; Cooper, G.M.; Bucek, A.; et al. BPA Alters Estrogen Receptor Expression in the Heart After Viral Infection Activating Cardiac Mast Cells and T Cells Leading to Perimyocarditis and Fibrosis. Front. Endocrinol. 2019, 10, 598.
- da Silva, E.Z.; Jamur, M.C.; Oliver, C. Mast cell function: A new vision of an old cell. J. Histochem. Cytochem. 2014, 62, 698–738.
- Plum, T.; Wang, X.; Rettel, M.; Krijgsveld, J.; Feyerabend, T.B.; Rodewald, H.R. Human Mast Cell Proteome Reveals Unique Lineage, Putative Functions, and Structural Basis for Cell Ablation. Immunity 2020, 52, 404–416.e405.
- Kritikou, E.; Depuydt, M.A.C.; de Vries, M.R.; Mulder, K.E.; Govaert, A.M.; Smit, M.D.; van Duijn, J.; Foks, A.C.; Wezel, A.; Smeets, H.J.; et al. Flow Cytometry-Based Characterization of Mast Cells in Human Atherosclerosis. Cells 2019, 8, 334.
- Ngkelo, A.; Richart, A.; Kirk, J.A.; Bonnin, P.; Vilar, J.; Lemitre, M.; Marck, P.; Branchereau, M.; Le Gall, S.; Renault, N.; et al. Mast cells regulate myofilament calcium sensitization and heart function after myocardial infarction. J. Exp. Med. 2016, 213, 1353–1374.
- Sperr, W.R.; Bankl, H.C.; Mundigler, G.; Klappacher, G.; Grossschmidt, K.; Agis, H.; Simon, P.; Laufer, P.; Imhof, M.; Radaszkiewicz, T.; et al. The human cardiac mast cell: Localization, isolation, phenotype, and functional characterization. Blood 1994, 84, 3876–3884.
- Valent, P.; Bettelheim, P. Cell surface structures on human basophils and mast cells: Biochemical and functional characterization. Adv. Immunol. 1992, 52, 333–423.
- Martini, E.; Kunderfranco, P.; Peano, C.; Carullo, P.; Cremonesi, M.; Schorn, T.; Carriero, R.; Termanini, A.; Colombo, F.S.; Jachetti, E.; et al. Single-Cell Sequencing of Mouse Heart Immune Infiltrate in Pressure Overload-Driven Heart Failure Reveals Extent of Immune Activation. Circulation 2019, 140, 2089–2107.
- Sridharan, G.; Shankar, A.A. Toluidine blue: A review of its chemistry and clinical utility. J. Oral Maxillofac. Pathol. 2012, 16, 251–255.
- Krystel-Whittemore, M.; Dileepan, K.N.; Wood, J.G. Mast Cell: A Multi-Functional Master Cell. Front. Immunol. 2015, 6, 620.
- Theoharides, T.C.; Tsilioni, I.; Ren, H. Recent advances in our understanding of mast cell activation—Or should it be mast cell mediator disorders? Expert Rev. Clin. Immunol. 2019, 15, 639–656.
- Gilfillan, A.M.; Austin, S.J.; Metcalfe, D.D. Mast cell biology: Introduction and overview. Adv. Exp. Med. Biol. 2011, 716, 2–12.
- Kovanen, P.T. Mast Cells as Potential Accelerators of Human Atherosclerosis-From Early to Late Lesions. Int. J. Mol. Sci. 2019, 20, 4479.
- Ribatti, D. The Staining of Mast Cells: A Historical Overview. Int. Arch. Allergy Immunol. 2018, 176, 55–60.
- Guimbal, S.; Cornuault, L.; Rouault, P.; Hollier, P.L.; Chapouly, C.; Bats, M.L.; Imbault, J.; Gadeau, A.P.; Couffinhal, T.; Renault, M.A. Mast Cells Are the Trigger of Small Vessel Disease and Diastolic Dysfunction in Diabetic Obese Mice. Arterioscler. Thromb. Vasc. Biol. 2021, 41, e193–e207.
- Teodosio, C.; Mayado, A.; Sánchez-Muñoz, L.; Morgado, J.M.; Jara-Acevedo, M.; Álvarez-Twose, I.; García-Montero, A.C.; Matito, A.; Caldas, C.; Escribano, L.; et al. The immunophenotype of mast cells and its utility in the diagnostic work-up of systemic mastocytosis. J. Leukoc. Biol. 2015, 97, 49–59.
- Moon, T.C.; Befus, A.D.; Kulka, M. Mast cell mediators: Their differential release and the secretory pathways involved. Front. Immunol. 2014, 5, 569.
- Bernstein, A.; Chabot, B.; Dubreuil, P.; Reith, A.; Nocka, K.; Majumder, S.; Ray, P.; Besmer, P. The mouse W/c-kit locus. Ciba Found. Symp. 1990, 148, 158–166, discussion 166–172.
- Katz, H.R.; Austen, K.F. Mast cell deficiency, a game of kit and mouse. Immunity 2011, 35, 668–670.
- Nocka, K.; Tan, J.C.; Chiu, E.; Chu, T.Y.; Ray, P.; Traktman, P.; Besmer, P. Molecular bases of dominant negative and loss of function mutations at the murine c-kit/white spotting locus: W37, Wv, W41 and W. EMBO J. 1990, 9, 1805–1813.
- Reith, A.D.; Rottapel, R.; Giddens, E.; Brady, C.; Forrester, L.; Bernstein, A. W mutant mice with mild or severe developmental defects contain distinct point mutations in the kinase domain of the c-kit receptor. Genes Dev. 1990, 4, 390–400.
- Russell, E.S.; Bernstein, S.E. Blood and blood formation. Biol. Lab. Mouse 1966, 2, 351–372.
- Nagle, D.L.; Kozak, C.A.; Mano, H.; Chapman, V.M.; Bućan, M. Physical mapping of the Tec and Gabrb1 loci reveals that the Wsh mutation on mouse chromosome 5 is associated with an inversion. Hum. Mol. Genet. 1995, 4, 2073–2079.
- Duttlinger, R.; Manova, K.; Chu, T.Y.; Gyssler, C.; Zelenetz, A.D.; Bachvarova, R.F.; Besmer, P. W-sash affects positive and negative elements controlling c-kit expression: Ectopic c-kit expression at sites of kit-ligand expression affects melanogenesis. Development 1993, 118, 705–717.
- Grimbaldeston, M.A.; Chen, C.C.; Piliponsky, A.M.; Tsai, M.; Tam, S.Y.; Galli, S.J. Mast cell-deficient W-sash c-kit mutant Kit W-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am. J. Pathol. 2005, 167, 835–848.
- Nigrovic, P.A.; Gray, D.H.; Jones, T.; Hallgren, J.; Kuo, F.C.; Chaletzky, B.; Gurish, M.; Mathis, D.; Benoist, C.; Lee, D.M. Genetic inversion in mast cell-deficient (Wsh) mice interrupts corin and manifests as hematopoietic and cardiac aberrancy. Am. J. Pathol. 2008, 173, 1693–1701.
- Litviňuková, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the adult human heart. Nature 2020, 588, 466–472.
- Cohen, C.D.; Rousseau, S.T.; Bermea, K.C.; Bhalodia, A.; Lovell, J.P.; Zita, M.D.; Čiháková, D.; Adamo, L. Myocardial Immune Cells: The Basis of Cardiac Immunology. J. Immunol. 2023, 210, 1198–1207.
- Marone, G.; de Crescenzo, G.; Adt, M.; Patella, V.; Arbustini, E.; Genovese, A. Immunological characterization and functional importance of human heart mast cells. Immunopharmacology 1995, 31, 1–18.
- Silver, R.B.; Reid, A.C.; Mackins, C.J.; Askwith, T.; Schaefer, U.; Herzlinger, D.; Levi, R. Mast cells: A unique source of renin. Proc. Natl. Acad. Sci. USA 2004, 101, 13607–13612.
- Ingason, A.B.; Mechmet, F.; Atacho, D.A.M.; Steingrímsson, E.; Petersen, P.H. Distribution of mast cells within the mouse heart and its dependency on Mitf. Mol. Immunol. 2019, 105, 9–15.
- Engels, W.; Reiters, P.H.; Daemen, M.J.; Smits, J.F.; van der Vusse, G.J. Transmural changes in mast cell density in rat heart after infarct induction in vivo. J. Pathol. 1995, 177, 423–429.
- Patella, V.; de Crescenzo, G.; Ciccarelli, A.; Marinò, I.; Adt, M.; Marone, G. Human heart mast cells: A definitive case of mast cell heterogeneity. Int. Arch. Allergy Immunol. 1995, 106, 386–393.
- Gorr, M.W.; Sriram, K.; Chinn, A.M.; Muthusamy, A.; Insel, P.A. Transcriptomic profiles reveal differences between the right and left ventricle in normoxia and hypoxia. Physiol. Rep. 2020, 8, e14344.
- Luitel, H.; Sydykov, A.; Schymura, Y.; Mamazhakypov, A.; Janssen, W.; Pradhan, K.; Wietelmann, A.; Kosanovic, D.; Dahal, B.K.; Weissmann, N.; et al. Pressure overload leads to an increased accumulation and activity of mast cells in the right ventricle. Physiol. Rep. 2017, 5, e13146.
- Widiapradja, A.; Manteufel, E.J.; Dehlin, H.M.; Pena, J.; Goldspink, P.H.; Sharma, A.; Kolb, L.L.; Imig, J.D.; Janicki, J.S.; Lu, B.; et al. Regulation of Cardiac Mast Cell Maturation and Function by the Neurokinin-1 Receptor in the Fibrotic Heart. Sci. Rep. 2019, 9, 11004.
- Panizo, A.; Mindan, F.J.; Galindo, M.F.; Cenarruzabeitia, E.; Hernandez, M.; Diez, J. Are mast cells involved in hypertensive heart disease? J. Hypertens. 1995, 13, 1201–1208.
- Sydykov, A.; Luitel, H.; Mamazhakypov, A.; Wygrecka, M.; Pradhan, K.; Pak, O.; Petrovic, A.; Kojonazarov, B.; Weissmann, N.; Seeger, W.; et al. Genetic Deficiency and Pharmacological Stabilization of Mast Cells Ameliorate Pressure Overload-Induced Maladaptive Right Ventricular Remodeling in Mice. Int. J. Mol. Sci. 2020, 21, 9099.
- Joseph, J.; Kennedy, R.H.; Devi, S.; Wang, J.; Joseph, L.; Hauer-Jensen, M. Protective role of mast cells in homocysteine-induced cardiac remodeling. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2541–H2545.
- Kwon, J.S.; Kim, Y.S.; Cho, A.S.; Cho, H.H.; Kim, J.S.; Hong, M.H.; Jeong, S.Y.; Jeong, M.H.; Cho, J.G.; Park, J.C.; et al. The novel role of mast cells in the microenvironment of acute myocardial infarction. J. Mol. Cell. Cardiol. 2011, 50, 814–825.
- Cildir, G.; Pant, H.; Lopez, A.F.; Tergaonkar, V. The transcriptional program, functional heterogeneity, and clinical targeting of mast cells. J. Exp. Med. 2017, 214, 2491–2506.
- He, A.; Fang, W.; Zhao, K.; Wang, Y.; Li, J.; Yang, C.; Benadjaoud, F.; Shi, G.P. Mast cell-deficiency protects mice from streptozotocin-induced diabetic cardiomyopathy. Transl. Res. 2019, 208, 1–14.
- Zeng, Z.; Shen, L.; Li, X.; Luo, T.; Wei, X.; Zhang, J.; Cao, S.; Huang, X.; Fukushima, Y.; Bin, J.; et al. Disruption of histamine H2 receptor slows heart failure progression through reducing myocardial apoptosis and fibrosis. Clin. Sci. 2014, 127, 435–448.
- Powell, P.C.; Wei, C.C.; Fu, L.; Pat, B.; Bradley, W.E.; Collawn, J.F.; Dell’Italia, L.J. Chymase uptake by cardiomyocytes results in myosin degradation in cardiac volume overload. Heliyon 2019, 5, e01397.
- Jiang, W.; Xiong, Y.; Li, X.; Yang, Y. Cardiac Fibrosis: Cellular Effectors, Molecular Pathways, and Exosomal Roles. Front. Cardiovasc. Med. 2021, 8, 715258.
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488.
- Varricchi, G.; Marone, G.; Kovanen, P.T. Cardiac Mast Cells: Underappreciated Immune Cells in Cardiovascular Homeostasis and Disease. Trends Immunol. 2020, 41, 734–746.
- Levick, S.P.; Melendez, G.C.; Plante, E.; McLarty, J.L.; Brower, G.L.; Janicki, J.S. Cardiac mast cells: The centrepiece in adverse myocardial remodelling. Cardiovasc. Res. 2011, 89, 12–19.
- Legere, S.A.; Haidl, I.D.; Legare, J.F.; Marshall, J.S. Mast Cells in Cardiac Fibrosis: New Insights Suggest Opportunities for Intervention. Front. Immunol. 2019, 10, 580.
- Batlle, M.; Pérez-Villa, F.; Lázaro, A.; Garcia-Pras, E.; Ramirez, J.; Ortiz, J.; Orús, J.; Roqué, M.; Heras, M.; Roig, E. Correlation between mast cell density and myocardial fibrosis in congestive heart failure patients. Transplant. Proc. 2007, 39, 2347–2349.
- Palaniyandi, S.S.; Inagaki, K.; Mochly-Rosen, D. Mast cells and epsilonPKC: A role in cardiac remodeling in hypertension-induced heart failure. J. Mol. Cell. Cardiol. 2008, 45, 779–786.
- Li, J.; Jubair, S.; Janicki, J.S. Estrogen inhibits mast cell chymase release to prevent pressure overload-induced adverse cardiac remodeling. Hypertension 2015, 65, 328–334.
- Zhao, X.Y.; Zhao, L.Y.; Zheng, Q.S.; Su, J.L.; Guan, H.; Shang, F.J.; Niu, X.L.; He, Y.P.; Lu, X.L. Chymase induces profibrotic response via transforming growth factor-beta 1/Smad activation in rat cardiac fibroblasts. Mol. Cell. Biochem. 2008, 310, 159–166.
- Schnee, J.M.; Hsueh, W.A. Angiotensin II, adhesion, and cardiac fibrosis. Cardiovasc. Res. 2000, 46, 264–268.
- Jahanyar, J.; Youker, K.A.; Loebe, M.; Assad-Kottner, C.; Koerner, M.M.; Torre-Amione, G.; Noon, G.P. Mast cell-derived cathepsin g: A possible role in the adverse remodeling of the failing human heart. J. Surg. Res. 2007, 140, 199–203.
- Jin, D.; Takai, S.; Sakaguchi, M.; Okamoto, Y.; Muramatsu, M.; Miyazaki, M. An antiarrhythmic effect of a chymase inhibitor after myocardial infarction. J. Pharmacol. Exp. Ther. 2004, 309, 490–497.
- Matsumoto, T.; Wada, A.; Tsutamoto, T.; Ohnishi, M.; Isono, T.; Kinoshita, M. Chymase inhibition prevents cardiac fibrosis and improves diastolic dysfunction in the progression of heart failure. Circulation 2003, 107, 2555–2558.
- Kawano, H.; Do, Y.S.; Kawano, Y.; Starnes, V.; Barr, M.; Law, R.E.; Hsueh, W.A. Angiotensin II has multiple profibrotic effects in human cardiac fibroblasts. Circulation 2000, 101, 1130–1137.
- Crabos, M.; Roth, M.; Hahn, A.W.; Erne, P. Characterization of angiotensin II receptors in cultured adult rat cardiac fibroblasts. Coupling to signaling systems and gene expression. J. Clin. Investig. 1994, 93, 2372–2378.
- Levick, S.P.; McLarty, J.L.; Murray, D.B.; Freeman, R.M.; Carver, W.E.; Brower, G.L. Cardiac mast cells mediate left ventricular fibrosis in the hypertensive rat heart. Hypertension 2009, 53, 1041–1047.
- McLarty, J.L.; Melendez, G.C.; Brower, G.L.; Janicki, J.S.; Levick, S.P. Tryptase/Protease-activated receptor 2 interactions induce selective mitogen-activated protein kinase signaling and collagen synthesis by cardiac fibroblasts. Hypertension 2011, 58, 264–270.
- Luxán, G.; Dimmeler, S. The vasculature: A therapeutic target in heart failure? Cardiovasc. Res. 2022, 118, 53–64.
- Wang, H.; da Silva, J.; Alencar, A.; Zapata-Sudo, G.; Lin, M.R.; Sun, X.; Ahmad, S.; Ferrario, C.M.; Groban, L. Mast Cell Inhibition Attenuates Cardiac Remodeling and Diastolic Dysfunction in Middle-aged, Ovariectomized Fischer 344 × Brown Norway Rats. J. Cardiovasc. Pharmacol. 2016, 68, 49–57.
- Dona, M.S.I.; Hsu, I.; Meuth, A.I.; Brown, S.M.; Bailey, C.A.; Aragonez, C.G.; Russell, J.J.; Krstevski, C.; Aroor, A.R.; Chandrasekar, B.; et al. Multi-omic analysis of the cardiac cellulome defines a vascular contribution to cardiac diastolic dysfunction in obese female mice. Basic Res. Cardiol. 2023, 118, 11.
- Neumann, J.; Kirchhefer, U.; Dhein, S.; Hofmann, B.; Gergs, U. The Roles of Cardiovascular H(2)-Histamine Receptors Under Normal and Pathophysiological Conditions. Front. Pharmacol. 2021, 12, 732842.
- Deliargyris, E.N.; Upadhya, B.; Sane, D.C.; Dehmer, G.J.; Pye, J.; Smith, S.C., Jr.; Boucher, W.S.; Theoharides, T.C. Mast cell tryptase: A new biomarker in patients with stable coronary artery disease. Atherosclerosis 2005, 178, 381–386.
- Heikkilä, H.M.; Lätti, S.; Leskinen, M.J.; Hakala, J.K.; Kovanen, P.T.; Lindstedt, K.A. Activated mast cells induce endothelial cell apoptosis by a combined action of chymase and tumor necrosis factor-alpha. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 309–314.
- Lätti, S.; Leskinen, M.; Shiota, N.; Wang, Y.; Kovanen, P.T.; Lindstedt, K.A. Mast cell-mediated apoptosis of endothelial cells in vitro: A paracrine mechanism involving TNF-alpha-mediated down-regulation of bcl-2 expression. J. Cell. Physiol. 2003, 195, 130–138.
- Theoharides, T.C.; Alysandratos, K.D.; Angelidou, A.; Delivanis, D.A.; Sismanopoulos, N.; Zhang, B.; Asadi, S.; Vasiadi, M.; Weng, Z.; Miniati, A.; et al. Mast cells and inflammation. Biochim. Biophys. Acta 2012, 1822, 21–33.
- Frangogiannis, N.G.; Lindsey, M.L.; Michael, L.H.; Youker, K.A.; Bressler, R.B.; Mendoza, L.H.; Spengler, R.N.; Smith, C.W.; Entman, M.L. Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation 1998, 98, 699–710.
- Bhattacharya, K.; Farwell, K.; Huang, M.; Kempuraj, D.; Donelan, J.; Papaliodis, D.; Vasiadi, M.; Theoharides, T.C. Mast cell deficient W/Wv mice have lower serum IL-6 and less cardiac tissue necrosis than their normal littermates following myocardial ischemia-reperfusion. Int. J. Immunopathol. Pharmacol. 2007, 20, 69–74.
- Levick, S.P.; Gardner, J.D.; Holland, M.; Hauer-Jensen, M.; Janicki, J.S.; Brower, G.L. Protection from adverse myocardial remodeling secondary to chronic volume overload in mast cell deficient rats. J. Mol. Cell. Cardiol. 2008, 45, 56–61.
- Paivandy, A.; Pejler, G. Novel Strategies to Target Mast Cells in Disease. J. Innate Immun. 2021, 1–17.
- Hara, M.; Ono, K.; Hwang, M.W.; Iwasaki, A.; Okada, M.; Nakatani, K.; Sasayama, S.; Matsumori, A. Evidence for a role of mast cells in the evolution to congestive heart failure. J. Exp. Med. 2002, 195, 375–381.
- Toshner, M.; Spiekerkoetter, E.; Bogaard, H.; Hansmann, G.; Nikkho, S.; Prins, K.W. Repurposing of medications for pulmonary arterial hypertension. Pulm Circ 2020, 10, 2045894020941494.
- Cha, Y.; Erez, T.; Reynolds, I.J.; Kumar, D.; Ross, J.; Koytiger, G.; Kusko, R.; Zeskind, B.; Risso, S.; Kagan, E.; et al. Drug repurposing from the perspective of pharmaceutical companies. Br. J. Pharmacol. 2018, 175, 168–180.
- Ahmad, S.; Ferrario, C.M. Chymase inhibitors for the treatment of cardiac diseases: A patent review (2010-2018). Expert Opin. Ther. Pat. 2018, 28, 755–764.
- Kanemitsu, H.; Takai, S.; Tsuneyoshi, H.; Nishina, T.; Yoshikawa, K.; Miyazaki, M.; Ikeda, T.; Komeda, M. Chymase inhibition prevents cardiac fibrosis and dysfunction after myocardial infarction in rats. Hypertens. Res. 2006, 29, 57–64.
- Jin, D.; Takai, S.; Yamada, M.; Sakaguchi, M.; Kamoshita, K.; Ishida, K.; Sukenaga, Y.; Miyazaki, M. Impact of chymase inhibitor on cardiac function and survival after myocardial infarction. Cardiovasc. Res. 2003, 60, 413–420.
- Oyamada, S.; Bianchi, C.; Takai, S.; Chu, L.M.; Sellke, F.W. Chymase inhibition reduces infarction and matrix metalloproteinase-9 activation and attenuates inflammation and fibrosis after acute myocardial ischemia/reperfusion. J. Pharmacol. Exp. Ther. 2011, 339, 143–151.
- Takahama, H.; Asanuma, H.; Sanada, S.; Fujita, M.; Sasaki, H.; Wakeno, M.; Kim, J.; Asakura, M.; Takashima, S.; Minamino, T.; et al. A histamine H2 receptor blocker ameliorates development of heart failure in dogs independently of β-adrenergic receptor blockade. Basic Res. Cardiol. 2010, 105, 787–794.
- Leary, P.J.; Tedford, R.J.; Bluemke, D.A.; Bristow, M.R.; Heckbert, S.R.; Kawut, S.M.; Krieger, E.V.; Lima, J.A.; Masri, C.S.; Ralph, D.D.; et al. Histamine H2 Receptor Antagonists, Left Ventricular Morphology, and Heart Failure Risk: The MESA Study. J. Am. Coll. Cardiol. 2016, 67, 1544–1552.
- Leary, P.J.; Hess, E.; Barón, A.E.; Branch, K.R.; Choudhary, G.; Hough, C.L.; Maron, B.A.; Ralph, D.D.; Ryan, J.J.; Tedford, R.J.; et al. H2 Receptor Antagonist Use and Mortality in Pulmonary Hypertension: Insight from the VA-CART Program. Am. J. Respir. Crit. Care Med. 2018, 197, 1638–1641.
- Leary, P.J.; Barr, R.G.; Bluemke, D.A.; Bristow, M.R.; Kronmal, R.A.; Lima, J.A.; Ralph, D.D.; Ventetuolo, C.E.; Kawut, S.M. H2 receptor antagonists and right ventricular morphology: The MESA right ventricle study. Ann Am Thorac Soc 2014, 11, 1379–1386.
More
Information
Subjects:
Pathology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
762
Revisions:
2 times
(View History)
Update Date:
28 Feb 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No