Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Joseph Sloop | -- | 4479 | 2024-02-26 16:00:27 | | | |
| 2 | Stefanie Casa | Meta information modification | 4479 | 2024-02-26 23:08:02 | | | | |
| 3 | Sirius Huang | Meta information modification | 4479 | 2024-02-27 02:05:22 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Henary, E.; Casa, S.; Dost, T.L.; Sloop, J.C.; Henary, M. The Role of Small Molecules Containing Fluorine Atoms. Encyclopedia. Available online: https://encyclopedia.pub/entry/55477 (accessed on 31 July 2026).
Henary E, Casa S, Dost TL, Sloop JC, Henary M. The Role of Small Molecules Containing Fluorine Atoms. Encyclopedia. Available at: https://encyclopedia.pub/entry/55477. Accessed July 31, 2026.
Henary, Emily, Stefanie Casa, Tyler L. Dost, Joseph C. Sloop, Maged Henary. "The Role of Small Molecules Containing Fluorine Atoms" Encyclopedia, https://encyclopedia.pub/entry/55477 (accessed July 31, 2026).
Henary, E., Casa, S., Dost, T.L., Sloop, J.C., & Henary, M. (2024, February 26). The Role of Small Molecules Containing Fluorine Atoms. In Encyclopedia. https://encyclopedia.pub/entry/55477
Henary, Emily, et al. "The Role of Small Molecules Containing Fluorine Atoms." Encyclopedia. Web. 26 February, 2024.
Copy Citation
The fluorine atom possesses many intrinsic properties that can be beneficial when incorporated into small molecules. These properties include the atom’s size, electronegativity, and ability to block metabolic oxidation sites. Substituents that feature fluorine and fluorine-containing groups are currently prevalent in drugs that lower cholesterol, relieve asthma, and treat anxiety disorders, as well as improve the chemical properties of various medications and imaging agents.
fluorine
dye
fluorophore
chromophore
pharmaceutical
imaging
1. Introduction
The value of fluorine-containing small molecules with pharmaceutical efficacy and the underlying importance of fluorine incorporation into these molecules is undeniable [1][2]. Every year, new fluoro-pharmaceuticals achieve FDA approval and are introduced into the market; this is an ongoing trend that has been observed over the past 20 years [3][4]. In 2021, nine fluorine-containing drugs were approved for use by the FDA, showing an increase in relevancy for the incorporation of fluorine in medicinal research [5]. The design, synthesis, and testing of medically viable fluorine-containing compounds have burgeoned over the past two decades, leading to numerous publications and patents.
Utilization of the fluorine atom and fluorine-containing functional groups in pharmaceuticals is very attractive to the medical field for a variety of reasons [2]. First, the fluorine atom is the second smallest “functional group” with a van der Waals radius of 1.47 Å [1][6]. Because fluorine’s size falls between that of a hydrogen atom (1.20 Å) and an oxygen atom (1.47 Å), it is often used as a bioisostere for these atoms [6][7][8]. It has been suggested that the trifluoromethyl group is comparable to an isopropyl group, even though its van der Waals volume is much smaller (-CF3: 39.8 Å3 vs. -CH(CH3)2: 56.2 Å3) [1]. The fact that the -CF3 substituent has rotational symmetry around the carbon–carbon bond, whereas the isopropyl group lacks rotational symmetry, helps account for this apparent discrepancy [9]. Modeling studies reveal the relative similarity in sizes between stilbene and perfluorinated stilbene while highlighting the dramatic impact fluorine has on electrostatic potentials in the stilbene molecule [10]. Because the fluorine atom is the smallest halogen, an additional advantage is found in its ability (as well as fluorinated methyl groups) to fit into smaller pockets of space in comparison to the other halogens and their corresponding groups.
Fluorine’s high electronegativity, 4.0 on the Pauling scale, offers it other advantages over similarly sized atoms and functional groups when incorporated into small molecules. The C-F bond is considered the strongest bond in organic chemistry due to the electronegativity difference between the two atoms [11][12]. In addition, the highly electronegative nature of fluorine often alters the dipole moment of the overall molecule. Another molecular property affected by the electronegativity of the fluorine atom is the pKa [13]. The acidity of fluorinated molecules increases due to inductive effects brought about by fluorine’s electronegativity [7][14].
Additionally, benefits arising from the incorporation of one or more fluorine atoms into a compound often include alterations in drug biodistribution and drug–receptor binding, as well as enhancement of potency [15]. A significant advantage found upon fluorine substitution in drugs and imaging agents is the ability to modulate lipophilicity by adding or taking away fluorine atoms from the molecule in medical and biomedical imaging applications [16]. The tuning of lipophilicity helps the compounds to be absorbed and transported in vivo faster and more easily. This stems from the greater lipophilicity of the C-F bond relative to the C-H bond [17]. Because the fluorine atom is the smallest halogen, it (as well as fluorinated methyl groups) fits into receptor pockets and it contributes to blocking sites from metabolic oxidation more than that of other halogens [18]. The electronegativity and lipophilic properties of fluorine atoms incorporated into medicinal compounds have strong impacts on how the body will react to the molecules in ways such as the clearance rate, biodistribution route, and toxicity of the molecule. Figure 1 summarizes the key effects that fluorine substitution has on small molecules with potential pharmaceutical value.
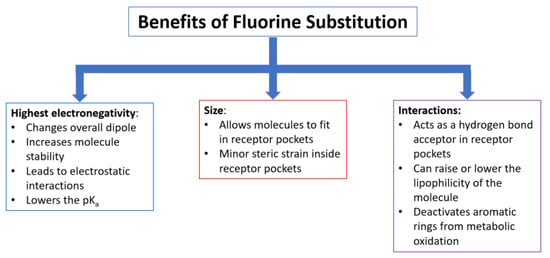
Figure 1. Effects of fluorine incorporation on drugs and imaging probes.
2. Common Reactions Incorporating Fluorine in Small Molecules
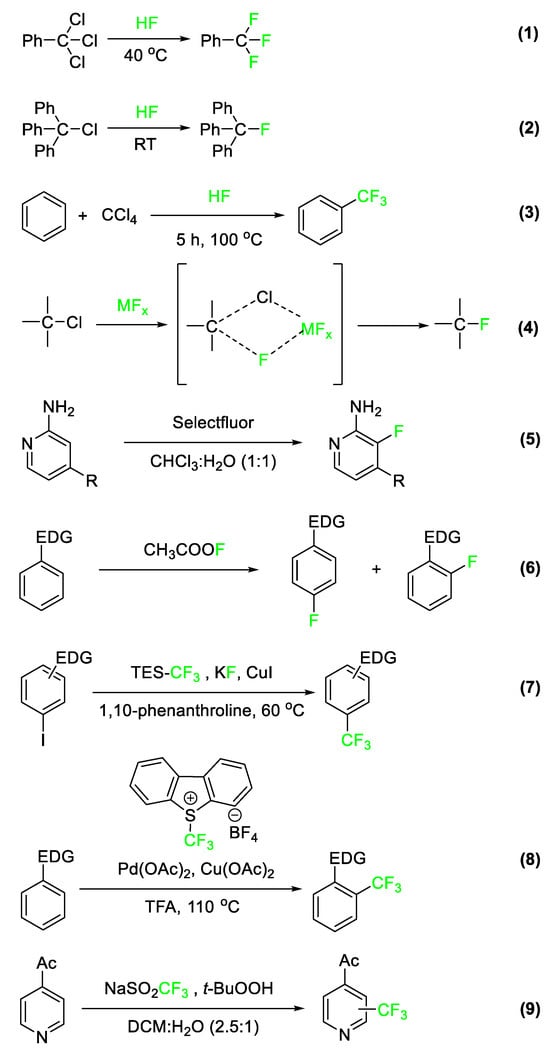
The introduction of fluorine and other fluorinated functional groups into the final drug architecture is most often accomplished via reactions with fluorinated precursors. Therefore, it is important to understand the reactions that are used to prepare these fluorinated precursor molecules. In Figure 2, a selection of processes used to prepare fluorobenzylic, fluoroalkyl, fluoroaromatic, and fluoroheteroaromatic systems is presented. For a more detailed discussion on current state-of-the-art organofluorine synthetic strategies, a review by Gouverneur et al. published in 2021 is illustrative [19]. The first reaction is performed at a large scale in industrial settings and involves hydrogen fluoride (HF) (Equation (1)). In this reaction, the fluoride anion serves as a nucleophilic substitute for other halogens occupying allylic and benzylic positions. [20]. Another reaction commonly used to incorporate fluorine is the Friedel–Crafts (FC) alkylation. Hydrogen fluoride, in this case, acts as both the FC catalyst and the fluorinating agent itself, resulting in the preparation of trifluoromethylated aromatic systems (Equation (2)). The carbon tetrachloride reagent in Figure 2, Equation (3), replaces a hydrogen atom on the benzene ring, followed by the substitution of fluorine for the three chlorine atoms.
Equation (4) is an example of the Swarts reaction. In this process, a metal fluoride species acts as a Lewis acid catalyst in removing a halide substituent and replacing it with a fluorine atom via the formation of a four-membered cyclic intermediate [21]. Equation (5) shows a green, highly regioselective fluorination of activated 2-aminopyridines using Selectfluor® [22]. Equation (6) shows electrophilic fluorination using acetyl hypofluorite (CH3COOF) as the fluorinating reactant [23].
Equations (7) and (8) are examples of electrophilic trifluoromethylations of activated aromatic and heteroaromatic substrates developed within the last two decades [20][24]. Equation (7) employs trifluoromethyltriethylsilane (TES-CF3) under Cu(I) catalysis to prepare trifluoromethylated aromatic and heteroaromatic molecules from the corresponding iodide starting material. The mechanism by which this reaction takes place is not fully understood but may involve a CF3-Cu complex. Equation (8) depicts aromatic trifluoromethylation by S-(trifluoromethyl)dibenzothiophenium tetrafluoroborate via a transition metal-catalyzed reaction. This reaction generates a product with the trifluoromethyl group introduced to the ortho position of benzene [25]. Finally, Equation (9) is an example of a highly regioselective heteroaromatic trifluoromethylation of a pyridine derivative [26].
3. Characteristics of Pharmaceuticals Featuring Fluorine and Examples
Fluorinated therapeutics represent compounds available as drugs that use fluorine atoms to change the molecule’s overall lipophilicity, drug potency, and other factors. For many potentially viable pharmaceutical compounds, the clearance rate and clearance mechanism are critical considerations when designing and synthesizing molecules. Due to the extraordinary properties exhibited by the fluorine atom, drug designers continue to incorporate this atom into lead compounds. In the next section, the benefits of the properties of fluorine and fluorine-containing functional groups will be discussed.
3.1. Metabolic Oxidation
A primary reason to incorporate the fluorine atom, especially on aromatic and heterocyclic ring systems, is to combat the effect of metabolic oxidation which occurs when drugs are taken into the body and the process eliminates foreign molecules from the body as quickly as possible. The addition of a fluorine atom or fluorine-containing group often stops this metabolic pathway as the modified fluorobenzene does not fit into the active site of the monooxygenase. An additional benefit to adding fluorine to the para position on the benzene ring is that the inductive electron-withdrawing effect of the fluorine atom deactivates other positions on the ring against this particular metabolic pathway [9]. Slowing the oxidation process that the P450 protein performs allows for longer drug retention and thus increases effectiveness. Examples in the literature show compounds that have very rapid bodily clearances, but when fluorine is incorporated into the para position on a phenyl ring, the rate at which the compound is cleared slows by as much as 108-fold [28]. And, while the main function of the P450 protein is to initiate the metabolic process, oxygenizing a pharmaceutically active compound can turn it toxic [29]. Fluorine and chlorine can block this process from happening, but due to its minimal size, electronic withdrawing nature, and steric perturbation in active sites, fluorine is preferred over chlorine. Many therapeutics highlighted herein share the theme of a p-fluorophenyl group. The protection afforded by the para-substituted halogen reduces the potential toxicity when oxygenized and can increase drug effectiveness due to interactions with activation sites in proteins [9][30]. The strategy of p-fluorination has been thoroughly investigated and has been frequently used in drug design [29].
3.2. Electronic Considerations
While deactivating a drug against metabolic oxidation is possibly the most important consequence of incorporating fluorine into drugs, researchers can utilize fluorine atoms to alter compounds to achieve other strategic effects. Electronic considerations such as electronegativity, pKa modification, and lipophilicity tailoring are also motives that chemists take advantage of when designing fluorine-containing pharmaceutical compounds [4][15]. Electronegativity leads to several underlying effects on a molecule, including molecule and bond stability, dipole magnitude and direction, and electrostatic interactions with receptor sites. Here, several drugs are described that do not contain fluorine atoms for their metabolic effects but rather for their other properties.
3.3. Size Considerations
As mentioned previously, the size of fluorine-containing functional groups is very unique. Structurally, they take up more room than hydrogen but less than a hydroxyl group. The size of the functional groups can help direct a compound into its target pocket. Once the molecule is inside the pocket, the electronic characteristics of the fluorine then assist in adding to the potency and effectiveness of the drug.
3.4. Examples of Pharmaceuticals Containing Fluorine
Numerous pharmaceuticals feature fluorine, including six examples seen in Figure 4: atorvastatin (Lipitor) 1, rosuvastatin (Crestor) 2, escitalopram (Lexapro) 3, fluticasone (Flonase®) 4, asciminib (ScemblixTM-AB001) 5, and atogepant (Qulipta®) 6. These drugs treat high cholesterol, depression, anxiety disorders, allergic rhinitis, chronic myelogenous leukemia (CML), and migraines, respectively [3][31]. These six drugs are used by patients in every demographic, and their functions affect millions of people worldwide; however, these compounds represent only a fraction of pharmaceuticals that contain at least one fluorine atom. In this section, pharmaceutical compounds will be described and categorized in detail, paying special attention to the features of the compounds containing fluorine atoms highlighted in Figure 4.
Atorvastatin 1 and rosuvastatin 2 are statin drugs used in the treatment of patients exhibiting high cholesterol [32]. Rosuvastatin 2, was developed from a desire to replace large, complex functional groups with simpler, achiral alternatives. The addition of the pyrimidine ring compared to other synthetic statins, such as atorvastatin 1, improves activity for inhibiting the targeted reductase. The p-fluorophenyl group on this molecule not only enhances its biological activity but also deactivates the ring against P450 monooxygenase.
Escitalopram 3 is a selective serotonin reuptake inhibitor used to treat major depression and anxiety disorders [33]. The drug targets the serotonin transporter and facilitates the reuptake of serotonin into the neurons for rapid antidepressant activity [34]. A binding study of 5-HT transporter (SERT) was conducted to determine affinity for the two potential binding sites and substituent selectivity [35]. The study determined that derivatives of escitalopram containing aromatic fluorine and the presence of the cyano group were significant for a twofold decrease in dissociation rate and had a high contribution to allosteric potency.
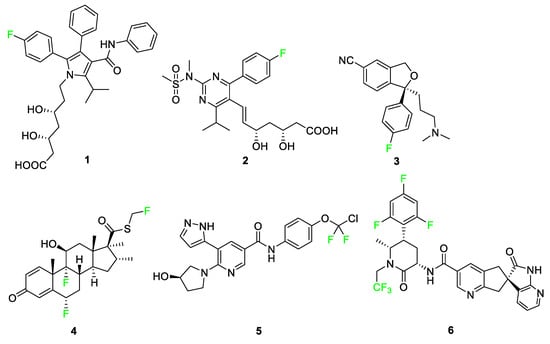
Fluticasone 4 is used for relieving nasal symptoms such as sneezing, itching, and runny nose provoked by allergies; recent studies use this compound in combination with other medications to improve its effects against allergic rhinitis [38][39][40]. Also, fluticasone is combined with salmeterol and prescribed under the medication named Advair for the treatment of asthma as well as other obstructive airway diseases [41][42]. Fluticasone 4 containing the electron-withdrawing fluorine atom demonstrated greater topical activity compared to other analogs and increased glucocorticoid and mineralocorticoid effects [43].
Asciminib 5 is the active ingredient used in the treatment of CML containing a chiral compound with a difluorinated moiety [44]. CML is a disease associated with the overproduction of white blood cells in the bone marrow that arises from the BCR-ABL protein mutation leading to abnormal signaling pathways resulting in the growth and generation of leukemic cells [36][45]. These types of tyrosine kinase inhibitors work by binding to the ATP-binding site of BCR-ABL, thus transforming the disease into a controllable state during treatment [36][46]. The fluorine atoms in asciminib 5 interact with the carbonyl carbon of leucine-359 of the active site pocket, making the position of the fluorine atoms advantageous for improving the bioactivity of the overall compound [5].
Atogepant 6 is an orally administered calcitonin gene-related peptide (CGRP) receptor antagonist that aids in the treatment of migraines [37][47]. Substitution of the 1,3,5-trifluorobenzene moiety in place of a nonfluorinated benzene ring produces a four-fold increase in the affinity of atogepant 6 compared to the nonfluorinated benzene ring derivative [48][49][50].
Approved by the FDA in 2003, aprepitant (Emend) 7 combats chemotherapy-induced nausea and vomiting and serves as an example where the addition of a fluorine atom blocks oxidative metabolism and lowers the oxidation potential elsewhere on the ring (Figure 5) [10][51]. Ongoing clinical trials show promising results for the use of this drug in cancer treatment [52]. The 3,5-bis-(trifluoromethyl)phenyl group is a common feature with other NK1 receptor antagonists and improves penetration of the drug into the central nervous system [51]. The fluorine atom blocks the 4-position of the benzene ring from oxidation as well as deactivates the remainder of the ring positions.
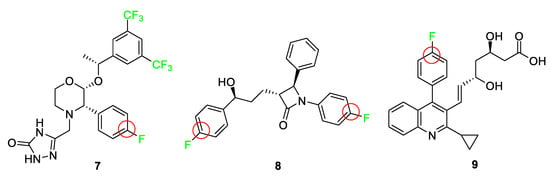
Ezetimibe (Zetia®) 8 [53] is a drug that combats high cholesterol by targeting Niemann–Pick C1-like 1 protein (NPC1L1), thus lowering low-density lipoprotein (LDL); it features two p-fluoro substituents to block other sites on the ring from metabolic oxidation and improves metabolic stability [12][56]. Figure 5 shows the structure of ezetimibe 8 and highlights the blocking of sites because of fluorine atoms. The compound utilizes the fluorine atoms as a defense against aromatic hydroxylation as well as yielding a derivative that exhibits improved pharmacokinetic properties, which increases the activity of the drug significantly. Another property of the fluorine atom substitution that ezetimibe 8 takes advantage of is the increase in polarity of the overall molecule. Molecules with higher polarity are more susceptible to glucuronidation, a process that generally inactivates drugs. However, in the case of ezetimibe 8, glucuronidation improves the activity of the drug by recirculating the drug to the activation site and increasing the residence time [21]. Therefore, through the utilization of fluorine, researchers are able to increase lipophilicity and polarity, as well as block the oxidation sites for ezetimibe 8, which in turn exhibited a 50-fold increase in activity over the parent compound.
Synthetic statins are a class of drugs that must contain 4-fluorophenyl groups as structural requirements for biological activity. Examples include atorvastatin 1, rosuvastatin 2, and pitavastatin (Livalo) 9, as shown in Figure 4 and Figure 5. These drugs combat cholesterol problems by inhibiting hydroxymethylglutaryl-coenzyme A (HMG-CoA) reductase. All of these compounds feature a p-fluorophenyl moiety, as studies have shown that this substituent greatly surpasses the biological activity of all other functional groups tested [54][55]. Pitavastatin 9, a recent statin-type drug to enter the market, is completely synthetic. This HMG-CoA reductase inhibitor and low-density lipoprotein cholesterol (LDL-C) receptor inducer improves its pharmacokinetics, efficiency, and bioavailability by incorporating a p-fluoro group [57]. The 2-cyclopropyl-4-(4-fluorophenyl)quinoline pharmacophore differentiates it from other statins, and the highly functionalized heterocycle provides superior resistance to metabolism and prolongs its duration of action [58]. A modified version of pitavastatin 9 has been used for positron emission tomography (PET) imaging in vivo by incorporating a fluorine-18 atom [59].
As seen in Figure 6, fulvestrant (Faslodex) 11 utilizes the replacement of an n-ethyl functional group with a pentafluoroethyl moiety [60]. ICI 164,384 10, the original precursor, was developed for the treatment of breast cancer, specifically to combat the negative side effects of the receptor modulator Tamoxifen which increased the risk of the metastasis of associated tumors. The parent molecule did not display high levels of potency during in vivo testing. The introduction of the -CF2CF3 group in step 1 of the 12-step synthesis of compound 11, resulted in a five-fold increase in intrinsic potency relative to the parent molecule 10 [60]. The addition of the terminal pentafluoroethyl group as opposed to the ethyl end-chain increases the strength of the bonds and makes the compound more stable as well as increases hydrogen bonding with the receptor, thus increasing metabolic stability during estrogen receptor (ER) binding [60].
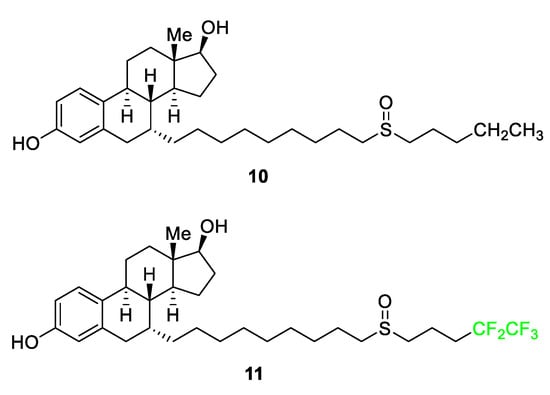
Figure 6. The parent molecule 10 and the FDA-approved drug derivative, fulvestrant 11 [60].
Multiple drugs use the added lipophilicity afforded by the substitution of a hydrogen atom with a fluorine atom; lipophilicity alteration can lead to greater drug uptake through cell membranes [61][62][63][64]. While fluorination of an aromatic or π-system increases lipophilicity, the addition of fluorine and trifluoromethyl groups on an n-alkyl chain does not have the same effect; some tailoring and chemical designs of aliphatic fluorination can decrease lipophilicity [65]. Vandetanib (Caprelsa) 12 [66], a drug that acts as an antagonist of the vascular endothelial growth factor receptor (VEGFR), is used as an oral kinase inhibitor for thyroid tumors. This is an example of a compound modified with fluorine to achieve the “Goldilocks” level of lipophilicity (Figure 7). Many derivatives of vandetanib 12 were tested to tailor the lipophilicity of the compound to increase its potency. Structure–activity relationship tests show that bromine at the C-4’ position was preferred, and that fluorine was optimal at the C-2’ position. The bromofluorophenyl group was analyzed as a residue matching these two cases, and it was shown that the fluorine atom leads the bromofluorophenyl group deep into the protein’s hydrophobic pocket, ultimately increasing the potency of the drug [66].
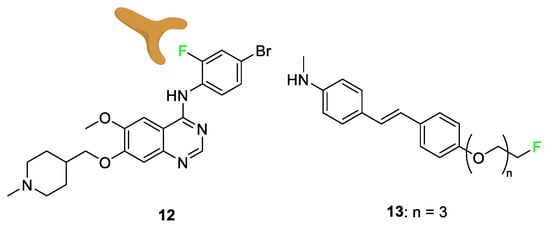
Compound 13, a specific AV-45 derivative with three additional polyethylene glycol (PEG) groups added to the compound, was tested alongside other chain lengths to determine how varying the hydrophilicity altered efficacy [67]. These compounds were being studied for amyloid beta (Aβ) plaque affinity in relation to combatting Alzheimer’s disease. Compound 13 displayed good Aβ binding coupled with high blood–brain barrier penetration. A range of fluoroethylene glycol (FPEG) lengths was studied to find the greatest uptake depending on the logP (lipophilicity). The result shows only a minimal change in uptake when modifying the PEG length below n = 5. However, when the FPEG group was replaced with a hydroxyl group, a significant increase in lipophilicity and, more importantly, a decrease in potency was observed [67].
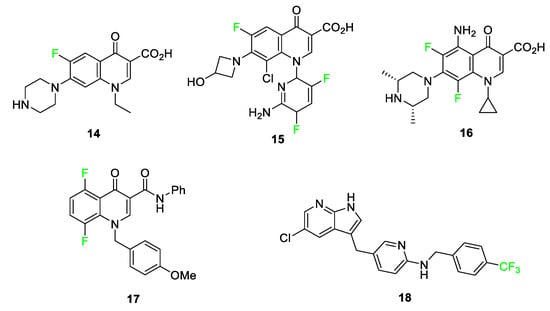
Fluorine-containing quinolones, pyrimidoquinolines, and pyridyl-substituted indoles exhibit anti-cancer properties [68]. The fluorine atom and trifluoromethyl substituents play major roles in the anti-cancer, antimicrobial, and antituberculosis effectiveness of these compounds [69][70]. Adding fluorine to this class of small molecules increases the hydrophobicity of the compounds, which in turn, aids in the penetration into hydrophobic protein pockets. Examples of common amino acids that attract the hydrophobic fluorine group include leucine and phenylalanine [68]. More specific examples of these compounds 14–18 are shown in Figure 8.
In Figure 9, nilotinib (TASIGNA®) 19, a derivative of Imatinib, is shown. The addition of the trifluoromethyl group resulted in the compound exhibiting 30 times the potency when compared to the nonfluorinated parent compound. Once inside the pocket of the Bcr-Abl tyrosine kinase inhibitor, the trifluoromethyl group interacts with the histidine and isoleucine side chain residues. By comparison, when an analog featuring a methyl substituent was tested in place of the trifluoromethyl, it showed a five-fold decrease in activity relative to compound 19 [55][72].
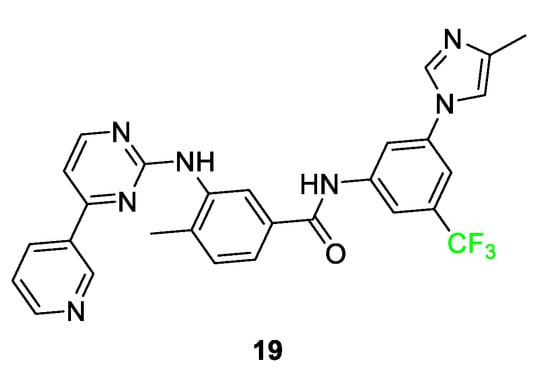
Figure 9. Nilotinib 19 is a compound that inhibits a tyrosine kinase inhibitor in patients with chronic myelogenous leukemia [72].
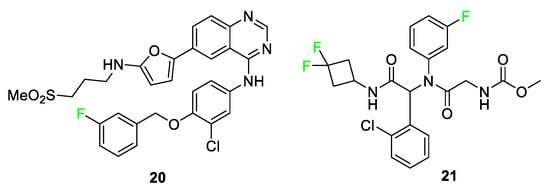
Other compounds that utilize the size of fluorine to advantage are lapatinib (TYKERB®) 20 and ivosidenib (TIBSOVO®) 21 (Figure 10) [73][74]. Lapatinib 20 is a human epidermal growth factor inhibitor and dual tyrosine kinase inhibitor for fighting breast cancer and other solid tumors. Groups larger than the featured m-fluorophenyl, e.g., the m-chlorophenyl group, displayed diminished drug activity for compound 20 analogs. Analogs that substituted -OH or -Br in place of the m-fluorophenyl group showed substantial decreases in inhibition. These findings led researchers to the conclusion that fluorine is essential to fit into the binding pocket as well as to the interactions that retain the drug in the pocket; X-ray crystallographic results support this conclusion [73]. Ivosidenib 21 is a drug developed to treat relapsed or refractory acute myeloid leukemia in adults with an isocitrate dehydrogenase 1 (IDH1) mutation. Fluorine substitution on the phenyl ring brought about desirable metabolic stability in observations from compound AGI-14100 containing two aromatic fluorine atoms; however, in the same study, compound 21 was determined to have more desirable properties upon changing the design to incorporate a nitrogen atom in the ring containing fluorine atom [74].
Thus far, this section has highlighted therapeutic agents and pharmaceutical compounds that feature fluorine atoms or fluorine-containing functional groups on small molecules. This special atom has become extremely important in drug development in recent decades and will continue to be an essential building block when designing and synthesizing drugs [4][75][76]. The characteristics of fluorine and the benefits of incorporation on pharmaceutical molecules, whether they be size considerations, electronic properties, or blocking of metabolic sites susceptible to the P450 monooxygenase, are undeniable in medicinal chemistry.
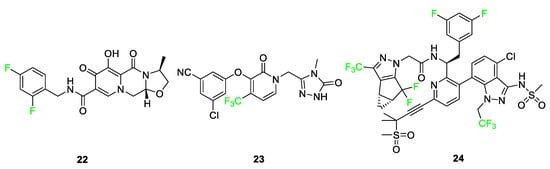
In the literature, fluorinated compounds including cabotegravir (Vocabria) 22, doravirine (PIFELTRO™) 23, and lenacapavir (SUNLENCA®) 24 are recent FDA-approved drugs that have been used in the development of HIV treatments (Figure 11) [14]. Cabotegravir 22 has been used in conjunction with rilpivirine as the cocktail under the name of Cabenuva [5][36][44]. The introduction of a fluorine atom to the benzene ring of cabotegravir compared to its analog improved its potency as an agent. In 2021, a study was conducted where it was used as an injectable in combination with rilpivirine to give it a long-lasting effect as treatment. Doravirine 23, like rilpivirine, is a non-nucleoside reverse transcriptase inhibitor (NNRTI) used in HIV treatment after it was FDA-approved in 2018 [47]. Its structure is a trifluorinated compound compared to an existing chlorine-containing analog; the modification to the structure shows improvements in half-life and plasma stability in studies conducted. Lenacapavir 24, FDA-approved in 2022, is used as a capsid protein inhibitor with pico-molar range potency and it showed significant viral suppression after some weeks when administered to patients. The introduction of fluorine to the structure anchors the conformation, thus improving the drug’s potency [47].
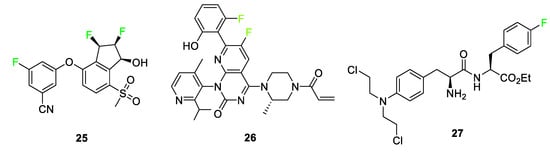
Anti-cancer medications are also highlighted as a sector where fluorinated compounds have been introduced (Figure 12). Belzutifan (WELIREG™) 25 is a hypoxia-inducible factor-2α inhibitor used for the treatment of Hippel–Lindau disease which is associated with the appearance of renal cell carcinoma, hemangioblastomas, and/or pancreatic neuroendocrine tumors [36]. Sotorasib 26 (LUMAKRAS®), an elective and irreversible covalent inhibitor B, is used to treat non-small cell lung cancer [36]. Sotorasib 26 interacts with a cysteine-12 residue of KRAS mutation, KRas G12C, to disrupt the signaling pathway. This KRAS mutation brings about the following tumors: lung adenocarcinoma, pancreatic ductal carcinoma, and colorectal carcinoma. The fluorine-containing version of the compound overcame the bioavailability issue observed for derivatives of the compound containing different halogens [5]. Melphalan flufenamide (Pepaxto®) 27, a drug approved in 2021 for use in the treatment of myeloma, is 10 times more pharmacologically active than melphalan [5][77]. The presence of the p-fluoro substituent on melphalan flufenamide 27 enhances metabolic stability over nonfluorinated melphalan analogs [78].
The bioactivities of a venerable class of compounds, sulfonamides, have seen a resurgence in research interest over the past decade [80]. Numerous examples of fluorinated sulfonamides show enhanced biological efficacy over their nonfluorinated counterparts. The enhancements in effectiveness can arise from resistance to oxidation, electronic alterations, or size brought about by fluorine substitution. Tetrafluoropyridyl sulfonamide 28 (Figure 13) was found to be active against both Gram-positive (B. subtillis and S. pneumoniae) and Gram-negative (P. aeruginosa and E. coli) bacteria strains as well as effective against fungi (A. fumigatus). This sulfonamide was also active in a cytotoxic assay against the MCF-7 breast cancer and HepG2 hepatic cancer cell lines. Activity exceeded that of both cisplatin and 5-fluorouracil. Molecular docking shows that sulfonamide exhibits good interactions with amino acid residues on the mitogen-activated kinase active site. The parafluorophenyl sulfonamide 29 was found to be active in a cytotoxic assay against the HepG2 hepatic cancer cell lines with activity on par with cisplatin. Molecular docking shows that sulfonamide 29 exhibits good interactions with amino acid residues on the mitogen-activated kinase active site.
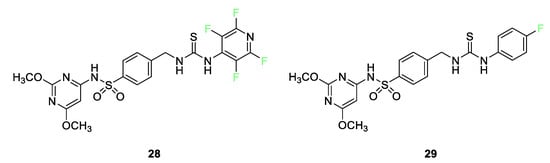
Figure 13. Tetrafluoropyridyl sulfonamide 28 and parafluorophenyl sulfonamide 29 [80].
Some fluorinated drugs have been found to be relevant in the treatment of patients with mild to moderate symptoms of severe acute respiratory syndrome coronavirus 2, SARS-CoV-2. Nirmatrelvir (PAXLOVID™) 30 is a protease inhibitor containing a -CF3 group that is used to disrupt virus replication (Figure 14) [36]. In COVID-19 patients, it has been used alongside ritonavir to maintain the concentration of nirmatrelvir 30 during treatment. This combination of drugs was the first orally administered treatment approved by the FDA for the treatment of COVID-19. Other drugs such as ensitrelvir (Xocova®) and favipiravir (Avigan) are two promising antiviral compounds currently undergoing studies for potential use in the treatment of SARS-CoV-2.
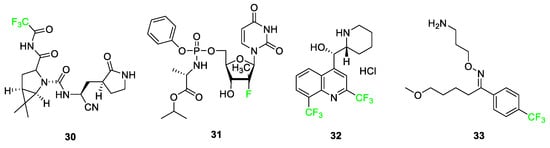
Figure 14. COVID-related fluorine-containing drugs: nirmatrelvir 30, sofosbuvir 31, mefloquine HCl 32, and fluvoxamine 33 [36].
Fluorinated drugs used to treat other diseases have been repurposed for potential uses in the treatment of SARS-CoV-2 such as sofosbuvir (SOVALDI®) 31, mefloquine HCl (Lariam®) 32, and fluvoxamine (Luvox®) 33, In 2013, sofosbuvir was used as a pro-antiviral drug for the treatment of the Hepatitis C virus, and in 2020, it was being studied for inhibition of RNA-dependent RNA polymerase in SARS-CoV-2 replication. Mefloquine HCl 32 was originally used as an antimalarial drug; studies conducted in 2021 adopted this drug for usage in the treatment of COVID-19 as the fluorinated version of the molecule increased its antiviral activity compared to derivative hydroxychloroquine (Figure 14). Fluvoxamine 33, originally used for obsessive-compulsive disorder (OCD) as a selective serotonin reuptake inhibitor, is being studied for its effect in reducing the need for hospitalization in COVID-19 patients [36].
In a recent review, Berrino et al. recounted the current state of research into the enhancement of carbonic anhydrase (CA) inhibition by fluorination of sulfonamides [81]. Several research teams have investigated [82][83][84][85] how aromatic fluorine substitution (compounds 34 and 35) as well as fluoroalkyl substitution (compounds 36 and 37) can improve CA II and CA IX inhibition (Figure 15).
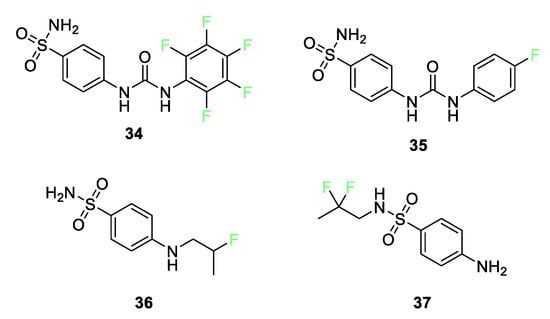
Figure 15. Fluorine-containing sulfonamides is used for inhibition of carbonic anhydrase.
The presence of fluorine in the para position of the aromatic rings in compounds 34 and 35 makes oxidation unlikely. As discussed earlier, in vitro inhibition studies show that compounds 34 and 35 inhibit the tumor-associated CA IX [82][83][84][85]. Compound 35 is in Phase 1b/II clinical trials as a CA IX inhibitor (with gemcitabine) for hypoxic solid tumor treatment. The fluorine(s) give the aryl group pharmacokinetic and pharmacodynamic properties not enjoyed by the nonfluorinated analogs. Additionally, X-ray studies of compound 35 have shown that the larger hydrophobic pocket of CA IX can better accommodate the tail of compound 35, leading to efficient hydrophobic interactions.
Substitution of fluorine on alkyl chains attached to either the aniline nitrogen or the sulfonylamino group can also enhance CA inhibition. Substitution of a 2-fluoropropyl moiety on the aniline nitrogen in compound 36 strongly enhances the inhibition potency relative to its nonfluorinated N-allylic precursor against CA II by a factor of 40 and against CA IX by a factor of 30. When the fluorinated chain is attached to the sulfonyl nitrogen, as in compound 37, the difluoro group enhances the inhibition of CA II and CA IX by a factor of 40.
References
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in Pharmaceuticals: Looking Beyond Intuition. Science 2007, 317, 1881–1886.
- Shah, P.; Westwell, A.D. The role of fluorine in medicinal chemistry. J. Enzym. Inhib. Med. Chem. 2007, 22, 527–540.
- Wang, Q.; Han, J.; Sorochinsky, A.; Landa, A.; Butler, G.; Soloshonok, V.A. The Latest FDA-Approved Pharmaceuticals Containing Fragments of Tailor-Made Amino Acids and Fluorine. Pharmaceuticals 2022, 15, 999.
- Inoue, M.; Sumii, Y.; Shibata, N. Contribution of Organofluorine Compounds to Pharmaceuticals. ACS Omega 2020, 5, 10633–10640.
- He, J.; Li, Z.; Dhawan, G.; Zhang, W.; Sorochinsky, A.E.; Butler, G.; Soloshonok, V.A.; Han, J. Fluorine-containing drugs approved by the FDA in 2021. Chin. Chem. Lett. 2022, 34, 107578.
- Arntson, K.E.; Pomerantz, W.C.K. Protein-Observed Fluorine NMR: A Bioorthogonal Approach for Small Molecule Discovery. J. Med. Chem. 2016, 59, 5158–5171.
- Smart, B.E. Fluorine substituent effects (on bioactivity). J. Fluor. Chem. 2001, 109, 3–11.
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451.
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330.
- Reichenbächer, K.; Süss, H.I.; Hulliger, J. Fluorine in crystal engineering—“The little atom that could”. Chem. Soc. Rev. 2005, 34, 22–30.
- O’Hagan, D. Understanding organofluorine chemistry. An introduction to the C–F bond. Chem. Soc. Rev. 2008, 37, 308–319.
- Aradi, K.; Kiss, L. Carbotrifluoromethylations of C−C Multiple Bonds (Excluding Aryl-and Alkynyltrifluoromethylations). Chem. A Eur. J. 2023, 29, e202203499.
- Alle, T.; Joyasawal, S.; Oukoloff, K.; Long, K.; Owyang, Z.; Francisco, K.R.; Cahard, D.; Huryn, D.M.; Ballatore, C. Structure-property relationships of fluorinated carboxylic acid bioisosteres. Bioorganic Med. Chem. Lett. 2023, 91, 129363.
- Han, S.; Lu, Y. Fluorine in anti-HIV drugs approved by FDA from 1981 to 2023. Eur. J. Med. Chem. 2023, 258, 115586.
- Johnson, B.M.; Shu, Y.-Z.; Zhuo, X.; Meanwell, N.A. Metabolic and Pharmaceutical Aspects of Fluorinated Compounds. J. Med. Chem. 2020, 63, 6315–6386.
- Casa, S.; Henary, M. Synthesis and Applications of Selected Fluorine-Containing Fluorophores. Molecules 2021, 26, 1160.
- Hyun, H.; Park, M.H.; Owens, E.A.; Wada, H.; Henary, M.; Handgraaf, H.J.M.; Vahrmeijer, A.L.; Frangioni, J.V.; Choi, H.S. Structure-inherent targeting of near-infrared fluorophores for parathyroid and thyroid gland imaging. Nat. Med. 2015, 21, 192–197.
- Zhou, P.; Zou, J.; Tian, F.; Shang, Z. Fluorine Bonding—How Does It Work In Protein−Ligand Interactions? J. Chem. Inf. Model. 2009, 49, 2344–2355.
- Britton, R.; Gouverneur, V.; Lin, J.-H.; Meanwell, M.; Ni, C.; Pupo, G.; Xiao, J.-C.; Hu, J. Contemporary synthetic strategies in organofluorine chemistry. Nat. Rev. Methods Primers 2021, 1, 47.
- Liang, T.; Neumann, C.N.; Ritter, T. Introduction of Fluorine and Fluorine-Containing Functional Groups. Angew. Chem. Int. Ed. 2013, 52, 8214–8264.
- Preparation of Highly Fluorinated Compounds. In Fluorine in Organic Chemistry; Blackwell Publishing Ltd.: Hoboken, NJ, USA, 2004; pp. 23–46.
- Zhou, G.; Tian, Y.; Zhao, X.; Dan, W. Selective Fluorination of 4-Substituted 2-Aminopyridines and Pyridin-2(1H)-ones in Aqueous Solution. Org. Lett. 2018, 20, 4858–4861.
- Lerman, O.; Tor, Y.; Hebel, D.; Rozen, S. A novel electrophilic fluorination of activated aromatic rings using acetyl hypofluorite, suitable also for introducing fluorine-18 into benzene. J. Org. Chem. 1984, 49, 806–813.
- Furuya, T.; Kamlet, A.S.; Ritter, T. Catalysis for fluorination and trifluoromethylation. Nature 2011, 473, 470–477.
- Sloop, J.C.; Holder, C.; Henary, M. Selective Incorporation of Fluorine in Pyrazoles. Eur. J. Org. Chem. 2015, 2015, 3405–3422.
- Yang, X.; Sun, R.; Li, S.; Zheng, X.; Yuan, M.; Xu, B.; Jiang, W.; Chen, H.; Fu, H.; Li, R. Regioselective Direct C–H Trifluoromethylation of Pyridine. Org. Lett. 2020, 22, 7108–7112.
- Liang, D.; Li, Y.; Gao, S.; Li, R.; Li, X.; Wang, B.; Yang, H. Amide-assisted radical strategy: Metal-free direct fluorination of arenes in aqueous media. Green Chem. 2017, 19, 3344–3349.
- Park, B.K.; Kitteringham, N.R.; O’Neill, P.M. Metabolism of Fluorine-Containing Drugs. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 443–470.
- Nassar, A.-E.F.; Kamel, A.M.; Clarimont, C. Improving the decision-making process in the structural modification of drug candidates: Enhancing metabolic stability. Drug Discov. Today 2004, 9, 1020–1028.
- Hagmann, W.K. The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem. 2008, 51, 4359–4369.
- Schoepfer, J.; Jahnke, W.; Berellini, G.; Buonamici, S.; Cotesta, S.; Cowan-Jacob, S.W.; Dodd, S.; Drueckes, P.; Fabbro, D.; Gabriel, T.; et al. Discovery of Asciminib (ABL001), an Allosteric Inhibitor of the Tyrosine Kinase Activity of BCR-ABL1. J. Med. Chem. 2018, 61, 8120–8135.
- Szarek, M.; Amarenco, P.; Callahan, A.; DeMicco, D.; Fayyad, R.; Goldstein, L.B.; Laskey, R.; Sillesen, H.; Welch, K.M. Atorvastatin Reduces First and Subsequent Vascular Events Across Vascular Territories: The SPARCL Trial. J. Am. Coll. Cardiol. 2020, 75, 2110–2118.
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506.
- Zhang, X.; Ma, X.; Cheng, W.; Han, Y.; Mu, Q.; Li, J.; He, S. Effects of escitalopram oxalate plus low-dose trazodone on psychological state and quality of life in patients with treatment-refractory depression. Am. J. Transl. Res. 2023, 15, 1905–1912.
- Larsen, M.A.B.; Plenge, P.; Andersen, J.; Eildal, J.N.N.; Kristensen, A.S.; Bøgesø, K.P.; Gether, U.; Strømgaard, K.; Bang-Andersen, B.; Loland, C.J. Structure–activity relationship studies of citalopram derivatives: Examining substituents conferring selectivity for the allosteric site in the 5-HT transporter. Br. J. Pharmacol. 2016, 173, 925–936.
- Chandra, G.; Singh, D.V.; Mahato, G.K.; Patel, S. Fluorine-a small magic bullet atom in the drug development: Perspective to FDA approved and COVID-19 recommended drugs. Chem. Pap. 2023, 77, 4085–4106.
- Ailani, J.; Lipton, R.B.; Goadsby, P.J.; Guo, H.; Miceli, R.; Severt, L.; Finnegan, M.; Trugman, J.M. Atogepant for the Preventive Treatment of Migraine. N. Engl. J. Med. 2021, 385, 695–706.
- Kariyawasam, H.H.; Scadding, G.K. Seasonal allergic rhinitis: Fluticasone propionate and fluticasone furoate therapy evaluated. J. Asthma Allergy 2010, 3, 19–28.
- Kumar, R.S.; Jain, M.K.; Kushwaha, J.S.; Patil, S.; Patil, V.; Ghatak, S.; Sanmukhani, J.; Mittal, R. Efficacy and Safety of Fluticasone Furoate and Oxymetazoline Nasal Spray: A Novel First Fixed Dose Combination for the Management of Allergic Rhinitis with Nasal Congestion. J. Asthma Allergy 2022, 15, 783–792.
- Zhang, Y.; Wei, P.; Chen, B.; Li, X.; Luo, X.; Chen, X.; Xiang, M.; Li, L.; Zhao, S.; Xiao, X.; et al. Intranasal fluticasone furoate in pediatric allergic rhinitis: Randomized controlled study. Pediatr. Res. 2021, 89, 1832–1839.
- Zhang, X.; Liu, M.; Mao, Y. Efficacy of Fluticasone and Salmeterol Dry Powder in Treating Patients with Bronchial Asthma and Its Effect on Inflammatory Factors and Pulmonary Function. Evid. Based Complement. Altern. Med. Ecam 2022, 2022, 8555417.
- Prabhudesai, P.; Singh, B.P.; Agrawal, G.; Singh, A.K.; Jadhav, A.Y.; Patil, S.R.; Bhagat, S.; Patil, S.; Barkate, H. Fluticasone Furoate/Vilanterol Use Trends and Characteristics in Patients with Obstructive Airway Disease: A Real-World Study of 10,374 Patients from India. Cureus 2023, 15, e34825.
- Crim, C.; Pierre, L.N.; Daley-Yates, P.T. A review of the pharmacology and pharmacokinetics of inhaled fluticasone propionate and mometasone furoate. Clin. Ther. 2001, 23, 1339–1354.
- Ali, S.; Zhou, J. Highlights on U.S. FDA-approved fluorinated drugs over the past five years (2018–2022). Eur. J. Med. Chem. 2023, 256, 115476.
- Lin, H.-C.; Saputra, F.; Audira, G.; Lai, Y.-H.; Roldan, M.J.M.; Alos, H.C.; Aventurado, C.A.; Vasquez, R.D.; Tsai, G.-J.; Lim, K.-H.; et al. Investigating Potential Cardiovascular Toxicity of Two Anti-Leukemia Drugs of Asciminib and Ponatinib in Zebrafish Embryos. Int. J. Mol. Sci. 2022, 23, 11711.
- Deeks, E.D. Asciminib: First Approval. Drugs 2022, 82, 219–226.
- Rizzo, C.; Amata, S.; Pibiri, I.; Pace, A.; Buscemi, S.; Palumbo Piccionello, A. FDA-Approved Fluorinated Heterocyclic Drugs from 2016 to 2022. Int. J. Mol. Sci. 2023, 24, 7728.
- Dubowchik, G.M.; Conway, C.M.; Xin, A.W. Blocking the CGRP Pathway for Acute and Preventive Treatment of Migraine: The Evolution of Success. J. Med. Chem. 2020, 63, 6600–6623.
- Martelletti, P.; Cipolla, F.; Capi, M.; Curto, M.; Lionetto, L. Atogepant. Calcitonin gene-related peptide (CGRP) receptor antagonist, Preventive treatment of migraine. Drugs Futur. 2020, 45, 285.
- Benedetto Tiz, D.; Bagnoli, L.; Rosati, O.; Marini, F.; Sancineto, L.; Santi, C. New Halogen-Containing Drugs Approved by FDA in 2021: An Overview on Their Syntheses and Pharmaceutical Use. Molecules 2022, 27, 1643.
- Di Fabio, R.; Alvaro, G.; Griffante, C.; Pizzi, D.A.; Donati, D.; Mattioli, M.; Cimarosti, Z.; Guercio, G.; Marchioro, C.; Provera, S.; et al. Discovery and Biological Characterization of (2R,4S)-1′-Acetyl-N-{(1R)-1-ethyl}-2-(4-fluoro-2-methylphenyl)-N-methyl-4,4′-bipiperidine-1-carboxamide as a New Potent and Selective Neurokinin 1 (NK1) Receptor Antagonist Clinical Candidate. J. Med. Chem. 2011, 54, 1071–1079.
- Wang, D.-S.; Hu, M.-T.; Wang, Z.-Q.; Ren, C.; Qiu, M.-Z.; Luo, H.-Y.; Jin, Y.; Fong, W.P.; Wang, S.-B.; Peng, J.-W.; et al. Effect of Aprepitant for the Prevention of Chemotherapy-Induced Nausea and Vomiting in Women: A Randomized Clinical Trial. JAMA Netw. Open 2021, 4, e215250.
- Van Heek, M.; Farley, C.; Compton, D.S.; Hoos, L.; Alton, K.B.; Sybertz, E.J.; Davis, H.R., Jr. Comparison of the activity and disposition of the novel cholesterol absorption inhibitor, SCH58235, and its glucuronide, SCH60663. Br. J. Pharmacol. 2000, 129, 1748–1754.
- Igel, M.; Sudhop, T.; von Bergmann, K. Pharmacology of 3-Hydroxy-3-Methylglutaryl-Coenzyme A Reductase Inhibitors (Statins), Including Rosuvastatin and Pitavastatin. J. Clin. Pharmacol. 2002, 42, 835–845.
- Hak Lee, S.; Chung, N.; Kwan, J.; Kim, D.-I.; Ho Kim, W.; Jeong Kim, C.; Seung Kim, H.; Hoon Park, S.; Seog Seo, H.; Gu Shin, D.; et al. Comparison of the efficacy and tolerability of pitavastatin and atorvastatin: An 8-week, multicenter, randomized, open-label, dose-titration study in korean patients with hypercholesterolemia. Clin. Ther. 2007, 29, 2365–2373.
- Xu, Q.; Deng, Y.; Xiao, J.; Liu, X.; Zhou, M.; Ren, Z.; Peng, J.; Tang, Y.; Jiang, Z.; Tang, Z.; et al. Three Musketeers for Lowering Cholesterol: Statins, Ezetimibe and Evolocumab. Curr. Med. Chem. 2021, 28, 1025–1041.
- Saito, Y. Pitavastatin: An overview. Atheroscler. Suppl. 2011, 12, 271–276.
- Saito, Y.; Yamada, N.; Teramoto, T.; Itakura, H.; Hata, Y.; Nakaya, N.; Mabuchi, H.; Tushima, M.; Sasaki, J.; Goto, Y.; et al. Clinical Efficacy of Pitavastatin, a New 3-Hydroxy-3-methylglutaryl Coenzyme A Reductase Inhibitor, in Patients with Hyperlipidemia. Arzneimittelforschung 2002, 52, 251–255.
- Yagi, Y.; Kimura, H.; Okuda, H.; Ono, M.; Nakamoto, Y.; Togashi, K.; Saji, H. Evaluation of pitavastatin as a positron emission tomography tracer for in vivo organic transporter polypeptide function. Nucl. Med. Biol. 2019, 74–75, 25–31.
- Wakeling, A.E.; Dukes, M.; Bowler, J. A Potent Specific Pure Antiestrogen with Clinical Potential. Cancer Res. 1991, 51, 3867–3873.
- Troup, R.I.; Jeffries, B.; Saudain, R.E.-B.; Georgiou, E.; Fish, J.; Scott, J.S.; Chiarparin, E.; Fallan, C.; Linclau, B. Skipped Fluorination Motifs: Synthesis of Building Blocks and Comparison of Lipophilicity Trends with Vicinal and Isolated Fluorination Motifs. J. Org. Chem. 2021, 86, 1882–1900.
- Jeffries, B.; Wang, Z.; Felstead, H.R.; Le Questel, J.-Y.; Scott, J.S.; Chiarparin, E.; Graton, J.; Linclau, B. Systematic Investigation of Lipophilicity Modulation by Aliphatic Fluorination Motifs. J. Med. Chem. 2020, 63, 1002–1031.
- Kürschner, M.; Nielsen, K.; von Langen, J.R.G.; Schenk, W.A.; Zimmermann, U.; Sukhorukov, V.L. Effect of Fluorine Substitution on the Interaction of Lipophilic Ions with the Plasma Membrane of Mammalian Cells. Biophys. J. 2000, 79, 1490–1497.
- Yerien, D.E.; Bonesi, S.; Postigo, A. Fluorination methods in drug discovery. Org. Biomol. Chem. 2016, 14, 8398–8427.
- Jeffries, B.; Wang, Z.; Graton, J.; Holland, S.D.; Brind, T.; Greenwood, R.D.R.; Le Questel, J.-Y.; Scott, J.S.; Chiarparin, E.; Linclau, B. Reducing the Lipophilicity of Perfluoroalkyl Groups by CF2–F/CF2–Me or CF3/CH3 Exchange. J. Med. Chem. 2018, 61, 10602–10618.
- Ryan, A.J.; Wedge, S.R. ZD6474–a novel inhibitor of VEGFR and EGFR tyrosine kinase activity. Br. J. Cancer 2005, 92, S6–S13.
- Kung, H.F.; Choi, S.R.; Qu, W.; Zhang, W.; Skovronsky, D. 18F Stilbenes and Styrylpyridines for PET Imaging of Aβ Plaques in Alzheimer’s Disease: A Miniperspective. J. Med. Chem. 2010, 53, 933–941.
- Ghorab, M.M.; Ragab, F.A.; Heiba, H.I.; Arafa, R.K.; El-Hossary, E.M. Docking study, in vitro anticancer screening and radiosensitizing evaluation of some new fluorine-containing quinoline and pyrimidoquinoline derivatives bearing a sulfonamide moiety. Med. Chem. Res. 2011, 20, 388–400.
- Kathrotiya, H.G.; Patel, M.P. Synthesis and identification of β-aryloxyquinoline based diversely fluorine substituted N-aryl quinolone derivatives as a new class of antimicrobial, antituberculosis and antioxidant agents. Eur. J. Med. Chem. 2013, 63, 675–684.
- Mistry, S.N.; Valant, C.; Sexton, P.M.; Capuano, B.; Christopoulos, A.; Scammells, P.J. Synthesis and Pharmacological Profiling of Analogues of Benzyl Quinolone Carboxylic Acid (BQCA) as Allosteric Modulators of the M1 Muscarinic Receptor. J. Med. Chem. 2013, 56, 5151–5172.
- Chen, D.; Zhang, Y.; Li, J.; Liu, Y. Exploratory Process Development of Pexidartinib through the Tandem Tsuji–Trost Reaction and Heck Coupling. Synthesis 2019, 51, 2564–2571.
- Kantarjian, H.M.; Giles, F.; Gattermann, N.; Bhalla, K.; Alimena, G.; Palandri, F.; Ossenkoppele, G.J.; Nicolini, F.-E.; O’Brien, S.G.; Litzow, M.; et al. Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is effective in patients with Philadelphia chromosome–positive chronic myelogenous leukemia in chronic phase following imatinib resistance and intolerance. Blood 2007, 110, 3540–3546.
- Kuang, Y.-H.; Shen, T.; Chen, X.; Sodani, K.; Hopper-Borge, E.; Tiwari, A.K.; Lee, J.W.K.K.; Fu, L.-W.; Chen, Z.-S. Lapatinib and erlotinib are potent reversal agents for MRP7 (ABCC10)-mediated multidrug resistance. Biochem. Pharmacol. 2010, 79, 154–161.
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers. ACS Med. Chem. Lett. 2018, 9, 300–305.
- Gupta, P.S. Roles of Fluorine in Drug Design and Drug Action. Lett. Drug Des. Discov. 2019, 16, 1089–1109.
- Dabur, M.; Loureiro, J.A.; Pereira, M.C. Fluorinated Molecules and Nanotechnology: Future ‘Avengers’ against the Alzheimer’s Disease? Int. J. Mol. Sci. 2020, 21, 2989.
- Dhillon, S. Melphalan Flufenamide (Melflufen): First Approval. Drugs 2021, 81, 963–969.
- Lehmann, F.; Wennerberg, J. Melflufen: A Journey from Discovery to Multi-Kilogram Production. In Complete Accounts of Integrated Drug Discovery and Development: Recent Examples from the Pharmaceutical Industry Volume 3; American Chemical Society: Washington, DC, USA, 2020; Volume 1369, pp. 157–177.
- Mei, H.; Han, J.; Fustero, S.; Medio-Simon, M.; Sedgwick, D.M.; Santi, C.; Ruzziconi, R.; Soloshonok, V.A. Fluorine-Containing Drugs Approved by the FDA in 2018. Chem. A Eur. J. 2019, 25, 11797–11819.
- El-Gaby, M.; Ammar, Y.A.; IH El-Qaliei, M.; Hussein, M.F.; Faraghally, F.A. Sulfonamides: Synthesis and the recent applications in Medicinal Chemistry. Egypt. J. Chem. 2020, 63, 5289–5327.
- Berrino, E.; Michelet, B.; Martin-Mingot, A.; Carta, F.; Supuran, C.T.; Thibaudeau, S. Modulating the Efficacy of Carbonic Anhydrase Inhibitors through Fluorine Substitution. Angew. Chem. Int. Ed. 2021, 60, 23068–23082.
- Vullo, D.; Scozzafava, A.; Pastorekova, S.; Pastorek, J.; Supuran, C.T. Carbonic anhydrase inhibitors: Inhibition of the tumor-associated isozyme IX with fluorine-containing sulfonamides. The first subnanomolar CA IX inhibitor discovered. Bioorg. Med. Chem. Lett. 2004, 14, 2351–2356.
- Pastorekova, S.; Vullo, D.; Casini, A.; Scozzafava, A.; Pastorek, J.; Nishimori, I.; Supuran, C.T. Carbonic anhydrase inhibitors: Inhibition of the tumor-associated isozymes IX and XII with polyfluorinated aromatic/heterocyclic sulfonamides. J. Enzym. Inhib. Med. Chem. 2005, 20, 211–217.
- Pacchiano, F.; Aggarwal, M.; Avvaru, B.S.; Robbins, A.H.; Scozzafava, A.; McKenna, R.; Supuran, C.T. Selective hydrophobic pocket binding observed within the carbonic anhydrase II active site accommodate different 4-substituted-ureido-benzenesulfonamides and correlate to inhibitor potency. Chem. Commun. 2010, 46, 8371–8373.
- Compain, G.; Martin-Mingot, A.; Maresca, A.; Thibaudeau, S.; Supuran, C.T. Superacid synthesis of halogen containing N-substituted-4-aminobenzene sulfonamides: New selective tumor-associated carbonic anhydrase inhibitors. Bioorg. Med. Chem. 2013, 21, 1555–1563.
More
Information
Subjects:
Chemistry, Medicinal
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.5K
Revisions:
3 times
(View History)
Update Date:
27 Feb 2024
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No