Biological therapies have transformed high-burden treatments. As the patent and exclusivity period for biological medicines draws to a close, there is a possibility for the development and authorization of biosimilars. These products boast comparable levels of safety, quality, and effectiveness to their precursor reference products. Biosimilars, although similar to reference products, are not identical copies and should not be considered generic substitutes for the original. Their development and evaluation involve a rigorous step-by-step process that includes analytical, functional, and nonclinical evaluations and clinical trials. Clinical studies conducted for biosimilars aim to establish similar efficacy, safety, and immunogenicity, rather than demonstrating a clinical benefit, as with the reference product.
1. Biological Medicines
The human body continually produces enzymes, hormones, antibodies, and other endogenous substances necessary for its survival. Over time, drug development has tried to compensate, through targeted approaches, for any deficiencies observed in certain diseases or disorders. More than two decades ago, biological medicines were introduced on the market
[1]. They are produced from living cells, using biotechnology techniques, unlike common medicines, which are produced from chemical synthesis in the laboratory
[2][3][4]. As previously mentioned, biological medicines play a critical role in the treatment of various diseases
[1][4].
Biological medicines possess a significantly more intricate and larger structure when compared to small-molecule drugs
[5]. Currently, they are divided into three main categories: (1) products with a high correlation to endogenous factors, often used as a substitute therapy; (2) monoclonal antibodies (mAbs) that bind to soluble or cell surface targets, blocking cell signaling pathways and their functional responses; (3) engineered proteins that mimic soluble receptors, receptor antagonists, and fusion proteins. More specifically, biological medicines are produced as hormones (as is the case of insulin, hormone deficiencies, and growth hormones), mAb (e.g., the management of autoimmune diseases and cancer), blood products (e.g., individuals with hemophilia), immunomodulators (e.g., interferon beta, for multiple sclerosis), enzymes (e.g., for the removal of blood clots), and vaccines for the prevention of various diseases
[3][6].
The production of most biological drugs is conducted through genetically modified host cells, which can be derived from plants, yeasts, bacteria, or animals. Each manufacturer has its own host cell bank, allowing it to produce a distinct cell line. Furthermore, each manufacturer is responsible for its own manufacturing process
[6]. First, the genetic code of the chosen protein is identified, which can be a hormone, an antibody, or a blood derivative, among others, and then a DNA sequence is fully synthesized. The resulting genetic code is added to different host cell lines. (e.g., yeast or bacteria) for them to produce this protein. Then, the strain that produces the most effective protein is selected and cultivated in bioreactors, a process called fermentation. Afterward, outside the bioreactor, the separation, purification, and stabilization of the protein occur. Then, it is processed into a drug
[7].
The problem with biological drugs lies in the fact that their activity can be impaired by the environmental and manufacturing conditions, making it difficult to achieve equivalent purity between different batches
[5]. Several factors contribute to this: inadequate selection of the cell line; the biophysical characteristics of proteins; changes in temperature or pH conditions during the cultivation phases; the handling and conservation of the product in the various stages of manufacture; the drug product formulation; the production scale; and the production site. Hence, batch discrepancies are circumvented through the implementation of rigorous manufacturing controls over the cellular systems used
[3][5].
1.1. Biosimilars
Following the approval of the first biosimilar drug (somatropin) in 2006 by the European Medicine Agency (EMA), the market of biosimilars has seen considerable expansion. Europe approved, for the first time, an mAb biosimilar (infliximab) in 2013. It is known that between 30 and 50% of new medicines approved in Europe are currently biosimilar
[8]. By August 2022, 1775 biological products were licensed in the US market, of which 82 were biosimilar
[9].
The European Medicine Agency’s guideline states that “a biosimilar is a biological medicinal product that contains a version of the active substance of an already authorised original biological medicinal product (reference medicinal product—RP) in the European Economic Area (EEA)”. It is necessary to establish a comprehensive comparability exercise to determine the similarity to the reference medicinal product in terms of quality characteristics, biological activity, safety, and efficacy
[10]. The biosimilar’s pharmacokinetic (PK) and pharmacodynamic (PD) properties are similar to those of an existing biological medicine (i.e., a medicine that has already received approval from the EMA and is used in the EU, according to approved therapeutic indications). When the approved biological medicine is no longer protected by its exclusivity period (generally 10 years) or by a patent, a biosimilar medicine can be introduced on the market
[4][11].
Biological medicines, including biosimilars, are known for their complex characteristics and high heterogeneity. It is therefore recognized that there may be some variability between RPs and biosimilars. However, small variations between the reference product and the biosimilar have no significant impact on the quality and safety of the product due to the stringent development and approval process for biosimilars. Biosimilars undergo extensive comparability studies to demonstrate their high similarity to the reference product in terms of quality, efficacy, and safety. These studies encompass detailed analyses of the physical, chemical, and biological characteristics of the products, along with clinical trials to assess their efficacy and safety in humans
[4][12].
It is relevant to clarify that a biosimilar is not a generic medicine in the definition of the generic term—the differences are presented in
Table 1 [13][14]. Although biosimilars and generics are both variants of already approved, branded drugs, they have distinct characteristics and the terms should not be correlated
[15]. Generics are chemically synthesized small molecules, which makes them chemically identical to RPs. In contrast, biosimilars are produced by a living cell. Even if two cells have the exact same sequence of amino acids in a protein, there is the possibility of natural variation in the glycosylation process or protein folding. In other words, generating a biological medicine equal to an RP is impossible. However, it is possible to produce a compound that has comparable excellence and physiological and scientific impacts, while ensuring its safety and effectiveness
[15][16].
Table 1. Main differences between biosimilars and generic medicines.
| |
Biosimilars |
Generics |
Ref. |
| Product characteristics |
- Large complex molecules
(up to 270,000 Da) |
- Small and simple molecules
(up to 300 Da) |
[17] |
| Production |
- Produced using live organisms (highly sensitive to manufacturing changes)
- 5–9 years
- High production costs |
- Produced by chemical synthesis
- 2–3 year
- Lower production costs |
[17][18] |
| Structural comparison to reference medication |
- Highly similar to the RP: same amino acid sequence;
- There may be differences in minor parts of the structure |
- Structurally identical to the reference medicine |
[5][13][17] |
| Development |
- Comparability studies between the biosimilar and the RP |
- Bioequivalence between the generic and the RP is evaluated |
[5][19] |
| Nomenclature |
- Rules vary from country to country |
- Same chemical name (active ingredient) as the reference medicine |
[11][13][14][20] |
| Requirements for approval |
- Animal and clinical studies (toxicity, PK, PD, and immunogenicity) |
- No animal or clinical studies (only bioequivalence studies)
- The active ingredient must be identical in strength, dosage form, and route of administration |
[13][17][20][21] |
| Post-authorization activities |
- Pharmacovigilance (PV) |
- Phase IV, risk management plan including PV |
[4][14][22][23][24] |
| Immunogenicity |
- Immunogenic |
- Mostly nonimmunogenic |
[20][22][23][25] |
| Equivalence |
- Data must demonstrate, in each indication, that clinically significant differences, related to safety and efficacy, are not verified
- Conclusive clinical studies may not be necessary for all indications |
- Demonstration of bioequivalence is enough to grant all approved indications for the RP, without requiring any additional clinical studies |
[13][14][17][20][24] |
| Interchangeability and substitution |
- EMA see biosimilars to be scientifically interchangeable (2022), but any decision on the use of biosimilars is the mandate of the EU member states
- FDA can designate a biosimilar interchangeable if a sponsor applies for this (but it is up to US State Law to permit substitution at pharmacies)
- Automatic substitution is generally the decision of each country |
- If permitted by state law, pharmacists may automatically substitute the generic for the reference medicine |
[14][20][25][26][27] |
To establish biosimilarity, it is essential to evaluate whether the quality attributes (QAs) of a biosimilar and the RP are comparable. QAs are measurable product characteristics that describe the main properties of a drug molecule. Unlike generics, biologicals are molecules with intrinsic variability, which makes their QAs heterogeneous and susceptible to changes throughout the manufacturing process. It is important to conduct a comprehensive analysis of any differences that exist, to ensure that these are not clinically [20] relevant to the point of compromising the product’s function, immunogenicity, and efficacy. Therefore, unlike a generic, a biosimilar must present clinical and non-clinical data that prove its similarity, i.e., comparability studies must be carried out [12][16].
1.2. Intended Copies, Biobetters, and Standalone Biologics
Biosimilars should not be confused with intended copies, biobetters, and standalone products, which are related but entirely different concepts
[28]. Physicians are not required to recommend biosimilars solely due to their low cost. This decision should be based on scientific evidence and an understanding of the differences between them
[29].
Intended copies are copies of an RP that do not align with the guidelines set forth by the EMA/FDA and WHO. Therefore, they are not accessible in heavily controlled markets, like the United States, Europe, and Australia, but are promoted in less regulated countries. Because they are cheaper, accessibility to biologicals in these countries has increased. For example, reditux and kikuzubam are intended copies of rituximab, available in India and some South American countries. The first one has undergone a phase III study to confirm its efficacy but has not undergone a direct comparison with the original rituximab. Kikuzubam was removed due to a lack of safety and confirmed toxicity. It has not been proven that intended copies offer the same effectiveness, quality, and level of security as the reference medicine.
Even if the amino acid sequence is indeed the same, the pharmacological profile of the molecule can be affected, either by the existence of impurities, cluster development, or the occurrence of post-translational modifications (PTMs), for example. There is a lack of clinical trials to compare the effectiveness and safety of these medicines or to determine whether they are equivalent or non-inferior based on an acceptable number of patients. They are also not announced on global biosimilar news websites and do not have a registered protocol on
clinicaltrials.gov (or it is not followed or not verified)
[14][21][29].
Biobetters, in turn, are deliberately modified versions of other biological drugs, to improve certain attributes of their pharmacological profile, such as the dosage regimen, safety, efficacy, or immunogenicity
[30][31]. Their manufacturing process is similar to that of biologicals; however, it involves using various advanced methods such as albumin replacement and pegylation (the addition of PEG), among others
[30][31]. Biobetters are considered to be distinctive biological entities due to their unique molecular structures and functions. As a result, they must undergo the standard approval process prescribed by regulatory agencies, rather than the biosimilar approval process
[15]. For example, insulin glargine is a biobetter that resulted from an alteration in the polypeptide chain of insulin; it was formulated to slow down the discharge of insulin molecules following subcutaneous administration
[14].
Darbepoetin alpha is a modified version of epoetin, with an extended elimination half-life, due to alterations in glycosylation
[14]. Another example is neulasta, a biobetter of neupogen, whose dosage frequency is once per cycle of chemotherapy, while neupogen is required once a day during the chemotherapy cycle. In addition, neulasta is more effective than neupogen, resulting in greater adherence. The lower dosage frequency, along with superior efficacy, means a lower economic load for each scheduled dose administration
[14][29].
In July 2021, Sorrento Therapeutics (a pharmaceutical corporation committed to creating innovative drugs for infectious diseases, chronic pain, and cancer) granted approval for the marketing of the biobetter version of infliximab (CMAB008) in China. The utilization of Chinese hamster ovary (CHO) cell lines in production is anticipated to ensure greater safety and reduced immunogenicity, in comparison to the marketed tumor necrosis factor-α Ab, produced in murine cell lines
[32].
Finally, standalone biologics are a novel class of medicines that are distinct from existing drugs. Their efficacy and safety are assessed by comparing them to a placebo or an appropriate comparator. In essence, these medications are the biological counterparts of me-too drugs and can be categorized as biosimilars
[14].
2. Development and Regulatory Approval of Biosimilars
2.1. Development of a Biosimilar
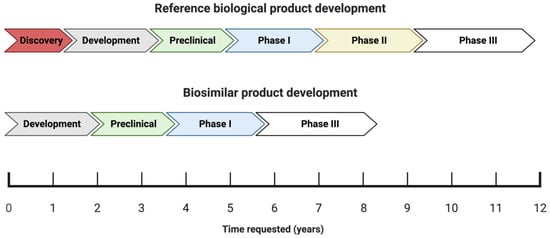
The process of developing and attaining approval for biosimilars differs significantly from that of new and innovative drugs (chemical or biological), as shown in
Figure 1 [18].
Figure 1. Schematic illustration of the development phases and the timeline (in years) of reference biologics versus biosimilars.
The process of obtaining approval for new biological medicines typically spans a period of approximately twelve years, which involves a rigorous research and development phase aimed at creating a suitable molecule. The molecule is then subjected to thorough analysis during the preclinical stage, a critical phase in the drug development process
[15]. The process of bringing a drug to market typically involves several established phases: I, II, III, and IV. Once a drug has been commercialized, phase IV begins. In the case of generic drugs, the process is simpler because the drug molecule has already been established and characterized. Therefore, all that is required is the production of the finished product and bioequivalence testing
[14]. Since biosimilars are essentially copies of existing molecules with established product characteristics, there is no need to carry out the initial discovery or efficacy phase (phase II), thus shortening the development time to eight or fewer years and reducing the development costs by 10–20%
[15][17][33].
In other words, biosimilar medicine development lies somewhere between innovative drugs and generics. Although the molecule is established at the beginning of the process, as with generics, its reproduction and characterization are not an easy process, which constitutes a drawback
[4][14]. Thus, biosimilar manufacturers face a huge challenge: the manufacturing process of the RP is proprietary, i.e., its details are not publicly available—biosimilar production, formulation, and administration are expected to be similar to those of the RP, without relying on the knowledge of the manufacturer in this case. In this way, the biosimilar manufacturer must extensively analyze the RP and use a method of “reverse engineering” in order to obtain a product with high similarity to the RP
[3][14][34][35]. Healthcare professionals should understand therapeutic equivalence and “comparability” to provide the best care. Regarding generics, therapeutically equivalent medications are defined as having an identical chemical composition and the same pharmacokinetic profile. On the other hand, “comparability” is used in the context of biosimilars and means that comparable efficacy and safety have been verified between them and the RP (but this does not mean that they are identical, and it does not guarantee therapeutic equivalence)
[36].
When a new biosimilar is designed, a well-established, step-by-step approach according to scientific principles and risk assessment must be employed
[18]. Then, “quality-by-design” (QbD) constitutes an important tool, which allows the biosimilar to achieve higher similarity regarding the RP. It is essential to select an appropriate RP, obtain the reference active principle, identify the RP’s “quality target product profile” (QTPP) and critical quality attributes (CQAs), and develop a manufacturing process that allows the matching of the RP’s attributes. Thus, to accomplish this approach, characterization and matching of the QTPP is necessary
[11][13][18].
First, the RP’s quality attributes (QAs) must be defined. It is crucial to measure the range of variation for quality attributes that directly affect the effectiveness or safety of the RP (CQAs). This can be achieved by using various batches of drugs to profile the quality of the proposed biosimilar’s target product
[18][21][37]. In other words, CQAs are chemical, physical, biological, and microbiological characteristics that are defined, measured, and monitored continuously, to guide the clinical profile and establish clinical comparability
[13][16][18].
Post-Translational Modifications (PTMs)
Several factors may affect the similarity between the suggested biosimilar and the RP, and PTMs are a huge challenge for the pharmaceutical industry.
mAbs are subject to various modifications throughout their production, purification, and storage processes, resulting in different forms. The genetic sequence dictates the amino acid arrangement in a protein, but its structure, stability, and function will be determined by the PTMs.
Proteins can undergo PTMs through the addition of functional groups, cleavage, or degradation. Recombinant mAbs often experience PTMs, such as N- and C-terminal modifications, deamination, glycosylation, glycation, phosphorylation, acetylation, sulfation, alkylation, methylation, proteolysis, oxidation, mismatched S-S bridges, and truncation. Among these, glycosylation has the most significant impact on biological function
[38].
Proteins undergo glycosylation, in which carbohydrate fractions are added during polypeptide synthesis or in the endoplasmic reticulum and Golgi apparatus of the cell. Proteins undergo two primary forms of glycosylation: O-linked and N-linked. O-linked glycosylation involves the addition of glycans to serine or threonine amino acid residues through an oxygen atom. On the other hand, N-linked glycosylation initiates with the attachment of a high-mannose-based structure to an asparagine amino acid residue within an Asn-X-Ser/Thr consensus sequence during the process of translation. The modification occurs downstream in the endoplasmic reticulum and Golgi apparatus, and X can represent any amino acid except proline (Pro)
[38].
The prevailing form of glycosylation observed in monoclonal antibody (mAb) therapeutics is N-linked oligosaccharides
[38][39]. The N-glycosylation of therapeutic antibodies involves PTMs affecting biological function and treatment effectiveness. The characterization of these glycans is required by manufacturers and regulatory agencies, but their complex structure and heterogeneity present challenges
[38].
Extensive analytical testing platforms are necessary to verify the protein’s identity, maintain a consistent manufacturing process, and ensure product quality. A single error in modifying or combining components could lead to adverse reactions in patients or a reduction in the effectiveness of the final product. Liquid chromatography (LC), mass spectrometry (MS), electrophoresis, and spectroscopy serve as effective tools in confirming the biological identity, sequence variations, and glycosylation patterns, as well as other PTMs and impurities, in antibody products
[8][39][40].
Peptide mapping is a technique used for the analysis of a biopharmaceutical’s primary structure and it is recognized by the International Council of Harmonization guidelines (ICHQ6B). Peptide mapping assumes a crucial role within the biopharmaceutical industry, serving to authenticate the identity of a protein therapeutic and monitor degradation events like oxidation or deamination. It offers a comprehensive analysis of the protein under scrutiny, making it a standard method for the characterization of mAbs
[39][40][41].
However, it should be taken into account that, as this technique involves sample preparation steps, it is subject to variations due to the differences between techniques, technicians, and/or laboratories. Differences in product batches or biosimilar products can make it difficult to compare and reproduce results over time, potentially affecting the quality, efficacy, and safety of the product.
2.2. How to Build the Evidence for Biosimilarity
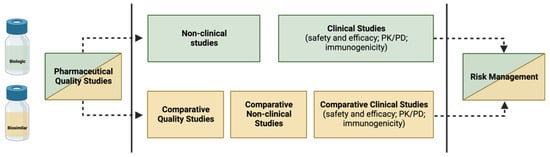
The purpose of the biosimilar investigation pathway is to show that the proposed biosimilar has a safety and efficacy profile that is neither better or worse than that of the RP, rather than to demonstrate superiority or inferiority between the two
[4]. The demonstration of biosimilarity is achieved through a step-by-step approach, as approved by the EMA and the FDA: first, a study of physicochemical and biological comparability (quality studies) is carried out, followed by non-clinical comparability (in vitro and in vivo studies) and, finally, clinical comparability
[5][10][13][42][43]. The clinical comparability study is generally conducted in successive steps, starting with PK and, if possible, PD studies, followed by at least one clinical trial of efficacy and tolerance
[5]. However, in most cases, quality studies performed in vitro are sufficient to confirm that the modifications are insignificant from a clinical perspective. It is important to note that the two products do not need to be identical; it is only necessary to show that there is no clinically significant change (i.e., the proposed biosimilar and the RP need to be comparable)
[14].
The (bio)physical characteristics of a biosimilar are heavily influenced by each step of its development process. Therefore, it is imperative that no step refutes or overcomes substantial distinctions encountered in the preceding steps, and that all three stages demonstrate adequacy to affirm the biosimilarity. Despite the potential depth of biosimilar development comparable to that of an RP, the primary focus lies in achieving comparability during the initial phases of development. In contrast, the focus for a new biological agent is on establishing clinical efficacy and tolerability
[14]—as shown in
Figure 2.
Figure 2. Building evidence for biosimilarity—comparison of data requirements for approval of a biosimilar versus an RP.
3.2.1. Demonstration of Analytical Similarity—Comparative Quality Studies
Analytical assessments constitute a repetitive and iterative process, whose aim is to evaluate the standard of the suggested biosimilar in comparison to the RP. Creating biosimilars demands substantial effort, with a particular emphasis on assays that offer a sensitive assessment of similarity. To ensure comparability in quality, extensive analytical characterization, receptor binding studies, and bioassays are necessary to confirm that the molecular structure and functionality are alike. These studies are carried out through in vitro assays, which are sensitive techniques capable of detecting distinct clinical characteristics between the biosimilar and the RP
[11][13]
Quality attributes (QAs) are compared using analytical methods such as surface plasmon resonance (SPR), enzyme-linked immunosorbent assays (ELISA), mass spectrometry, and flow cytometry. The QAs to be evaluated include the physicochemical properties, biological activity, immunochemical properties, purity and impurities, quantity, strength, thermal stability profiles, and other modifications, such as oxidation and deamination
[11][13][18].
The PK comparison involves evaluating the PK parameters, as well as analyzing the composition, physical properties, primary structures (amino acid sequence and disulfide bond), and higher-order structures (e.g., local and three-dimensional conformation) of the biosimilar. As mentioned above, it is important to verify that the amino acid sequence matches the RP and to compare the N- and C-terminal amino acid sequences, free SH groups, and disulfide bonds. If there are PTMs, such as glycosylation, oxidation, and deamination, these must also be defined. If their presence is verified, it is necessary to compare the carbohydrate structures, including the glycan profiles and glycosylation patterns. The assessment of the biological effects relies on the characteristics of the substance and generally comprises mAb–antigen binding, Fcγ receptor binding, and FcRn binding
[13]. The characterization of the purity and impurities related to the product and its manufacturing process helps to guarantee its safety, and they must be determined and compared qualitatively and quantitatively, through a combination of analytical procedures
[18].
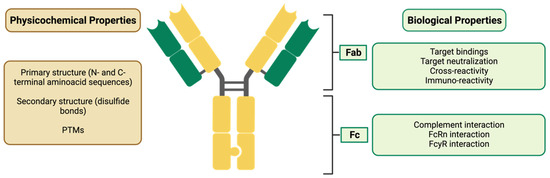
In the case of biosimilar mAbs (for which some of their QAs are represented in Figure 3), assessments of the biological activity to bind to the Ag, the connection to the Fcg receiver, the FcRn binding, the antibody-dependent cellular cytotoxicity (ADCC), and the complement-dependent cytotoxicity (CDC) must also be carried out. In addition, the affinity and Ag binding specificity of the biosimilar and the RP must be compared.
Figure 3. Characterization of a biosimilar mAb, evidencing its physicochemical and biological properties. Fab: fragment antigen binding; Fc: fragment crystallizable; FcRn: neonatal Fc receptor; FcγR: Fc-gamma receptor.
When considering the efficacy and safety of monoclonal antibodies (mAbs), the importance of differences in biological activity related to the Fc regions will vary depending on the mAb’s mechanism of action. If the mAb exerts ADCC activity, it is important to carefully consider the difference in FcgRIIIa binding and ADCC activity. On the other hand, if the mAb’s mode of action does not include ADCC activity (e.g., an mAb against soluble monomer Ag), it is possible that the difference in binding will not significantly impact the mAb’s efficacy and safety
[11].
The folding and conformation of the CH2 domain (Abs are flexible macromolecules consisting of two identical light chains and two heavy chains, also identical, alloys through dissulfide bridges. The heavy chains have a variable domain (VH), with great diversification in their aminoacid composition and three constant domains (CH1, CH2 and CH3), which present homogeneous amino acid sequences responsible for the effector actions of immunoglobulins (CH2 and CH3). The light chains are formed by a constante domain (CL) and a variable domain (VL). The CH2 and CH3 domains the Fc region (crystalline fragment–effector), while VH, VL, CH1 and CL constitute the Fab) within the Fc region of the IgGs depends on the N-glycans attached. Glycosylation causes the CH2 domains of IgGs to have higher stability, unlike deglycosylation, which causes instability, making them more prone to unfolding and disaggregation. It is also known that N-glycosylation has an impact on ADCC and CDC activity, through the modulation of the connection to the Fcγ receiver.
3.2.2. Establishing Non-Clinical Biosimilarity
The purpose of the non-clinical comparability exercise is to evaluate how alike the biosimilar and the RP are in regard to their mechanisms of action, their functional activity, and their quality characteristics
[44]. Physicochemical and laboratory analyses are conducted in vitro during PD studies in which ligands bind to the physiological targets and the physiological effects on cells are evaluated. They may activate or inhibit the receptor; thus, the cell function may increase or decrease. The preclinical comparison of the PK and PD can help to minimize residual uncertainty regarding their similarity
[18].
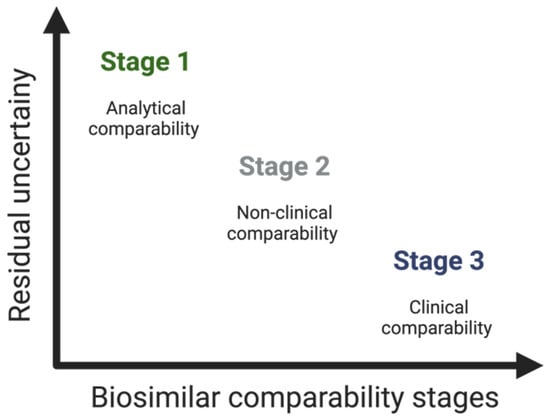
3.2.3. Clinical Considerations—The Supporting Role of Phase I and Phase III Clinical Studies
Clinical studies constitute the third stage of the comparability study
[45]. As mentioned, the establishment of biosimilarity focuses, above all, on preclinical aspects and, particularly, on the quality of the biosimilar. Thus, the number and scope of clinical studies executed rely on the level of uncertainty regarding biosimilarity, determined by previous analytical assessments (and non-clinical in vivo testing, if executed)—see
Figure 4 [13][18].
Figure 4. Residual uncertainty along the stages of the comparability study of biosimilars.
Briefly, in this third step of the comparability study, the aim is, through the analysis of analytical data or previous studies, to rule out clinically relevant PK/PD, clinical safety (immunogenicity), efficacy, extrapolation, and PV differences—and thus confirm the biosimilarity of the proposed biosimilar in relation to the RP
[16][18][22].
Pharmacokinetic and Pharmacodynamic Studies
The process of developing a biosimilar typically starts with a study that aims to prove that the proposed biosimilar is similar in terms of its PK and PD properties when compared to the RP
[18]. The study design depends on several factors, namely the clinical context, safety, and PK of the RP. Therefore, it is only carried out after being extensively characterized
[11][13].
PK assessments are needed to compare the biodisponibility of the drug, which includes absorption, disposition, time dependence, and binding to blood components. PD studies, in turn, ensure that the biosimilar’s efficacy in the target tissue is equivalent to that of the RP and that the mechanism of action is identical. In some cases, comparative PK/PD studies might be enough to show that the clinical outcomes are similar
[13].
Efficacy Studies
Based on the current regulatory guidelines, certain situations do not require comparative clinical efficacy studies. The FDA states that a comparative clinical study is necessary “if there is residual uncertainty about whether there are clinically meaningful differences” between the proposed biosimilar and the RP, “based on structural and functional characterization, animal testing, human PK and PD data, and clinical immunogenicity assessment”
[43]. The type and amount of clinical data needed depend on the complexity of the therapeutic mechanism, as well as the availability of an endpoint that correlates with effectiveness. The EMA has waived the need for rigorous comparative efficacy, safety, and immunogenicity studies in certain situations, while still emphasizing the importance of comparative PK/PD studies
[10][42].
Efficacy studies make it possible to analyze the significant differences that exist in terms of treatment efficacy, i.e., their main goal is not necessarily to prove effectiveness, but rather to confirm that the clinical performance is comparable
[13]. These studies require randomized parallel-group comparative clinical trials (preferably double-blind trials), as well as appropriate efficacy endpoints. The detection of potential differences related to a product should be sensitive enough to ensure that any impact caused by individual- or disease-related factors is decreased.
Safety Evaluation
The safety issues related to the biosimilar play a major role in comparability studies. According to the usual procedures in the development of biological medicines, the biosimilar’s safety profile is built across the entire clinical program—during phase I PK/PD studies and phase III direct comparison studies
[13].
To establish the similarity between a biosimilar and its RP, it is necessary to assess and compare the type, severity, and frequency of any adverse events (AEs) that may occur. Additionally, any potential safety risks arising from variations in the manufacturing process must be taken into consideration
[13].
3.3. Regulatory Concerns
In the last fifteen years, the EMA and/or the FDA have given their approval to approximately one hundred biosimilars, and this number is predicted to grow even more in the next years. For their approval, it is necessary to ensure robust regulation
[15][16]. Regulatory bodies and manufacturers are mainly responsible for guaranteeing that biosimilars intended for use closely resemble the RP in both structure and function. This is essential to prevent any adverse effects on the efficacy and safety of the biosimilar. By means of thorough evaluation and demonstration, any structural discrepancies that may have an impact on the clinical outcomes must be eliminated
[4].
Biosimilars have very particular and exclusive approval conditions, and implementing an abbreviated licensing pathway can present several challenges
[18]. Therefore, the EMA and the FDA have developed specific and consistent regulatory guidelines that must be followed to approve the final product
[12][42]. However, divergences exist between the regulatory agencies, so the number of biosimilars approved in the various markets is very different
[3]. The EMA was the pioneer among regulatory agencies in establishing a system for the approval of biosimilars in 2003. Then, in 2006, the first biosimilar (Ominotrope
®) was approved
[10]. Almost a decade later, the EMA was followed by the FDA, which adopted the same principles and approved the first biosimilar in 2015
[10]. Both agencies recognized the complexity of developing biosimilars, highlighting the fact that each biosimilar candidate has distinct and particular characteristics, making it crucial to design targeted development programs on a case-by-case basis. The WHO has made recommendations to promote global growth regarding these products, requiring at least the existence of clinically relevant parameters, including PK, PD, safety, efficacy, and immunogenicity data, to consider biosimilarity
[13][18][46].
US and EU laws state that an RP must be approved according to the legislation established by local authorities. While, in the US, the proposed biosimilar must demonstrate its similarity to the RP approved there in order to be accepted, for EU approval, it must be similar to the RP with approval in the European Economic Area (EEA)
[17][18]. These regulatory agencies have implemented measures to allow the use of foreign comparators in comparative clinical studies, if scientifically justified, due to the complexity and cost involved in developing biosimilars. From a regulatory and legislative perspective, the acceptability of reliance on clinical data generated using a comparator of foreign origin depends on the successful establishment of the “scientific bridge”. The “scientific bridge” between the product of local origin and that of foreign origin must consist of a comprehensive assessment of the analytical similarity of the proposed biosimilar with both comparators. In addition, a three-arm PK similarity analysis must be conducted. This analysis should confirm the bioequivalence between both comparator arms and also the candidate biosimilar in contrast to the respective comparators
[18][30].
Another regulatory consideration, exclusive to the US, is the determination of interchangeability. Under US guidelines, an interchangeable biosimilar is a product that is biosimilar to the RP, and it is hypothesized that the intended therapeutic result will be attained in all individuals
[47]. When the RP is substituted with a biosimilar, the risk regarding efficacy and safety cannot be greater than if there is no substitution. Prerequisites for interchangeability are not required in the EU regulatory framework
[10].
To ensure the successful acceptance of biosimilars, it is important to establish clear naming criteria and effective safety monitoring. In this regard, to achieve unique names, a four-letter suffix is added to the international nonproprietary name (INN). This helps to prevent misconceptions among pharmacists and ensures that adverse events are accurately attributed to the correct manufacturer. Proper PV is essential for the safe and effective use of biosimilars
[18][30].
 +1 credit
+1 credit