Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Andrew Willetts | -- | 2497 | 2024-02-26 09:20:17 | | | |
| 2 | Lindsay Dong | Meta information modification | 2497 | 2024-02-26 09:44:45 | | | | |
| 3 | Lindsay Dong | Meta information modification | 2497 | 2024-02-26 09:45:26 | | | | |
| 4 | Lindsay Dong | Meta information modification | 2497 | 2024-03-18 04:07:27 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Willetts, A. Role of Dioxygen in Microbial Bio-Oxygenation. Encyclopedia. Available online: https://encyclopedia.pub/entry/55439 (accessed on 29 July 2026).
Willetts A. Role of Dioxygen in Microbial Bio-Oxygenation. Encyclopedia. Available at: https://encyclopedia.pub/entry/55439. Accessed July 29, 2026.
Willetts, Andrew. "Role of Dioxygen in Microbial Bio-Oxygenation" Encyclopedia, https://encyclopedia.pub/entry/55439 (accessed July 29, 2026).
Willetts, A. (2024, February 26). Role of Dioxygen in Microbial Bio-Oxygenation. In Encyclopedia. https://encyclopedia.pub/entry/55439
Willetts, Andrew. "Role of Dioxygen in Microbial Bio-Oxygenation." Encyclopedia. Web. 26 February, 2024.
Copy Citation
Dioxygen (O2, more commonly referred to as molecular oxygen) is the only element in the Earth’s environment that is paramagnetic in its ground-state. Dioxygen-dependent enzymes (principally mono- and dioxygenases) play in relevant aspects of bio-oxygenation. This is reflected by the multiple strategic roles that dioxygen -dependent microbial enzymes play both in generating valuable synthons for chemoenzymatic synthesis and in facilitating reactions that help to drive the global geochemical carbon cycle.
dioxygen
molecular oxygen
Biodegradation
1. Introduction
Dioxygen (O2, more commonly referred to as molecular oxygen) is the only element in the Earth’s environment that is paramagnetic in its ground-state [1]. This energetically favoured state is determined by the molecule preferentially existing as a triplet diradical (3Σ), with its two least strongly held electrons, the ones most responsible for its chemistry, occupying two separate spin-paired anti-bonding π* orbitals [2]. This in turn dictates that a concerted reaction between O2 and carbon in organic compounds is spin-forbidden [3]. However, evolution has generated a significant number of enzymes able to oxygenate organic substrates by the prior activation of molecular oxygen to a 1-electron reduced form (O2−), either by coopting an organic radical (e.g., a flavin nucleotide), or deploying a transition element (characterised by partially filled outer-bonding orbitals), which may (e.g., heme-coordinated iron) or may not (e.g., nonheme iron) be coordinated into an organic cofactor [4][5].
While the first indirect evidence of the involvement of molecular oxygen in an enzyme-catalysed reaction emerged in the mid-1930s from studies of an FMN-dependent NADPH oxidoreductase (Old Yellow Enzyme) isolated from the bottom of fermenting yeast Saccharomyces carlesbergensis [6], it was over two decades later before direct confirmation of bio-oxygenation reactions catalysed by microbial enzymes was advanced as a result of similar programmes of research conducted by Osamu Hayaishi and Howard Mason. Hayashi used mass spectroscopy to established that an enzyme he confirmed contained a nonheme atom of the transition element iron (Fe3+) was able to incorporate both atoms of 18O2 in biotransforming catechol to cis,cis-muconate (Figure 1, [7]). He initially termed the enzyme, sourced from an environmental microbial isolate, ‘pyrocatechase’. Concurrently, Mason used the same technique to confirm that an enzyme he called ‘mushroom phenolase’, which was devoid of any form of iron but did contain a bound atom of the transition element copper (Cu2+), was able to biotransform 3,4-dimethylphenol to 4,5-dimethylcatechol by incorporating a single atom of 18O2 [8]. In an attempt to distinguish between the two enzyme types, Mason subsequently suggested that the terms ‘mixed-function oxidase’ and ‘oxygen transferase’ should be used to discriminate between the respective modes of action of ‘mushroom phenolase’ and ‘pyrocatechase’ [9]. However, this proposal was not received favourably by Hayaishi, who countered by advancing the corresponding names ‘monooxygenase’ and ‘dioxygenase’ to better reflect the differing outcomes of these two types of O2-dependent enzymes [10]. Significant in that respect, Hayaishi’s functionally more relevant terms have stood the test of time, and are still widely used decades later to describe many of the other disparate examples that have been subsequently recognised [11]. Semantic disagreements apart, Mason and Hayaishi’s pioneering studies promoted a substantial body of ongoing research to better characterise the biochemistry deployed by the two enzyme types in facilitating their respective interactions with molecular oxygen. However, a fully comprehensive understanding for each enzyme type has proved illusive, with a number of aspects still currently remaining works in progress. This is well illustrated by the reviewing the evolution of the perceived mode of action of 2,5-diketocamphane 1,2-monooxygenase (2,5-DKCMO). This monooxygenase is one of a suite of inducible enzymes coded for exclusively by corresponding genes located on the CAM plasmid of Pseudomonas putida ATCC 17453 [12][13][14][15] that collectively serve to biodegrade camphor to central intermediary metabolites (Figure 2, [16]).
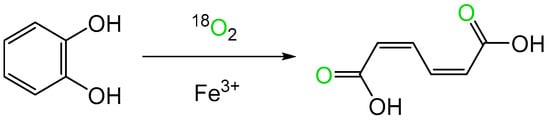
Figure 1. Bio-oxygenation of catechol to cis,cis-muconate by ‘pyrocatechase’ (catechol 1,2-dioxygenase) from a cell-free extract of a microbial soil isolate.
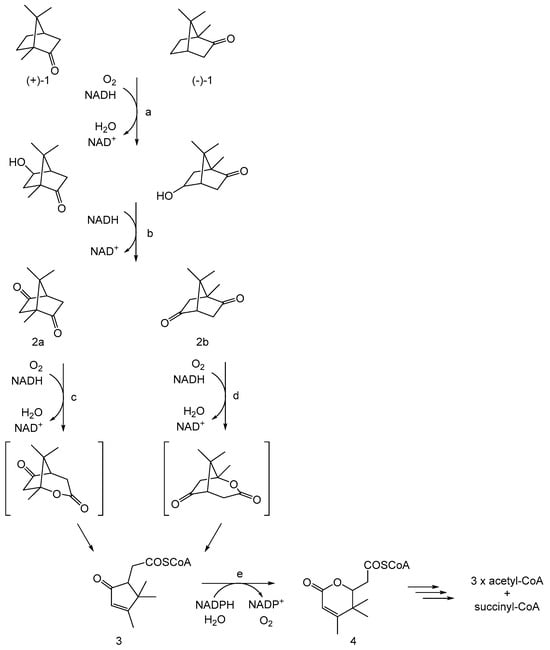
Figure 2. CAM plasmid-coded pathway of (rac)-camphor degradation by Pseudomonas putida ATCC 17453: adapted from [16]. a = cytochrome P450 monooxygenase (camCAB); b = hydroxycamphor dehydrogenase (camD); c = 2,5-diketocamphane 1,2-monooxygenase (camE25-1 + camE25-2); d = 3,6-diketocamphane 1,6-monooxygenase (camE36); e = 2-oxo-Δ3-4,5,5,-trimethylcyclopentenylacetyl-CoA monooxygenase (camG); (+)-1 and (-)-1 = (R)- and (S)-enantiomers of camphor, respectively; 2a = 2,5-diketocamphane; 2b = 3,6-diketocamphane; 3 = 2-oxo-Δ3-4,5,5-trimethylcyclopentenylacetyl-CoA; 4 = 3,4,4-trimethyl-Δ2-pimelyl-CoA lactone.
2. The Rise and Subsequent Fall of a Metal Ion-Dependent Monooxygenase
2,5-DKCMO is notable for being the O2-dependent activity that serves a key role in triggering the initial ring-opening step in the pathway of bicyclic (+)-camphor catabolism. Additionally, it has one indisputable claim to fame. It was the first enzyme reported to function as a biological Baeyer–Villiger monooxygenase (BVMO, [17][18][19]). BVMOs are so named because they bio-oxygenate ketones into corresponding lactones/esters, a transformation directly equivalent to the peracid-catalysed chemical reaction first reported by Adolf Baeyer and Victor Villiger in 1899 [20]. Although the lactonization by various bacteria and fungi of the D-ring of 4-androstene-3,17-dione to testololactone had been recognised by 1953 [21][22], the nature of the enzyme(s) responsible remained uncharacterised for several more years. Then, subsequently, definitive proof of the role of molecular oxygen in another microbial lactonization, the initial ring-opening step of camphor degradation by P. putida ATCC 17453, was obtained in late 1961 by Irwin Gunsalus at the University of Illinois [23].
Triggered by this initial observed outcome, Gunsalus then focussed his attention on establishing in more detail the mode of action of the ketolactonase that resulted in it being able to catalyse the spin-forbidden O2-dependent biotransformation of the DKC enantiomer. At the time, the precedents set by other O2-dependent enzymes had identified a number of alternative cofactor-dependent strategies for enabling inert molecular oxygen to participate in one electron-state biochemistry. These included the seminal recognised deployment of nonheme iron (catechol 1,2-dioxygenase, [7]), or copper (3,4-dimethylphenol monooxygenase, [8]), as well as other less directly confirmed options that were dependent on either heme-coordinated iron (tryptophan 2,3-dioxygenase, [24]), or FAD (D-amino acid oxidase, [25]). The then current protocols for discriminating between these alternative possibilities were based on either characteristic absorbance spectra (both flavin nucleotide- and heme iron-dependent enzymes), or the effect of metal chelating agents (iron- and copper-dependent enzymes, [26]).
Gunsalus initially established [27][28][29][30][31] that the lactonizing activity was dependent on the combined activities of two enzymes (E1 and E2) which upon purification were both confirmed to bind FMN. E1 was monomeric, (estimated MW 50,000 kDa), bound NADH but not NADPH, had an absorption spectrum that did not exhibit a characteristic heme-generated Soret band in response to CO, was not inhibited by divalent metal ion chelating agents (bipyridyl = Fe2+; NaN3 = Cu2+), and was able to donate electrons to decolourize methylene blue. Conversely, E2 was also reported to be monomeric (estimated MW 80,000 kDa), did not exhibit a Soret band or the ability to reduce methylene blue, but was strongly inhibited by bipyridyl but not NaN3. Gunsalus interpreted this initial data to conclude that both E1 and E2 were flavoproteins, both physically and functionally linked as a multienzyme complex in which the flavin nucleotide served as a molecular bridge or conduit to channel reducing power between the two activities. E1 was termed NADH oxidase, to reflect Mason’s terminology [9], and functionally served to reduce FMN to FMNH2 independent of any involvement of either Cu2+, Fe2+, or heme iron.
3. The Fallow Years
After a hiatus of over a decade, interest in 2,5-DKCMO was rekindled at the University of Aberystwyth by Peter Trudgill, one of Gunsalus’ former collaborators at Illinois. Significantly, in the intervening years several relevant things had changed, including better ways of purifying and characterising enzymes (e.g., gel-filtration and ligand-affinity chromatography, plus SDS-PAGE and Phast gel electrophoresis). Also highly relevant was that studies driven by Vincent Massey [32], David Ballou [33][34], and Thomas Bruice [35][36] had led to a better understanding of the interactions between reduced nicotinamide nucleotides and flavin nucleotides that serve to activate molecular oxygen by forming key C4a-peroxy- and C4a-hydroperoxyflavin moieties which could then drive bio-oxygenations reactions (Figure 3, [37]).
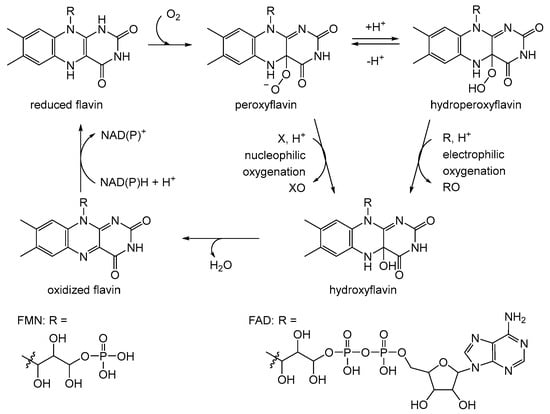
Figure 3. Generalised summary of the reactions undergone by flavin nucleotide cofactors of canonical flavoprotein monooxygenases in incorporating molecular oxygen as a participating reactant. X = ketone; XO = lactone/aldehyde; R = organosulfide; RO = sulfoxide.
4. The Relevance of the Correlation with Other Classes of Monooxygenase
At around the same time, an interdisciplinary team at the University of Exeter led by Stanley Roberts began using the NADH-dependent 2,5-DKCMO as a biocatalyst to generate key synthons for valuable chemoenzymatic syntheses [38][39][40][41][42][43]. While NADPH-dependent cyclohexanone monooxygenase (CHMO) from the Acinetobacter sp. strain NCIB 9871 had previously been developed successfully as such a lactone-generating enzyme [44], the significantly cheaper cost of NADH was the principal driver for this Exeter initiative. A complementary screening programme run by Raffaella Villa was also undertaken to include other known potentially useful NADH-dependent bio-oxygenating enzymes [45]. One important outcome from this approach was to establish a number of functional and homology similarities between the ketolactonase and a number of previously well-studied bacterial luciferases. Strongly supporting this recognised relationship was the novel reported ability of the luciferases, whose natural evolved substrate is the aliphatic aldehyde dodecanal [46], to lactonize the same abiotic alicyclic ketones as 2,5-DKCMO. As a result, for the first time, these enzyme types were grouped together as Type II BVMOs [47] to distinguish them from the corresponding NADPH-dependent Type I flavoprotein enzymes such as CHMO.
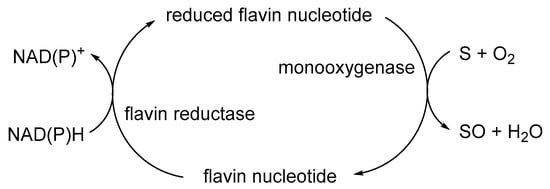
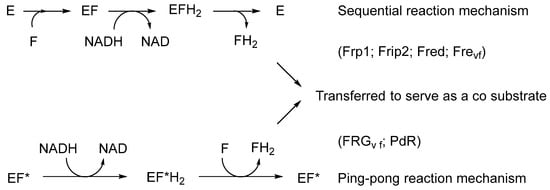
The initial catalyst to better comprehending that biochemistry was a review by Holly Ellis [48] that served to relate the FMN-dependence of known bacterial luciferases to a small number of other functionally equivalent recently recognised bacterial monooxygenases. Ellis introduced the term two-component flavin-dependent monooxygenases (fd-TCMOs) to define the new grouping. Most significantly, fd-TCMOs are not true flavoproteins. Both structurally and functionally, they are clearly unrelated to the canonical flavoprotein monooxygenases such as CHMO from the Acinetobacter sp. strain NCIB 9871 that deploy a bound flavin nucleotide cofactor [44]. Rather, fd-TCMOs operate as two clearly distinct half-reactions, with a flavin reductase (FR) serving to reduce FMN as a substrate to FMNH2 which is then transferred to serve as a cosubstrate for a monooxygenase which catalyses the bio-oxygenating activity of the second half-reaction (Figure 4). Some of these FRs are flavoproteins that operate a ping-pong mechanism to reduce a second external FMN substrate, whereas others are not, being nonflavoprotein reductases that reduce the FMN substrate by a sequential mechanism (Figure 5).
Figure 4. Generalised summary of the two clearly distinct reductive and oxidative half-reactions that characterise flavin-dependent two-component monooxygenases in incorporating molecular oxygen as a participating reactant.
Figure 5. The different types (bound vs. unbound flavin) and different reaction mechanisms (sequential vs. ping-pong) of flavin reductases. E = nonflavoprotein flavin reductase; EF* = flavoprotein flavin reductase; F = FMN.
5. Confirming the Role of 2,5-DKCMO as a Two-Component Monooxygenase
Direct evidence to support the new status of 2,5-DKCMO then followed from a study undertaken by a multinational research group led by Peter Lau that deployed genomic tools to further characterise the biochemistry of some of the key enzymes of the camphor biodegradation pathway of P. putida ATCC 17453 [49]. Notably, the group published a seminal 2013 study which specifically achieved two principal goals with respect to 2,5-DKCMO [50]. Firstly, for the first time, the complete sequence of the DNA of the 533 kb double-stranded linear CAM plasmid [12][13][14][15] was established, and homology searching was used to assign functions to every one of its orfs. This was an important outcome because it confirmed directly that the plasmid encodes relevant genes for all the characterised enzymes of both the (+)-camphor (2,5-DKCMO-dependent) and (-)-camphor (3,6-DKCMO-dependent) degradation pathways (Figure 2). Further, for the first time it became apparent that 2,5-DKCMO functions as a mixture of two very similar isoenzymic forms (60% overall similarity) coded for by two genes (camE25-1 and camE25-2) located on opposite stands of the plasmid. Secondly, the 2013 study identified and characterised Fred, a chromosomal DNA-coded 2 × 18 kDa homodimeric nonflavoprotein reductase that reduced FMN as an external substrate (Figure 5).
6. As the Area of Light Expands, So Does the Perimeter of Darkness
Further insights into the newly recognised status of 2,5-DKCMO as an fd-TCMO then emerged from subsequent studies that scrutinised the nature and extent of those FR activities able to support the ketolactonase throughout the successive trophophasic and idiophasic stages of growth of P. putida ATCC 17453 in a camphor-based minimal medium [51][52]. While supporting the finding of the 2013 report [50] that Fred was an inducible chromosome-coded dimeric (2 × 18 kDa) nonflavoprotein reductase, these new investigations revealed that it was only present in detectible levels in late trophophase (primary metabolism) and subsequent idiophase (secondary metabolism). In this respect, it resembles the equivalent ‘tailoring FRs’ involved in the late stage synthesis of some antibiotics [53][54], and a wide range of other microbial secondary metabolites [55][56][57][58][59][60]. Circumstantial evidence also suggests that Fred may correspond to the NADH oxidase activity of camphor-grown P. putida ATCC 17453 previously reported by both Gunsalus [27][28][29][30][31][61][62][63][64][65][66] and Trudgill [67][68][69][70][71][72][73], who both focussed their research exclusively camphor-grown cells entering idiophasic growth, a time frame chosen specifically to optimise biomass yield [72]. Trudgill conclusively confirmed by two different methods that the MW of the active form of the enzyme was 36 kDa, but significantly never subjected it to SDS-PAGE analysis.
Conversely, the equivalent role throughout trophophase (primary metabolism) was fulfilled by a combination of three newly recognised competent reductases. Two of these, Frp1 (27.0 kDa) and Frp2 (28.5 kDa), were constitutive monomeric nonflavoprotein ferric (flavin) reductases coded for by chromosomal DNA. Structurally, both corresponded closely with other well-characterised ferric (flavin) reductases and the related ferrodoxin reductases, a group of functionally related FRs that have been confirmed to be widely distributed in other prokaryotes [74][75].
By way of contrast, the third relevant activity after extensive purification was confirmed by sequence homology to be putidaredoxin reductase (PdR, MW 45.6 kDa), an inducible flavoprotein coded for by the camA gene on the CAM plasmid, and characterised by a tightly bound FAD cofactor [76][77][78][79]. This particular reductase was already known to function as one of the three activities (camABC) that collectively serve as cytochrome P450 monooxygenase (cytP450MO), the well-studied multienzyme complex that hydroxylates camphor [80][81][82], thereby initiating the terpene biodegradation pathway in P. putida ATCC 17453 (Figure 2).
The proven ability of PdR to satisfy the FMNH2 requirement of 2,5-DKCMO has additional strategic significance because it supports the concept that this key bio-oxygenating enzyme, responsible for initiating the ring cleavage of the bicyclic terpene (+)-camphor, can function independently of the chromosomal DNA of P. putida ATCC 17453. This contrasts with the previous proposals made by both Gunsalus and Trudgill that the monooxygenase is dependent on a 36 kDa chromosome-coded NADH oxidase as its source of FMNH2 (vide supra). This aspect of the functional independence of 2,5-DKCMO from chromosomal DNA was then supported further by using the known inhibitory effect of Zn2+ on ferric (flavin) reductases [83][84][85][86].
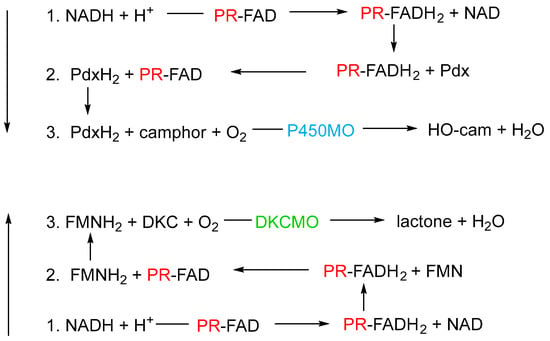
Currently there still remain a number of incompletely understood or unresolved aspects of the bio-oxygenating activity of 2,5-diketocamphane 1,2-monooxygenase from camphor-grown P. putida ATCC 17453. One important issue is to develop a more comprehensive understanding of the biochemical logistics that enable PdR to serve both as a redox partner for 2,5-DKCMO and as an integral member of the trimeric cytP450MO bio-oxygenating consortium (Figure 6).
Figure 6. Schematic of the two contrasting roles of putidaredoxin reductase (PR) in camphor-grown P. putida ATCC 17453. Reaction 1 is common to both roles. In both cases, the sequence of reactions progresses from one to three. P450MO = cytochrome P450 mono-oxygenase; Pdx = putidaredoxin; DKCMO = 2,5-diketocamphane 1,2-mono-oxygenase; DKC = 2,5-diketocamphane.
References
- Torres, M.E.S.; Sancedo-Vazquez, J.P.; Kroneck, P.M.H. The magic of dioxygen. In Sustsaining Life on Planet Earth: Metalloenzymes Mastering Dioxygen and Other Chewy Gases; Kroneck, P.M.H., Torres, M.E.S., Eds.; Springer International Publishing: Basel, Switzerland, 2015; pp. 1–12.
- Borden, W.T.; Hoffmann, R.; Stuyver, T.; Chen, B. Dioxygen: What makes this triplet diradical kinetically persistent? J. Am. Chem. Soc. 2017, 139, 9010–9018.
- Holtmann, D.; Hollmann, F. The oxygen dilemma: A severe challenge for the application of monooxygenases? ChemBioChem 2016, 7, 1391–1398.
- Phintha, A.; Chaiyen, P. Unifying and versatile features of flavin-dependent monooxygenases: Diverse catalysis by a common C4a-(hydro)peroxyflavin. J. Biol. Chem. 2023, 299, 105413.
- Torres Pazmino, D.E.; Winkler, M.; Glieder, A.; Fraaije, M.W. Monooxygenases as biocatalysts: Classification, mechanistic aspects and biotechnological applications. J. Biotechnol. 2010, 146, 9–24.
- Theorell, H. Preparation in pure state of the effect group of yellow enzyme. Biochem. Z. 1935, 275, 344–346.
- Hayaishi, O.; Katagiri, M.; Rothberg, S. Mechanism of the pyrocatechase reaction. J. Am. Chem. Soc. 1955, 77, 5450–5451.
- Mason, H.S.; Fowlks, W.L.; Peterson, E. Oxygen transfer and electron transport by the phenolase complex. J. Am. Chem. Soc. 1955, 77, 2914–2915.
- Mason, H. Mechanisms of oxygen metabolism. Science 1957, 125, 1185–1188.
- Hayaishi, O.; Katagiri, M.; Rothberg, S. Studies on oxygenases. J. Biol. Chem. 1957, 229, 376–386.
- Hayaishi, O. An odyssey with oxygen. Biochem. Biophys. Res. Commun. 2005, 338, 2–6.
- Chakrabarty, A.M.; Gunsalus, C.F.; Gunsalus, I.C. Transduction and clustering of genes in fluorescent pseudomonads. Proc. Nat. Acad. Sci. USA 1968, 60, 168–175.
- Rheinwald, J.G.; Chakrabarty, A.M.; Gunsalus, I.C. A transmissible plasmid controlling camphor oxidation in Pseudomonas putida. Proc. Nat. Acad. Sci. USA 1973, 70, 885–889.
- Shaham, M.; Chakrabarty, A.M.; Gunsalus, I.C. Camphor plasmid-mediated chromosomal transfer in Pseudomonas putida. J. Bacteriol. 1973, 116, 944–949.
- Chakrabarty, A.M. Plasmids in Pseudomonas. Ann. Rev. Genet. 1976, 10, 7–30.
- Willetts, A. Characterised flavin-dependent two-component monooxygenases from the CAM plasmid of Pseudomonas putida ATCC 17453 (NCIMB 10007). Ketolactonases by another name. Microorganisms 2019, 7, 1.
- Bradshaw, W.H.; Conrad, H.E.; Corey, E.J.; Gunsalus, I.C.; Lederer, D. Microbial degradation of (+)-camphor. J. Am. Chem. Soc. 1959, 81, 5507.
- Conrad, H.E.; Corey, E.J.; Hedegaard, J.; Paisley, N.S.; Gunsalus, I.C. Terpenoid metabolism: Oxidation and ring cleavage reactions. In Proceedings of the 4th International Congress of Biochemistry, Moscow, Russia, 25 August–2 September 1961; Volume 29, p. 284.
- Hedegaard, J.; Conrad, H.E.; Gundsalus, I.C. Camphor oxidation: Pathways and enzyme induction. Bacteriol. Proc. 1961, 61, 183, Abstract P66.
- Baeyer, A.; Villiger, V. Einwirkung des caro’schen reagens auf ketone. Ber. Dtsch. Chem. Ges. 1899, 32, 3625–3633.
- Fried, J.; Thoma, R.H.; Klingberg, A. Oxidation of steroids by microorganisms. III. Side chain degradation, ring D cleavage, and dehydrogenation of the A ring. J. Am. Chem. Soc. 1953, 75, 5764–5765.
- Peterson, D.H.; Eppstein, S.H.; Meister, P.D.; Murray, H.C.; Leigh, H.M.; Weintraub, A.M.; Reineke, L.M. Microbial transformation of steroids, IX. Degradation of C21 steroids to C19 lactones and testololactone. J. Am. Chem. Soc. 1953, 75, 5768–5769.
- Conrad, H.E.; DuBus, R.; Gunsalus, I.C. An enzyme system for cyclic lactonization. Biochem. Biophys. Res. Commun. 1961, 6, 293–297.
- Klingenberg, M. Pigments of rat liver microsomes. Arch. Biochem. Biophys. 1958, 75, 376–386.
- Warburg, O.; Christian, W. Isolation of the prosthetic group of the amino acid oxidase. Biochem. Z. 1938, 298, 150–168.
- Auld, D.S. Use of chelating agents to inhibit enzymes. Meth. Enzymol. 1988, 158, 110–114.
- Conrad, H.E.; DuBus, R.; Gunsalus, I.C. Enzymatic lactonization: A possible role for mixed function oxidase action. Fed. Proc. 1962, 21, 52, Abstract 031–038.
- Bertland, A.U.; Johnson, S.; Gunsalus, I.C. Induced enzymes in terpene metabolism: An FMN-coupled DPNH oxidase in pseudomonads. Bacteriol. Proc. 1963, 105, 44, Abstract P44.
- Conrad, H.E.; DuBus, R.; Lieb, I.C.; Gunsalus, I.C. Enzymatic keto-lactonization: Inhibition of mixed function oxidation. In Proceedings of the 6th International Congress of Biochemistry, New York, NY, USA, 26 July–1 August 1964. 301 Abstract 32.
- Conrad, H.E.; Trudgill, P.W.; Gunsalus, I.C. Oxidative enzyme systems in the metabolism of camphor enantiomers. In Proceedings of the 148th Meeting of the American Society of Chemistry, Chicago, IL, USA, 10–14 June 1964. Abstract Q3.
- Conrad, H.E.; DuBus, R.; Namtvedt, M.J.; Gunsalus, I.C. Mixed function oxidation. II. Separation and properties of the enzymes catalysing camphor lactonization. J. Biol. Chem. 1965, 240, 495–503.
- Massey, V. Activation of molecular oxygen by flavins and flavoproteins. J. Biol. Chem. 1994, 269, 22459–22462.
- Entsch, B.; Ballou, D.P.; Massey, V. The mechanism of action of xanthine oxidase. J. Biol. Chem. 1974, 249, 4363–4382.
- Vermilion, J.; Ballou, D.; Massey, V.; Coon, M.J. Separate roles for FMN and FAD in catalysis by liver microsomal NADPH-cytochrome P-450 reductase. J. Biol. Chem. 1981, 256, 266–277.
- Ball, S.; Bruice, T.C. 4a-Hydroperoxyflavin N-oxidation of tertiary amines. J. Am. Chem. Soc. 1979, 101, 4017–4023.
- Bruice, T.C. Oxygen-flavin chemistry. Isr. J. Chem. 1984, 24, 54–61.
- Robbins, J.M.; Ellis, H.R. Investigations of two-component flavin-dependent monooxygenase systems. Meth. Enzymol. 2019, 620, 399–422.
- Grogan, G.; Roberts, S.M.; Willetts, A. Camphor-grown Pseudomonas putida, a multifunctional biocatalyst for oxygenations. Biotechnol. Lett. 1993, 15, 913–918.
- Grogan, G.; Roberts, S.M.; Wan, P.; Willetts, A. Some Baeyer-Villiger oxygenations using monooxygenases from Pseudomonas putida. J. Chem. Soc. Chem. Commun. 1993, 699–701.
- Gagnon, R.; Grogan, G.; Roberts, S.M.; Villa, R.; Willetts, A. Enzymatic Baeyer-Villiger oxidations of some bicyclic ketones using monooxygenases from Pseudomonas putida NCIMB 10007: Enantioselective preparation a precursor of azadirachtin. J. Chem. Soc. Perkin Trans. I 1995, 1505–1511.
- Beecher, J.E.; Grogan, G.; Roberts, S.M.; Wan, P.; Willetts, A. Oxidative biotransformations by microorganisms: Enantioselective biooxidations by the enantiocomplementary diketocamphane monooxygenase isoenzymes from Pseudomonas putida NCIMB 10007. Biotechnol. Lett. 1996, 18, 571–576.
- Alphand, V.; Furstoss, R.; Pedragosa-Moreau, S.; Roberts, S.M.; Willetts, A. Comparison of microbiological and enzymatically mediated Baeyer-Villiger oxidations: Synthesis of optically active caprolactones. J. Chem. Soc. Perkin Trans. I 1996, 1867–1871.
- Pasta, P.; Carrea, G.; Gagerro, N.; Grogan, G.; Willetts, A. Enantioselective oxidations catalysed by diketocamphane monooxygenase from Pseudomonas putida with macromolecular NAD in a membrane reactor. Biotechnol. Lett. 1996, 18, 1123–1127.
- Chen, Y.; Peoples, O.; Walsh, C. Acinetobacter cyclohexanone monooxygenase: Gene cloning and sequence determination. J. Bacteriol. 1988, 170, 781–789.
- Villa, R.; Willetts, A. Oxidations by microbial NADH plus FMN-dependent luciferases from Photobacterium phospohoreum and Vibrio fischeri. J. Mol. Catal. B Enzym. 1997, 2, 193–197.
- Hastings, J.W.; Nealson, K.H. Bacterial bioluminescence. Ann. Rev. Microbiol. 1971, 31, 549–595.
- Willetts, A. Structural studies and synthetic applications of Baeyer-Villiger monooxygenases. Trends Biotechnol. 1998, 15, 55–62.
- Ellis, H.R. The FMN-dependent two-component monooxygenase systems. Arch. Biochem. Biophys. 2010, 497, 1–12.
- Leisch, H.; Shi, R.; Grosse, S.; Morley, K.; Bergeron, H.; Cygler, M.; Iwaki, H.; Hasegawa, Y.; Lau, P.C. Clonin, Baeyer-Villiger biooxidations and structures of the camphor pathway monooxygenases of Pseudomonas putida ATCC 17453. Appl. Environ. Microbiol. 2012, 78, 2200–2212.
- Iwaki, H.; Grosse, S.; Bergeron, H.; Leisch, H.; Morley, K.; Hasegawa, Y.; Lau, P.C. Camphor pathway redox: Functional recombinant expression of 2,5- and 3,6 diketocamphane monooxygenases in Pseudomonas putida ATCC 17453 with their cognate flavin reductase catalysing Baeyer-Villiger reactions. Appl. Environ. Microbiol. 2013, 79, 3282–3293.
- Willetts, A.; Kelly, D.R. Multiple native flavin reductases in camphor-metabolising Pseudomonas putida NCIMB 10007 (ATCC 217453). Functional interaction with two-component diketocamphane monooxygenase isoenzymes. Microbiology 2014, 160, 1784–1794.
- Willetts, A.; Kelly, D.R. Flavin-dependent redox transfers by two-component diketocamphane monooxygenases of camphor-grown Pseudomonas putida NCIMB 10007 (ATCC 17453). Microorganisms 2016, 4, 38.
- Kendrew, S.G.; Harding, D.A.; Hopwood, E.N.; Marsh, E.N. Identification of a flavin:NADH oxidoreductase involved in the biosynthesis of actinorhodin. Purification and characterisation of the recombinant enzyme. J. Biol. Chem. 1995, 270, 17339–17343.
- Parry, R.J.; Li, W. An NADPH:FAD oxidoreductase from the valinimycin producer Streptromyces viridifaciens. J. Biol. Chem. 1997, 272, 23303–23311.
- Hertwerk, C. The biosynthetic logic of polyketide diversity. Angew. Chem. Int. Ed. 2009, 48, 4688–4716.
- Begani, J.; Lakhani, J.; Harwani, D. Current strategies to induce secondary metabolites from microbial biosynthetic cryptic gene clusters. Annal. Microbiol. 2018, 68, 419–432.
- Scott, T.A.; Piel, J. The hidden enzymology of bacterial natural product biosynthesis. Nat. Rev. Chem. 2019, 3, 404–425.
- Russell, A.H.; Truman, A.W. Genome mining strategies for ribosomally synthesised post-transcriptionally modified peptides. Comput. Struct. Biotechnol. J. 2020, 18, 1838–1851.
- Scherlach, K.; Hertweck, C. Mining and unearthing hidden biosynthetic potential. Nat. Commun. 2021, 12, 3864.
- Walsh, C.T. Tailoring enzyme strategies and functional groups in biosynthetic pathways. Nat. Prod. Rep. 2023, 40, 326–386.
- Conrad, H.E.; Lieb, K.; Gunsalus, I.C. Mixed function oxidation. III. An electron transport complex in camphor lactonization. J. Biol. Chem. 1965, 240, 4029–4037.
- Gunsalus, I.C.; Conrad, H.E.; Trudgill, P.W. Generation of active oxygen for mixed function oxidation. In Oxidases and Related Redox Systems; King, T.E., Mason, H.S., Morrish, M., Eds.; Wiley: New York, NY, USA, 1965; pp. 417–447.
- Gunsalus, I.C.; Trudgill, P.W.; Cushman, D.; Conrad, H.E. Stereospecific biological oxygenation of terpenoids. In Symposium on Recent Advances in the Chemistry of Terpenoids; National Chemistry Laboratory: Poona, India, 1965; 6 Abstract 3.
- Trudgill, P.W.; DuBus, R.; Gunsalus, I.C. Mixed function oxidation. V. Flavin interaction with reduced diphosphopyridine nucleotide dehydrogenase, one of the enzymes participating in camphor lactonization. J. Biol. Chem. 1966, 241, 1194–1205.
- Trudgill, P.W.; DuBus, R.; Gunsalus, I.C. Mixed function oxidation. VI. Purification of a tightly linked electron transport complex in camphor lactinization. J. Biol. Chem. 1966, 241, 4288–4290.
- Yu, C.A.; Gunsalus, I.C. Monooxygenases. VII. Camphor lactonase I and the role of the protein components. J. Biol. Chem. 1969, 244, 6149–6152.
- Trudgill, P.W. Bacteria and the challenge of cyclic molecules. In Experiences in Biochemical Perception; Ornston, L.N., Sligar, S.G., Eds.; Academic Press: New York, NY, USA, 1982; pp. 59–73.
- Trudgill, P.W. Microbial degradation of alicyclic hydrocarbons. In Developments in Biodegradation of Hydrocarbons—I; Watkinson, R.J., Ed.; Applied Science Publishers: London, UK, 1982; pp. 47–84.
- Taylor, D.G.; Trudgill, P.W. Studies of 2,5-diketocamphane monooxygenase from Pseudomonas putida ATCC 17453. In Proceedings of the 8th International Symposium on Flavins and Flavoproteins; Bray, R.C., Engel, P.C., Mayhew, S.G., Eds.; de Gruyter: Berlin, Germany, 1984; pp. 343–344.
- Trudgill, P.W. Microbial degradation of the alicyclic ring. In Microbial Degradation of Organic Compounds; Gibson, D.T., Ed.; Marcel Dekker: New York, NY, USA, 1984; pp. 131–180.
- Taylor, D.G.; Trudgill, P.W. Camphor revisited: Studies of 2,5-diketocamphane 1,2-monooxygenase from Pseudomonas putida ATCC 17453. J. Bacteriol. 1986, 165, 489–497.
- Trudgill, P.W. Terpenoid metabolism by Pseudomonas. In The Bacteria; Sokatch, J.R., Ed.; Academic Press: Orlando, FL, USA, 1986; Volume 10, pp. 483–525.
- Jones, K.H.; Smith, R.T.; Trudgill, P.W. Diketocamphane enantiomer-specific ‘Baeyer-Villiger’ monooxygenases from camphor-grown Pseudomonas putida ATCC 17453. J. Gen. Microbiol. 1993, 139, 797–805.
- Schroder, I.; Johnson, E.; de Vies, S. Microbial ferric iron reductases. FEMS Microbiol. Rev. 2003, 27, 427–447.
- Cain, T.J.; Smith, A.T. Ferric ion reductases and their contribution to unicellular ferrous iron uptake. J. Inorg. Biochem. 2021, 218, 111407.
- Roome, P.W.; Philley, J.C.; Peterson, J.A. Purification and properties of putidaredoxin reductase. J. Biol. Chem. 1983, 258, 2593–2598.
- Koga, H.; Yamaguchi, E.; Matsunaga, K.; Aramaki, H.; Horiuchi, T. Cloning and nucleotide sequences of NADH-putidaredoxin reductase gene (camA) and putidaredoxin gene (camB) involved in cytochrome P-450cam hydroxylase of Pseudomonas putida. J. Biochem. 1989, 106, 831–836.
- Peterson, J.A.; Lorence, M.C.; Amameh, B. Putidaredoxin reductase and putidaredoxin. Cloning sequence determation, and heterologous expression of the proteins. J. Biol. Chem. 1990, 265, 6066–6076.
- Sevrioukova, I.F.; Li, H.; Poulos, T.L. Crystal structure of putidaredoxin reductase from Pseudomonas putida, the final structural component of the cytochrome P450cam monooxygenase. J. Mol. Biol. 2004, 336, 889–902.
- Brewer, C.B.; Peterson, J.A. Single turnover kinetics of the reaction between oxycytochrome P-450cam and reduced putidaredoxin. J. Biol. Chem. 1988, 263, 791–798.
- Kuznetsov, Y.V.; Blair, E.; Farmer, P.J.; Poulos, T.L.; Pifferritti, A.; Sevrioukova, I.F. The putidaredoxin reductase-putidaredoxin electron transfer complex. J. Biol. Chem. 2005, 280, 16135–16142.
- Hannemann, F.; Bichet, A.; Ewen, K.M.; Bernhardt, R. Cytochrome P450 systems—Biological variations of electron transport systems. Biochim. Biophys. Acta 2007, 1770, 330–334.
- Baghdianz, A. Role du zinc sur l’application de la composante du ‘pigment’ de Pseudomonas fluorescens. Arch. Sci. 1952, 5, 47–48.
- Chakrabarty, A.M.; Roy, S.C. Effects of trace elements on the production of pigments by a pseudomonad. Biochem. J. 1964, 93, 228–231.
- Huyer, M.; Page, W.J. Zn2+ increases siderophore production in Azotobacter vinelandii. Appl. Environ. Microbiol. 1988, 54, 2626–2631.
- Huyer, M.; Page, W.J. Ferric reductase activity in Azotobacter vinelandii and its inhibition by Zn2+. J. Bacteriol. 1989, 171, 4031–4037.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
535
Revisions:
4 times
(View History)
Update Date:
18 Mar 2024
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No