Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mark Ellsworth Kleinman | -- | 4469 | 2024-02-26 01:37:08 | | | |
| 2 | Fanny Huang | Meta information modification | 4469 | 2024-02-27 06:17:25 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Dubey, S.K.; Dubey, R.; Kleinman, M.E. Histone Loss in Aging and Senescence. Encyclopedia. Available online: https://encyclopedia.pub/entry/55419 (accessed on 26 July 2026).
Dubey SK, Dubey R, Kleinman ME. Histone Loss in Aging and Senescence. Encyclopedia. Available at: https://encyclopedia.pub/entry/55419. Accessed July 26, 2026.
Dubey, Sushil Kumar, Rashmi Dubey, Mark Ellsworth Kleinman. "Histone Loss in Aging and Senescence" Encyclopedia, https://encyclopedia.pub/entry/55419 (accessed July 26, 2026).
Dubey, S.K., Dubey, R., & Kleinman, M.E. (2024, February 26). Histone Loss in Aging and Senescence. In Encyclopedia. https://encyclopedia.pub/entry/55419
Dubey, Sushil Kumar, et al. "Histone Loss in Aging and Senescence." Encyclopedia. Web. 26 February, 2024.
Copy Citation
Aging is a complex and multifactorial process characterized by a combination of aging hallmarks that contribute to declines at the molecular, cellular, and systemic levels in an organism. The dysregulation of the cellular epigenome during aging and senescence is a complex phenomenon that manifests through various elements, including global histone levels, histone positioning on the DNA sequence, post-translational modifications (PTMs) of histones, histone variants, DNA methylation, and noncoding RNAs.
histones
nucleosome occupancy
aging
senescence
1. Introduction
With demographic shifts toward aging populations, the incidence of age-related diseases such as cancer, diabetes, cardiovascular disorders, osteoarthritis, and macular degeneration is on the rise in many countries [1][2][3]. There is a massive unmet need to significantly improve researchers' fundamental scientific understanding of aging biology in order to develop new therapeutic approaches to age-related diseases. Achieving this goal hinges on decoding the intricate molecular mechanisms of aging and identifying new experimental models to mitigate or reverse age-associated phenotypes.
Aging is a complex and multifactorial process characterized by a combination of aging hallmarks that contribute to declines at the molecular, cellular, and systemic levels in an organism. The hallmarks of aging, as outlined by López-Otín et al. [4], are categorized into three groups: primary hallmarks, antagonistic hallmarks, and integrative hallmarks. The primary hallmarks of aging that are deleterious to the cell include genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, and disabled macroautophagy [5][6][7][8][9][10][11]. The antagonistic hallmarks represent a more intricate response of the cell to aging and include deregulated nutrient sensing, cellular senescence, and mitochondrial dysfunction. The cellular nutrient sensing pathways that adapt to the nutrition availability change with age. Deregulated nutrient sensing is closely associated with mitochondrial dysfunction, peroxisomal dysfunction, and the deregulation of protein synthesis and glucose, nucleotide, and lipid metabolisms [12][13]. Mitochondrial dysfunction can also induce oxidative stress, which can initially benefit the cell by eliciting a protective gene response but may prove detrimental in the long run [14][15]. Collaborating closely with mitochondria, peroxisomes play a crucial role in preserving the oxidative balance within the cell [16]. While the peroxisomes typically generate elevated levels of reactive oxygen species and reactive nitrogen species, they also are armed with a battery of antioxidant enzymes and nonenzymatic free radical scavengers [17]. In addition to regulating ROS metabolism, peroxisomes perform crucial functions in lipid metabolism. However, a growing body of evidence indicates a decline in peroxisomal function with age, linking oxidative stress and disrupted lipid metabolism resulting from dysregulated peroxisomal function to various age-related diseases [17][18]. The accumulation of cholesterol is associated with peroxisomal dysfunction, and cholesterol oxide derivatives, known as oxysterols, are known to induce significant dysfunction in organelles, especially mitochondria. Oxysterols play pivotal roles in the disruption of redox homeostasis, inflammatory status, lipid metabolism, and ultimately, cell death induction during aging [19][20]. While specific phytosterols, also known as “plant cholesterol”, have been identified for their health benefits, recent research indicates that imbalances in these diet-derived phytosterols have substantial implications in neurodegeneration and cognitive decline [21]. Thus, antagonistic hallmarks seem to be beneficial to the cell when meticulously regulated but tend to become deleterious only at high levels. Integrative hallmarks arise when damage from primary and antagonistic aging accumulates, leading to stem cell exhaustion, altered intercellular communication, chronic inflammation, and dysbiosis [22][23][24]. Although these hallmarks appear to be independent entities, they are integrative, and a complex interplay of different regulatory pathways is inherent in driving the complex biological process of aging.
Earlier studies proposed that the accumulated burden of somatic postnatal mutations is the primary cause of aging [25][26][27]. As various theories on aging, particularly those centered on genetic changes within cells, evolved, a growing body of evidence also underscored the importance of epigenetic factors in the aging process [8][28][29]. While an organism’s genome maintains a relatively steady state throughout its lifetime, the epigenome undergoes extensive reprogramming. Thus, there is a growing interest in research challenging the prevailing idea that genetic aberrations primarily drive the aging process [30]. There are significant data to support the notion that normal cellular aging may occur despite fewer mutations, and conversely, cells with a higher mutation rate do not necessarily display premature aging [31][32][33]. The mounting evidence from yeast to humans indicates that the breakdown of epigenetic information, rather than genetic mutations, plays a pivotal role in aging [30][34][35][36]. The dysregulation of the cellular epigenome during aging and senescence is a complex phenomenon that manifests through various elements, including global histone levels, histone positioning on the DNA sequence, post-translational modifications (PTMs) of histones, histone variants, DNA methylation, and noncoding RNAs.
Eukaryotes have evolved a complex genome architecture composed of DNA and histones that synergistically regulate cell function and differentiation. The fundamental architecture of the chromatin, known as “beads-on-a-string”, refers to the nucleosome core particle. This nucleosome core comprises 147 base pairs of genomic DNA wrapped around a core histone octamer made of H2A, H2B, H3, and H4 [37]. The linker histone H1 stabilizes the chromatin by binding to specific sites on the DNA, anchoring the nucleosomes and facilitating the formation of higher-order chromatin structures. This compaction of chromatin around the core and linker histones is a key mechanism in eukaryotes to package and organize their genome efficiently. The higher-order chromatin structure can be classified as euchromatin and heterochromatin, depending on the level of compaction. Euchromatin corresponds to the less condensed regions of chromatin that allow for easy access to regulatory molecules such as transcription factors. On the other hand, heterochromatin represents transcriptionally inactive and highly condensed chromatin sites. The organization of these two distinct chromatin regions is closely associated with the histone content and organization of the nucleosome landscape. Nucleosomes are highly dynamic, both in their composition and positioning on the genome [38]. The genome is constantly reorganized through nucleosome eviction and histone sliding processes that are mediated by factors such as chromatin remodeling enzymes, histone chaperones, and polymerases [38][39][40]. Furthermore, nucleosomes in transcriptionally active or repressed sites carry multiple PTMs on histone proteins, which ultimately define the epigenomic state of the cell [41][42]. Among the vast repertoire of histone PTMs, acetylation, methylation, phosphorylation, and ubiquitination are the main modifications regulating the chromatin structure and gene transcription [41][43][44]. The accumulating evidence has shown that age-related dysregulation of the epigenome involves the breakdown of the nucleosome landscape, leading to changes in the organization of nuclear components, activation of previously silenced genes, and aberrant gene expression patterns [5][8].
2. Age-Related Histone Loss and Altered Nucleosome Occupancy in Non-Mammalian Models
For over 30 years, scientists have been studying global histone loss and its implications in cellular viability and aging in various yeast models. Early studies by Grunstein et al. provided compelling evidence that the depletion of histones H4 and H2B in yeast led to transcriptional defects, the disruption of the chromatin structure, and cell cycle arrest [45][46]. Subsequent investigations showed that the loss of histones H3 or H4 in yeast altered the transcriptional landscape with the derepression of many previously silenced genes [47][48]. Perhaps the most radical observation made by Hu et al. revealed an ~50% global depletion of histones and nucleosome repositioning in budding yeast during replicative aging. Histone removal from nucleosomes significantly enhanced DNA accessibility to transcriptional machinery, precipitating a sharp derepression of gene expression. Consequently, the yeast cells showed an overall upregulation in gene transcription and an increased genomic instability [49]. Similar to yeast studies, an age-dependent loss of histone H3 was reported in Caenorhabditis elegans when worm lysates were examined from young and old adults [50]. Thus, the mounting evidence from studies using yeast strains and other nonmammalian models via conditional histone knockouts or replicative aging has significantly enhanced researchers' scientific understanding of the biologic effects of histone loss, altered nucleosome occupancy, differential gene expression, and cellular senescence.
3. Histone Loss in Mammalian Models of Replicative and Chronological Aging
Mammalian cells progressively succumb to the adverse effects of aging assessed through two primary modalities: replicative life span and chronological life span [51][52]. Comprehensive investigations into these aging processes in yeast have demonstrated the emergence of aging hallmarks similar to those found in mammals [51][52][53]. Hence, evidence of the age-related effects on histone expression profiling of yeast cells captured the attention of researchers, prompting further investigations into histone levels in mammalian cells. An early study of histone depletion in human cells using pulse-chase labeling revealed a gradual decrease in H1 linker histones in senescent human diploid fibroblast cultures. This reduction was linked to a decline in the biosynthesis of H1 histone with progressive in vitro aging [54]. Houde and colleagues [55] provide a novel perspective on H1 expression changes in cultured human fibroblasts that exhibit a progressive cell cycle elongation. Their findings revealed that cells in early (28–35 mean population doubling; MPD) and late (65–70 MPD) passages, maintained in a confluent state at 0 and 6 weeks, exhibited a similar shift in the gene expression of H1 variants. Notably, this involved a reduction in H1B and a concurrent increase in H10 and H1A. These data suggest that changes in the expression levels of histone H1 variants occurred as a function of time of density-dependent growth rather than replicative age. Building on Mitsui et al.’s [54] discovery, subsequent studies focused on in vitro and in vivo aging in human skin fibroblasts and discovered a reduction in the nucleosome occupancy and an altered chromatin structure with aging [56][57]. While histone expression profiles in aging were being investigated in yeast, it took considerable time before Karsleder and colleagues delved into the correlation between histone levels and mammalian aging [58]. Their study using human diploid fibroblast (HDF) cell lines IMR90 and WI38 as models for replicative senescence revealed a significant decrease in histones H3 and H4 (43% and 47%, respectively) of late passage cells. The dysregulation of histone expression was primarily attributed to stress signals associated with telomere shortening. The telomeric region, affected by age-associated cellular damage, led to a DNA damage response and progressive genome-wide epigenetic changes with successive cell cycles that ultimately impacted histone biosynthesis. Chronic replicative stress only affected the H3 and H4 histones in HDFs, while the H1, H2A, and H2B histones remained unchanged [58]. Although the concept of histone loss in aging was initially suggested in actively replicating cells, Liu et al. studied the histone patterns of quiescent muscle stem cells (QSC) in young (2 months) and aged (24 months) mice and found a comparable decrease in the histone levels of H1, H2B, H3, and H4 [36]. This finding was quite significant in that histone loss also occurred during chronological aging in a quiescent cell type with minimal turnover.
While some aging models demonstrate a selective loss of canonical histones, recent research in mammals has revealed a global reduction in histone levels associated with aging. In activated naive CD4+ T cells from both young (20–35 years) and old (65–85 years) adults, a transcriptome analysis via RNA-seq showed that aged T cells tended to have globally reduced expressions of core histone genes compared to young cells, and this decrease was dependent on the cell cycle state of the activated, aged T cells. The global downregulation of histones, confirmed at both the mRNA and protein levels, was observed to delay the S-phase progression in aged T cells, thereby triggering a replication-stress response [59]. Similarly, a global loss of histones was observed in aged retinal pigment epithelium (RPE) cells, where all five canonical histones were depleted. To provide a comprehensive perspective, Dubey et al. assessed the histone expression in mouse RPE cells undergoing chronological aging and primary human RPE cell lines that reached senescence through replicative aging. In the RPE cells obtained from young (2–3 months) and aged (20–24 months) mice, a substantial reduction was observed in all core and linker histones. Furthermore, a replicative aging model using primary human RPE cells revealed a gradual decrease in the histone levels as the cells aged [60]. Together, these studies demonstrate that both actively proliferating and postmitotic cells in a quiescent state develop histone loss with aging. Also, a decreased histone expression in aged cells may be global or specific depending on the cell type and experimental model.
Much like aging, diverse senescence pathways can contribute to epigenetic dysregulation in cells [61]. Cellular senescence is an innate stress response, wherein cells enter a stable and irreversible state of cell cycle arrest even under optimal growth conditions. Senescent cells exhibit distinct changes in their morphology, chromatin structure, gene expression, and the emergence of a senescence-associated secretory phenotype (SASP), a chronic inflammatory state [62][63]. These alterations collectively cause the cell to exit the cell cycle permanently. Senescence is induced by multiple mechanisms including replicative senescence, senescence due to DNA damage, stress-induced senescence, and programmed developmental senescence [62][64]. In human RPE cells that attained replicative exhaustion and entered the senescent state, there was a substantial decrease in all canonical histones [60]. Furthermore, histone loss was observed in two different senescent models utilizing human umbilical vein endothelial cells (HUVECs) [65]. In the first model, senescence was induced through natural passaging until the cells reached a point of replicative exhaustion, while the second model involved prolonged exposure to TNF-alpha over 26 days. In both instances, senescent cells displayed a reduction in many histone isoforms along with the downregulation of genes responsible for regulating the cell cycle and DNA repair mechanisms. Regardless of the upstream stimuli prompting senescence, the process typically converges in downstream effects, with histone depletion emerging as one of the significant consequences. These findings underscore the importance of histone depletion as a critical factor driving the process of cellular senescence [65].
While the acquisition of senescence has been viewed as an alternative pathway to prevent cancer, the prolonged accumulation of senescent cells can paradoxically promote cancer development [66][67]. The emergence of SASP factors during senescence can impact the surrounding cells via alterations of the cellular microenvironments, establishing chronic inflammation and fostering conditions conducive to cancer [68]. Interestingly, histone depletion associated with aging and senescence has been inversely linked to malignancy, suggesting that histone loss can facilitate cell proliferation arrest, ultimately contributing to tumor suppression [69]. Unlike histone loss, histone mutations, known as oncohistones, are implicated in promoting cancers [70]. However, given that many age-dependent changes in the cellular epigenome resemble those observed in cancer, the epigenetic reprogramming occurring during aging may predispose individuals to cancer development [71]. Therefore, gaining a deeper understanding of age-related epigenomic changes holds the potential to elucidate the underlying causes of cancer.
4. Alterations in Nucleosome Landscape in Aging
The age-related loss of histone expression appears to be a widespread phenomenon observed in organisms ranging from yeast to mammals. However, there is limited research on how the distribution of nucleosomes across the genome changes as organisms age. Celona et al. studied the impacts of histone loss and alterations in the nucleosome occupancy in yeast and mammalian cells. Their proposed model suggested that the loss of nucleosomes predominantly occurs in specific localized regions [72]. Mammalian cells with ablated high mobility group box 1 (HMGB1), a DNA-binding protein, showed a significant decrease in histones and nucleosomes. Their findings demonstrated that the shRNA- and siRNA-mediated ablation of HMGB1 in HeLa cells, or its yeast equivalent (Nhp6a/b), did not compromise the cell viability despite the histone loss. However, the HMGB1-deficient cells displayed a greater sensitivity to DNA damage, underwent a global increase in transcription, and showed specific alterations in the transcriptional profile of certain genes. This study proposed a chromatin model in which the loss of nucleosomes within the cells is not widespread, but rather localized, particularly in regions that inherently exhibit a lower tendency to form nucleosomes [72]. This model aligns with the genomic DNA code regulating nucleosome positioning, as postulated by Kaplan et al., and emphasizes the role of DNA sequence information in governing the arrangement of nucleosomes [73]. Thus, chromatin regions characterized by an innately lower tendency for histone deposition are more histone deficient.
Similar to the findings of Celona and colleagues, a subsequent study demonstrated that rather than a global redistribution of nucleosomes, aging causes nucleosome loss at specific sites [74]. Bochkis et al. used high-throughput methods such as RNA-seq, MNase-seq, and ChIP-seq to map nucleosome changes in young (3 months) and aged (21 months) mouse livers. They discovered age-related differences in the nucleosome distribution, which in turn influenced the accessibility of chromatin to transcriptional factors. A reduction in the nucleosome density correlated with the elevated transcription of key genes involved in lipid metabolic pathways in aged livers, potentially leading to metabolic dysfunction and hepatic steatosis. Furthermore, there were specific sites with an increased nucleosome occupancy with age, including the serum response factor (SRF) gene that impacted its target genes involved in liver proliferation, lipid metabolism, and histone expression. The interrogation of histone isoforms revealed an irregular expression pattern displaying both upregulation and downregulation within aged hepatic tissue [74]. Although this study focused on the decline in SRF activity in the aging liver due to altered nucleosome occupancy, it is important to note that other mechanisms have been reported for repressed SRF activity including nuclear exclusion, protein kinase C-δ induced phosphorylation, and the inactivation of SRF in senescent cells [75][76]. In line with Bochkis et al.’s research, Chen and colleagues investigated H3 nucleosomal profiles in chronologically aged mouse tissues [77]. A comprehensive H3 nucleosomal map was generated using ChIP-Seq data from four tissues (heart, liver, cerebellum, and olfactory bulb) and one primary cell type (primary neural stem cells obtained from the subventricular zone) in mice from different age groups (3, 12, and 29 months). While they did not find significant changes in the global H3 levels, localized alterations in the H3 occupancy were observed in all the four tissue types and cultured neural stem cells during aging. These changes can manifest as an increased or decreased occupancy in specific regions with subsequent chromatin remodeling in aging tissues. Notably, the distal regions located 5–500 kb away from the annotated transcriptional start sites exhibited a significant remodeling of H3 occupancy, both upstream and downstream. The sites proximal to genes, particularly within intronic regions, consistently demonstrated robust nucleosome enrichment. Within aging chromatin, the distinct changes observed in distally located nucleosomes suggest a differential occupancy of nucleosomes in regulatory elements, particularly the forkhead transcription factors, which are critical regulators of DNA remodeling. Additionally, the repositioning of nucleosomes triggered transcriptional changes in inflammatory transcription factors such as STAT6 and IRF8. Together, these studies suggest that compared to the observations in actively replicating yeast and mammalian cells, histone depletion in chronologically aging tissues is relatively less and limited to specific genomic sites [77].
5. Multiple Mechanisms of Histone Loss during Aging
Although aging is associated with histone loss and the reorganization of nucleosomes, the effects of histone depletion, nucleosome repositioning, and the factors driving histone reduction are still largely uncharted territories [4]. Studies in yeast offered critical insights into the genetic and epigenetic mechanisms behind the age-related dysregulation of histone expression. These mechanisms included telomere loss, DNA damage, changes in histone chaperones, reduced histone production, and alterations in histone marks, all of which affected the chromatin structure [78]. Despite limited research in mammals, similar causal links between decreasing histone levels and various genetic and epigenetic factors have been established in aging cells and tissues (Figure 1). For instance, the experiments on lung fibroblast cell lines undergoing replicative aging revealed that chronic exposure to DNA damage signals, such as those arising from telomere shortening, impacted histone biosynthesis. [58]. The presence of short telomeres and the consequent DNA damage signaling resulted in the depletion of the stem–loop binding protein (SLBP), a stabilizer of histone mRNA. This, in turn, reduced the biosynthesis of H3 and H4 and destabilized Asf1, a histone chaperone, leading to genome-wide epigenetic changes that amplified the damage signal during repeated cell cycles. This self-enforced regulatory loop constantly changed the chromatin environment at the telomeres, facilitating the incursion of damage markers beyond the threshold of cell tolerance, ultimately leading to irreversible cell cycle exit and senescence. The ectopic expression of telomerase enzymes in aging cells to increase the telomere length normalized the histone expression levels similar to those of young cells. Furthermore, this process partially restored SLBP and ASF1 expression, likely by alleviating the DNA damage signaling associated with short telomeres [58].
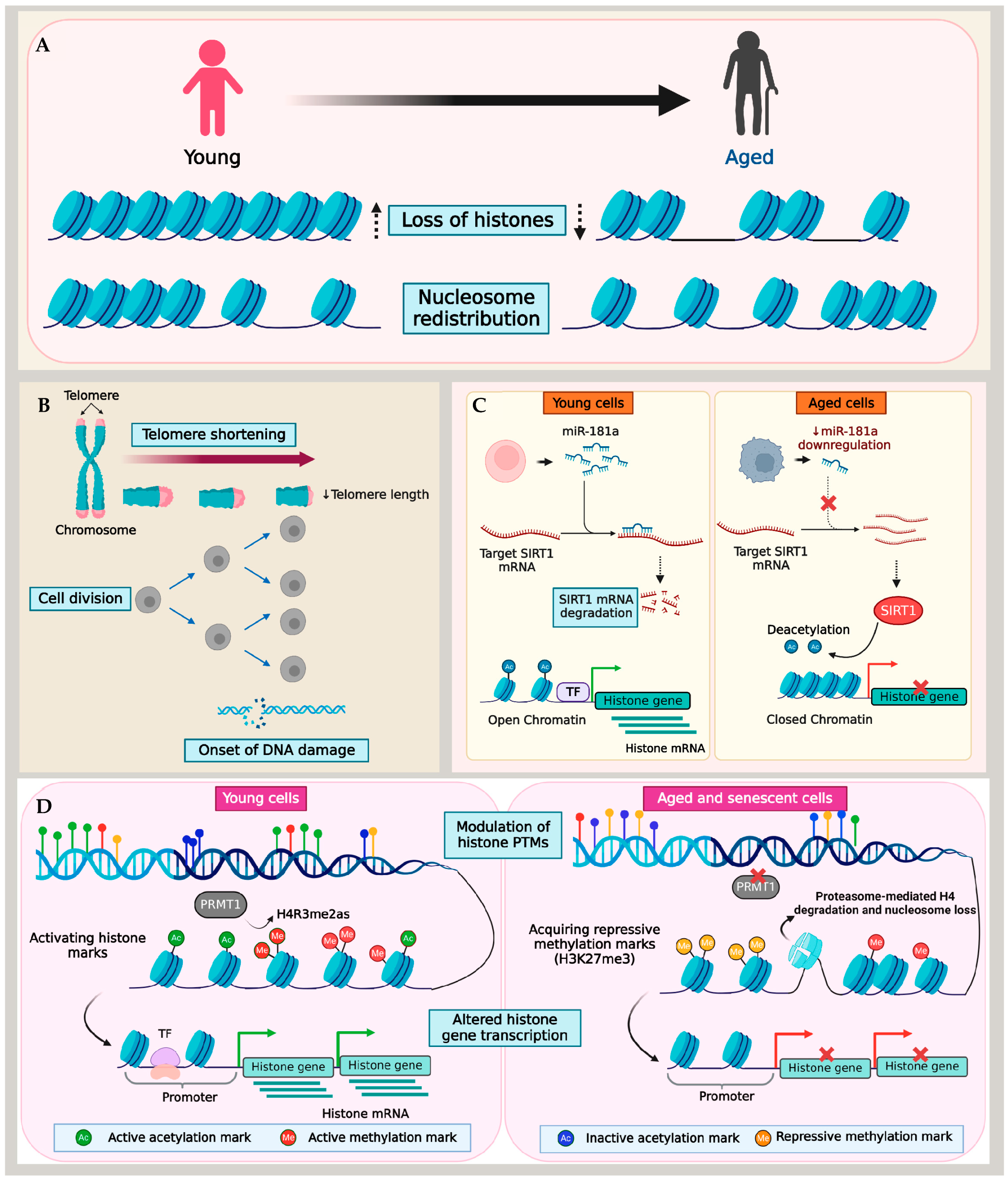
Figure 1. Mechanism of histone loss in aging and senescence. (A) Aging and senescence are characterized by the loss of histones and nucleosome redistribution. Different models show histone loss due to (B) telomere shortening and DNA damage, (C) miRNA-mediated changes to histone transcription, and (D) the imbalance of activating and repressive histone PTMs.
Telomere shortening and dysfunction are hallmarks of cellular aging and senescence. Despite their crucial role in maintaining chromosomal stability, the regulatory mechanisms governing telomeres during cellular aging remain poorly understood. Situated at the termini of linear chromosomes, telomeres form complexes of TTAGGG nucleotide repeats and proteins that regulate their functions, shielding them from recognition by the cell’s repair mechanism as double-stranded DNA breaks (DSBs). These proteins, including the telomerase (TERT) enzyme, histones, and the Shelterin complex, are critical for regulating the telomere length and preventing chromosomal end fusion [79][80].
Telomeres typically exhibit a heterochromatin structure, and a widespread phenomenon known as the telomere position effect results in low expression levels or the transcriptional silencing of genes within or near telomeres [81]. Additionally, telomeres tend to spatially organize at the nuclear periphery, a zone of transcriptional repression, in a cell cycle-dependent manner [82][83][84], and therefore, experience the transcriptional repression of genes. However, as cells approached senescence, a spatial overlap of lamina intranuclear structures with telomeres was observed [85]. During senescence, as the nuclear lamina’s organization gets disrupted, telomeres tend to form large aggregates lacking TERT. These telomere aggregates accumulated histone γ-H2AX, a classical marker of DSBs and telomere shortening, in senescent cells [85].
Furthermore, the formation of senescence-associated heterochromatin foci (SAHF), representing facultative heterochromatin domains, correlates with telomere shortening in cells entering senescence [86][87]. The SAHF contain domains with di- or tri-methylated lysine 9 of histone H3 (H3K9me2/3), a histone H2A variant (macroH2A), and heterochromatin protein 1 (HP1) proteins [87][88][89]. Additionally, epigenetic modifications such as histone methylation in the telomere region and TERT demethylation in humans play significant roles in maintaining the heterochromatin, transcriptional silencing at telomeres, and telomerase inactivation. Preserving the telomere structure and ensuring transcriptional silencing are critical to preventing premature aging [90][91].
Telomere shortening assumes that each successive cell division acts as a mitotic counting mechanism, eventually leading cells to attain replicative senescence [4][92]. In contrast, cells in a quiescent state transition into senescence despite negligible telomere shortening [93][94]. Therefore, the primary causes of age-associated histone loss in quiescent cells are likely driven by mechanisms beyond telomere dysfunction. Muscle stem cells provide a well-studied model for cellular quiescence and aging [95][96]. Liu and colleagues reported an epigenetic mechanism for histone loss in aging quiescent muscle stem cells (QSCs). Using the ChIP-Seq approach, the QSCs derived from young and aged mice were profiled for histone methylation marks, including H3K4me3, H3K27me3, and H3K36me3. As the QSCs underwent aging, there was an overall reduction in the expression of histone genes that acquired H3K27me3, a transcription repressive mark, at their transcription start sites. This finding underscores the critical role played by the repressive methylation mark in the regulation of histones within QSCs, simultaneously emphasizing the involvement of histone PTMs in histone regulation.
A similar methylation regulatory mechanism of histones was reported in fibroblast cell lines [97]. In this study, histone expression was examined in IMR90 fibroblast cells after inducing premature senescence via protein arginine methyltransferase 1 (PRMT1) knockdown. PRMT1 is the predominant arginine methyltransferase in humans, which mediates the asymmetric dimethylation of histone H4 at arginine 3 (H4R3me2as), a critical modification essential for histone H4 stability. Studies over the past decades have demonstrated the reduced expression of PRMT1 in replicative aging and senescent cells [98][99]. Lin et al. demonstrated that premature senescence in fibroblasts induced by PRMT1 knockdown caused a reduction in PRMT1-mediated H4 dimethylation, leading to the destabilization of H4. Consequently, H4 showed increased binding to PA200, causing the ubiquitin-independent degradation of H4 by PA200-capped proteasomes [97][100]. A similar H4 degradation was also observed in fibroblasts exposed to different senescence-associated signals such as oxidative stress, DNA damage, or oncogenic signaling. Importantly, H4 degradation preceded the depletion of other histones, resulting in a reduced nucleosomal occupancy and impacted cell proliferation by enhancing the transcription of genes responsible for cell cycle inhibition, senescence-related genes, and apoptosis regulators [97]. It is critical to note that different histone marks have contrasting roles in regulating genes in the context of aging and lifespan across species and even within different tissues of an organism [101][102]. Therefore, recognizing the precise functions of various histone PTMs in aging tissues is critical for unraveling the complexity of epigenetic processes in aging.
Though a large part of the age-associated histone depletion in cells appears to stem from altered histone PTM marks, telomere dysfunction, and DNA damage, a recent study reported the miRNA-mediated downregulation of histones in aged T cells. Initially, Ucar et al. detected a significant compaction of chromatin in several histone genes (HIST1H3D, HIST1H3E, and HIST4H4) and histone modifiers in T cells with age [103]. By combining ATAC-seq and RNA-seq data, their study showed that immune cells undergo alterations in their epigenome as individuals age. This involves the closing of the chromatin structure at the promoter and enhancer regions of genes that are actively expressed, including histones. This finding was consistent with the reduced expression of core histones demonstrated in a subsequent study by Kim et al. using T cells from young (20–35 years) and old (65–85 years) adults [59]. Naive T cells from aged individuals with a reduced miR-181a level as well as miR-181ab1-deficient murine T cells in an actively dividing state showed the downregulation of histone expression, which induced replication stress and a consequential slow-down of the cell cycle. In the absence of miR-181a, its target molecule SIRT1—a histone deacetylase enzyme—binds directly to the histone promoter region. This interaction led to a reduction in histone acetylation, thereby inducing the repression of histone genes [59].
Although different studies provide insight into the ramifications of age-related genetic and epigenetic alterations of the nucleosome landscape, a key factor is that this mechanism is cell-type dependent. Interestingly, Ivanov and colleagues reported that histone loss in senescent cells occurred in both fibroblasts and epidermal melanocytes [69]. Cells approaching senescence and those already in a senescent state undergo significant stress, marked by changes in both the morphology and physiology. Genomic DNA damage, particularly DSBs, along with the deterioration of nuclear integrity in senescent cells, leads to damaged DNA fragments leaving the nucleus. Ivanov et al. identified these fragments in the cytoplasm as cytoplasmic chromatin fragments (CCFs), which are strongly positive for γ-H2AX and H3K27me3. In the cytoplasm, an autophagic proteolytic activity targets these CCFs, progressively depleting the total histones in a process dependent on the lysosomal activity [69]. This process of lysosomal ubiquitin-mediated histone proteolysis, elucidated by Ivanov and colleagues in senescent cells, represents an important mechanism of post-translational histone degradation. Together, these studies have enhanced researchers' understanding of the intricate genetic and epigenetic pathways that control histone downregulation in the context of aging. Despite these insights, a consensus on the mechanism or function of histone loss during aging remains elusive, indicating the need for further research in this area.
References
- United Nations. World Population Ageing 2020 Highlights; United Nations Department of Economic and Social Affairs: New York, NY, USA, 2020.
- Jaul, E.; Barron, J. Age-Related Diseases and Clinical and Public Health Implications for the 85 Years Old and Over Population. Front. Public Health 2017, 5, 335.
- Swenor, B.K.; Ehrlich, J.R. Ageing and vision loss: Looking to the future. Lancet Glob. Health 2021, 9, e385–e386.
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278.
- Pal, S.; Tyler, J.K. Epigenetics and aging. Sci. Adv. 2016, 2, e1600584.
- Rodriguez-Rodero, S.; Fernandez-Morera, J.L.; Menendez-Torre, E.; Calvanese, V.; Fernandez, A.F.; Fraga, M.F. Aging genetics and aging. Aging Dis. 2011, 2, 186–195.
- Wheeler, H.E.; Kim, S.K. Genetics and genomics of human ageing. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 43–50.
- Booth, L.N.; Brunet, A. The Aging Epigenome. Mol. Cell 2016, 62, 728–744.
- Blackburn, E.H.; Epel, E.S.; Lin, J. Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science 2015, 350, 1193–1198.
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435.
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42.
- van der Rijt, S.; Molenaars, M.; McIntyre, R.L.; Janssens, G.E.; Houtkooper, R.H. Integrating the Hallmarks of Aging Throughout the Tree of Life: A Focus on Mitochondrial Dysfunction. Front. Cell Dev. Biol. 2020, 8, 594416.
- Amorim, J.A.; Coppotelli, G.; Rolo, A.P.; Palmeira, C.M.; Ross, J.M.; Sinclair, D.A. Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nat. Rev. Endocrinol. 2022, 18, 243–258.
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007, 6, 280–293.
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905.
- Pascual-Ahuir, A.; Manzanares-Estreder, S.; Proft, M. Pro- and Antioxidant Functions of the Peroxisome-Mitochondria Connection and Its Impact on Aging and Disease. Oxid. Med. Cell Longev. 2017, 2017, 9860841.
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O.; Van Veldhoven, P.P. Aging, age-related diseases and peroxisomes. Subcell. Biochem. 2013, 69, 45–65.
- Terlecky, S.R.; Koepke, J.I.; Walton, P.A. Peroxisomes and aging. Biochim. Biophys. Acta 2006, 1763, 1749–1754.
- Vigne, S.; Pot, C. Implication of Oxysterols and Phytosterols in Aging and Human Diseases. Adv. Exp. Med. Biol. 2024, 1440, 231–260.
- Zarrouk, A.; Vejux, A.; Mackrill, J.; O’Callaghan, Y.; Hammami, M.; O’Brien, N.; Lizard, G. Involvement of oxysterols in age-related diseases and ageing processes. Ageing Res. Rev. 2014, 18, 148–162.
- Spinedi, M.; Clark, C.; Zullo, L.; Kerksiek, A.; Pistis, G.; Castelao, E.; von Gunten, A.; Preisig, M.; Lütjohann, D.; Popp, J. Cholesterol-metabolism, plant sterols, and long-term cognitive decline in older people—Effects of sex and APOEe4. iScience 2024, 27, 109013.
- Blokzijl, F.; de Ligt, J.; Jager, M.; Sasselli, V.; Roerink, S.; Sasaki, N.; Huch, M.; Boymans, S.; Kuijk, E.; Prins, P.; et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 2016, 538, 260–264.
- Mogilenko, D.A.; Shpynov, O.; Andhey, P.S.; Arthur, L.; Swain, A.; Esaulova, E.; Brioschi, S.; Shchukina, I.; Kerndl, M.; Bambouskova, M.; et al. Comprehensive Profiling of an Aging Immune System Reveals Clonal GZMK+ CD8+ T Cells as Conserved Hallmark of Inflammaging. Immunity 2021, 54, 99–115.e112.
- Bosco, N.; Noti, M. The aging gut microbiome and its impact on host immunity. Genes. Immun. 2021, 22, 289–303.
- Szilard, L. On the Nature of the Aging Process. Proc. Natl. Acad. Sci. USA 1959, 45, 30–45.
- Failla, G. The aging process and cancerogenesis. Ann. N. Y. Acad. Sci. 1958, 71, 1124–1140.
- Hughes, K.A.; Charlesworth, B. A genetic analysis of senescence in Drosophila. Nature 1994, 367, 64–66.
- Partridge, L. Evolutionary theories of ageing applied to long-lived organisms. Exp. Gerontol. 2001, 36, 641–650.
- Benayoun, B.A.; Pollina, E.A.; Brunet, A. Epigenetic regulation of ageing: Linking environmental inputs to genomic stability. Nat. Rev. Mol. Cell Biol. 2015, 16, 593–610.
- Yang, J.H.; Hayano, M.; Griffin, P.T.; Amorim, J.A.; Bonkowski, M.S.; Apostolides, J.K.; Salfati, E.L.; Blanchette, M.; Munding, E.M.; Bhakta, M.; et al. Loss of epigenetic information as a cause of mammalian aging. Cell 2023, 186, 305–326.e327.
- De Majo, F.; Martens, L.; Hegenbarth, J.C.; Ruhle, F.; Hamczyk, M.R.; Nevado, R.M.; Andres, V.; Hilbold, E.; Bar, C.; Thum, T.; et al. Genomic instability in the naturally and prematurely aged myocardium. Proc. Natl. Acad. Sci. USA 2021, 118, e2022974118.
- Kaya, A.; Lobanov, A.V.; Gladyshev, V.N. Evidence that mutation accumulation does not cause aging in Saccharomyces cerevisiae. Aging Cell 2015, 14, 366–371.
- Narayanan, L.; Fritzell, J.A.; Baker, S.M.; Liskay, R.M.; Glazer, P.M. Elevated levels of mutation in multiple tissues of mice deficient in the DNA mismatch repair gene Pms2. Proc. Natl. Acad. Sci. USA 1997, 94, 3122–3127.
- Kennedy, B.K.; Gotta, M.; Sinclair, D.A.; Mills, K.; McNabb, D.S.; Murthy, M.; Pak, S.M.; Laroche, T.; Gasser, S.M.; Guarente, L. Redistribution of silencing proteins from telomeres to the nucleolus is associated with extension of life span in S. cerevisiae. Cell 1997, 89, 381–391.
- Sinclair, D.A.; Mills, K.; Guarente, L. Accelerated aging and nucleolar fragmentation in yeast sgs1 mutants. Science 1997, 277, 1313–1316.
- Liu, L.; Cheung, T.H.; Charville, G.W.; Hurgo, B.M.; Leavitt, T.; Shih, J.; Brunet, A.; Rando, T.A. Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep. 2013, 4, 189–204.
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260.
- Zhou, K.; Gaullier, G.; Luger, K. Nucleosome structure and dynamics are coming of age. Nat. Struct. Mol. Biol. 2019, 26, 3–13.
- Lai, W.K.M.; Pugh, B.F. Understanding nucleosome dynamics and their links to gene expression and DNA replication. Nat. Rev. Mol. Cell Biol. 2017, 18, 548–562.
- Gurard-Levin, Z.A.; Quivy, J.P.; Almouzni, G. Histone chaperones: Assisting histone traffic and nucleosome dynamics. Annu. Rev. Biochem. 2014, 83, 487–517.
- Stillman, B. Histone Modifications: Insights into Their Influence on Gene Expression. Cell 2018, 175, 6–9.
- Allahverdi, A.; Yang, R.; Korolev, N.; Fan, Y.; Davey, C.A.; Liu, C.F.; Nordenskiold, L. The effects of histone H4 tail acetylations on cation-induced chromatin folding and self-association. Nucleic Acids Res. 2011, 39, 1680–1691.
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395.
- Swygert, S.G.; Peterson, C.L. Chromatin dynamics: Interplay between remodeling enzymes and histone modifications. Biochim. Biophys. Acta 2014, 1839, 728–736.
- Han, M.; Chang, M.; Kim, U.J.; Grunstein, M. Histone H2B repression causes cell-cycle-specific arrest in yeast: Effects on chromosomal segregation, replication, and transcription. Cell 1987, 48, 589–597.
- Kim, U.J.; Han, M.; Kayne, P.; Grunstein, M. Effects of histone H4 depletion on the cell cycle and transcription of Saccharomyces cerevisiae. EMBO J. 1988, 7, 2211–2219.
- Gossett, A.J.; Lieb, J.D. In vivo effects of histone H3 depletion on nucleosome occupancy and position in Saccharomyces cerevisiae. PLoS Genet. 2012, 8, e1002771.
- Wyrick, J.J.; Holstege, F.C.; Jennings, E.G.; Causton, H.C.; Shore, D.; Grunstein, M.; Lander, E.S.; Young, R.A. Chromosomal landscape of nucleosome-dependent gene expression and silencing in yeast. Nature 1999, 402, 418–421.
- Hu, Z.; Chen, K.; Xia, Z.; Chavez, M.; Pal, S.; Seol, J.H.; Chen, C.C.; Li, W.; Tyler, J.K. Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes. Dev. 2014, 28, 396–408.
- Ni, Z.; Ebata, A.; Alipanahiramandi, E.; Lee, S.S. Two SET domain containing genes link epigenetic changes and aging in Caenorhabditis elegans. Aging Cell 2012, 11, 315–325.
- Longo, V.D.; Shadel, G.S.; Kaeberlein, M.; Kennedy, B. Replicative and chronological aging in Saccharomyces cerevisiae. Cell Metab. 2012, 16, 18–31.
- Janssens, G.E.; Veenhoff, L.M. Evidence for the hallmarks of human aging in replicatively aging yeast. Microb. Cell 2016, 3, 263–274.
- Denoth Lippuner, A.; Julou, T.; Barral, Y. Budding yeast as a model organism to study the effects of age. FEMS Microbiol. Rev. 2014, 38, 300–325.
- Mitsui, Y.; Sakagami, H.; Murota, S.; Yamada, M. Age-related decline in histone H1 fraction in human diploid fibroblast cultures. Exp. Cell Res. 1980, 126, 289–298.
- Houde, M.; Shmookler Reis, R.J.; Goldstein, S. Proportions of H1 histone subspecies in human fibroblasts shift during density-dependent growth arrest independent of replicative senescence. Exp. Cell Res. 1989, 184, 256–261.
- Ishimi, Y.; Kojima, M.; Takeuchi, F.; Miyamoto, T.; Yamada, M.; Hanaoka, F. Changes in chromatin structure during aging of human skin fibroblasts. Exp. Cell Res. 1987, 169, 458–467.
- Macieira-Coelho, A. Chromatin reorganization during senescence of proliferating cells. Mutat. Res. 1991, 256, 81–104.
- O’Sullivan, R.J.; Kubicek, S.; Schreiber, S.L.; Karlseder, J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat. Struct. Mol. Biol. 2010, 17, 1218–1225.
- Kim, C.; Jin, J.; Ye, Z.; Jadhav, R.R.; Gustafson, C.E.; Hu, B.; Cao, W.; Tian, L.; Weyand, C.M.; Goronzy, J.J. Histone deficiency and accelerated replication stress in T cell aging. J. Clin. Investig. 2021, 131, e143632.
- Dubey, S.K.; Dubey, R.; Jung, K.S.; Prajapati, S.; Tian, W.; Kleinman, M.E. Age-related global histone loss and altered histone acetylation in retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2023, 64, 4444.
- Zhu, X.; Chen, Z.; Shen, W.; Huang, G.; Sedivy, J.M.; Wang, H.; Ju, Z. Inflammation, epigenetics, and metabolism converge to cell senescence and ageing: The regulation and intervention. Signal Transduct. Target. Ther. 2021, 6, 245.
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246.
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593.
- Schmeer, C.; Kretz, A.; Wengerodt, D.; Stojiljkovic, M.; Witte, O.W. Dissecting Aging and Senescence-Current Concepts and Open Lessons. Cells 2019, 8, 1446.
- Kandhaya-Pillai, R.; Miro-Mur, F.; Alijotas-Reig, J.; Tchkonia, T.; Schwartz, S.; Kirkland, J.L.; Oshima, J. Key elements of cellular senescence involve transcriptional repression of mitotic and DNA repair genes through the p53-p16/RB-E2F-DREAM complex. Aging 2023, 15, 4012–4034.
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118.
- Wang, L.; Lankhorst, L.; Bernards, R. Exploiting senescence for the treatment of cancer. Nat. Rev. Cancer 2022, 22, 340–355.
- Domen, A.; Deben, C.; Verswyvel, J.; Flieswasser, T.; Prenen, H.; Peeters, M.; Lardon, F.; Wouters, A. Cellular senescence in cancer: Clinical detection and prognostic implications. J. Exp. Clin. Cancer Res. 2022, 41, 360.
- Ivanov, A.; Pawlikowski, J.; Manoharan, I.; van Tuyn, J.; Nelson, D.M.; Rai, T.S.; Shah, P.P.; Hewitt, G.; Korolchuk, V.I.; Passos, J.F.; et al. Lysosome-mediated processing of chromatin in senescence. J. Cell Biol. 2013, 202, 129–143.
- Amatori, S.; Tavolaro, S.; Gambardella, S.; Fanelli, M. The dark side of histones: Genomic organization and role of oncohistones in cancer. Clin. Epigenetics 2021, 13, 71.
- Gautrey, H.E.; van Otterdijk, S.D.; Cordell, H.J.; Newcastle 85+ Study Core, T.; Mathers, J.C.; Strathdee, G. DNA methylation abnormalities at gene promoters are extensive and variable in the elderly and phenocopy cancer cells. FASEB J. 2014, 28, 3261–3272.
- Celona, B.; Weiner, A.; Di Felice, F.; Mancuso, F.M.; Cesarini, E.; Rossi, R.L.; Gregory, L.; Baban, D.; Rossetti, G.; Grianti, P.; et al. Substantial histone reduction modulates genomewide nucleosomal occupancy and global transcriptional output. PLoS Biol. 2011, 9, e1001086.
- Kaplan, N.; Moore, I.K.; Fondufe-Mittendorf, Y.; Gossett, A.J.; Tillo, D.; Field, Y.; LeProust, E.M.; Hughes, T.R.; Lieb, J.D.; Widom, J.; et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 2009, 458, 362–366.
- Bochkis, I.M.; Przybylski, D.; Chen, J.; Regev, A. Changes in nucleosome occupancy associated with metabolic alterations in aged mammalian liver. Cell Rep. 2014, 9, 996–1006.
- Wheaton, K.; Riabowol, K. Protein kinase C delta blocks immediate-early gene expression in senescent cells by inactivating serum response factor. Mol. Cell Biol. 2004, 24, 7298–7311.
- Ding, W.; Gao, S.; Scott, R.E. Senescence represses the nuclear localization of the serum response factor and differentiation regulates its nuclear localization with lineage specificity. J. Cell Sci. 2001, 114, 1011–1018.
- Chen, Y.; Bravo, J.I.; Son, J.M.; Lee, C.; Benayoun, B.A. Remodeling of the H3 nucleosomal landscape during mouse aging. Transl. Med. Aging 2020, 4, 22–31.
- Hauer, M.H.; Seeber, A.; Singh, V.; Thierry, R.; Sack, R.; Amitai, A.; Kryzhanovska, M.; Eglinger, J.; Holcman, D.; Owen-Hughes, T.; et al. Histone degradation in response to DNA damage enhances chromatin dynamics and recombination rates. Nat. Struct. Mol. Biol. 2017, 24, 99–107.
- de Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110.
- Bar, C.; Blasco, M.A. Telomeres and telomerase as therapeutic targets to prevent and treat age-related diseases. F1000Research 2016, 5, F1000 Faculty Rev-89.
- Gottschling, D.E.; Aparicio, O.M.; Billington, B.L.; Zakian, V.A. Position effect at S. cerevisiae telomeres: Reversible repression of Pol II transcription. Cell 1990, 63, 751–762.
- Andrulis, E.D.; Neiman, A.M.; Zappulla, D.C.; Sternglanz, R. Perinuclear localization of chromatin facilitates transcriptional silencing. Nature 1998, 394, 592–595.
- Galy, V.; Olivo-Marin, J.C.; Scherthan, H.; Doye, V.; Rascalou, N.; Nehrbass, U. Nuclear pore complexes in the organization of silent telomeric chromatin. Nature 2000, 403, 108–112.
- Weierich, C.; Brero, A.; Stein, S.; von Hase, J.; Cremer, C.; Cremer, T.; Solovei, I. Three-dimensional arrangements of centromeres and telomeres in nuclei of human and murine lymphocytes. Chromosome Res. 2003, 11, 485–502.
- Raz, V.; Vermolen, B.J.; Garini, Y.; Onderwater, J.J.; Mommaas-Kienhuis, M.A.; Koster, A.J.; Young, I.T.; Tanke, H.; Dirks, R.W. The nuclear lamina promotes telomere aggregation and centromere peripheral localization during senescence of human mesenchymal stem cells. J. Cell Sci. 2008, 121, 4018–4028.
- Narita, M.; Narita, M.; Krizhanovsky, V.; Nunez, S.; Chicas, A.; Hearn, S.A.; Myers, M.P.; Lowe, S.W. A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell 2006, 126, 503–514.
- Narita, M.; Nunez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716.
- Kreiling, J.A.; Tamamori-Adachi, M.; Sexton, A.N.; Jeyapalan, J.C.; Munoz-Najar, U.; Peterson, A.L.; Manivannan, J.; Rogers, E.S.; Pchelintsev, N.A.; Adams, P.D.; et al. Age-associated increase in heterochromatic marks in murine and primate tissues. Aging Cell 2011, 10, 292–304.
- Zhang, R.; Poustovoitov, M.V.; Ye, X.; Santos, H.A.; Chen, W.; Daganzo, S.M.; Erzberger, J.P.; Serebriiskii, I.G.; Canutescu, A.A.; Dunbrack, R.L.; et al. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev. Cell 2005, 8, 19–30.
- Kozak, M.L.; Chavez, A.; Dang, W.; Berger, S.L.; Ashok, A.; Guo, X.; Johnson, F.B. Inactivation of the Sas2 histone acetyltransferase delays senescence driven by telomere dysfunction. EMBO J. 2010, 29, 158–170.
- Smeal, T.; Claus, J.; Kennedy, B.; Cole, F.; Guarente, L. Loss of transcriptional silencing causes sterility in old mother cells of S. cerevisiae. Cell 1996, 84, 633–642.
- Harley, C.B. Telomere loss: Mitotic clock or genetic time bomb? Mutat. Res. 1991, 256, 271–282.
- Munro, J.; Steeghs, K.; Morrison, V.; Ireland, H.; Parkinson, E.K. Human fibroblast replicative senescence can occur in the absence of extensive cell division and short telomeres. Oncogene 2001, 20, 3541–3552.
- Marthandan, S.; Priebe, S.; Hemmerich, P.; Klement, K.; Diekmann, S. Long-term quiescent fibroblast cells transit into senescence. PLoS ONE 2014, 9, e115597.
- Sousa-Victor, P.; Gutarra, S.; Garcia-Prat, L.; Rodriguez-Ubreva, J.; Ortet, L.; Ruiz-Bonilla, V.; Jardi, M.; Ballestar, E.; Gonzalez, S.; Serrano, A.L.; et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 2014, 506, 316–321.
- Tumpel, S.; Rudolph, K.L. Quiescence: Good and Bad of Stem Cell Aging. Trends Cell Biol. 2019, 29, 672–685.
- Lin, C.; Li, H.; Liu, J.; Hu, Q.; Zhang, S.; Zhang, N.; Liu, L.; Dai, Y.; Cao, D.; Li, X.; et al. Arginine hypomethylation-mediated proteasomal degradation of histone H4-an early biomarker of cellular senescence. Cell Death Differ. 2020, 27, 2697–2709.
- Zhang, X.S.; Wang, T.; Lin, X.W.; Denlinger, D.L.; Xu, W.H. Reactive oxygen species extend insect life span using components of the insulin-signaling pathway. Proc. Natl. Acad. Sci. USA 2017, 114, E7832–E7840.
- Lim, Y.; Lee, E.; Lee, J.; Oh, S.; Kim, S. Down-regulation of asymmetric arginine methylation during replicative and H2O2-induced premature senescence in WI-38 human diploid fibroblasts. J. Biochem. 2008, 144, 523–529.
- Jiang, T.X.; Ma, S.; Han, X.; Luo, Z.Y.; Zhu, Q.Q.; Chiba, T.; Xie, W.; Lin, K.; Qiu, X.B. Proteasome activator PA200 maintains stability of histone marks during transcription and aging. Theranostics 2021, 11, 1458–1472.
- Peleg, S.; Feller, C.; Ladurner, A.G.; Imhof, A. The Metabolic Impact on Histone Acetylation and Transcription in Ageing. Trends Biochem. Sci. 2016, 41, 700–711.
- Yi, S.J.; Kim, K. New Insights into the Role of Histone Changes in Aging. Int. J. Mol. Sci. 2020, 21, 8241.
- Ucar, D.; Marquez, E.J.; Chung, C.H.; Marches, R.; Rossi, R.J.; Uyar, A.; Wu, T.C.; George, J.; Stitzel, M.L.; Palucka, A.K.; et al. The chromatin accessibility signature of human immune aging stems from CD8+ T cells. J. Exp. Med. 2017, 214, 3123–3144.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
929
Revisions:
2 times
(View History)
Update Date:
27 Feb 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No