Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ankit Ganeshpurkar | -- | 3542 | 2024-02-22 18:07:41 | | | |
| 2 | Jason Zhu | Meta information modification | 3542 | 2024-02-23 03:00:39 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Dhudum, R.; Ganeshpurkar, A.; Pawar, A. AI Applications in Drug Discovery. Encyclopedia. Available online: https://encyclopedia.pub/entry/55362 (accessed on 09 August 2026).
Dhudum R, Ganeshpurkar A, Pawar A. AI Applications in Drug Discovery. Encyclopedia. Available at: https://encyclopedia.pub/entry/55362. Accessed August 09, 2026.
Dhudum, Rushikesh, Ankit Ganeshpurkar, Atmaram Pawar. "AI Applications in Drug Discovery" Encyclopedia, https://encyclopedia.pub/entry/55362 (accessed August 09, 2026).
Dhudum, R., Ganeshpurkar, A., & Pawar, A. (2024, February 22). AI Applications in Drug Discovery. In Encyclopedia. https://encyclopedia.pub/entry/55362
Dhudum, Rushikesh, et al. "AI Applications in Drug Discovery." Encyclopedia. Web. 22 February, 2024.
Copy Citation
The drug discovery and development process is very lengthy, highly expensive, and extremely complex in nature. Considering the time and cost constraints associated with conventional drug discovery, new methods must be found to enhance the declining efficiency of traditional approaches. Artificial intelligence (AI) has emerged as a powerful tool that harnesses anthropomorphic knowledge and provides expedited solutions to complex challenges. Advancements in AI and machine learning (ML) techniques have revolutionized their applications to drug discovery and development.
artificial intelligence
machine learning
deep learning
1. AI in Drug Target Identification
Drug development is a long, expensive, and risky process that requires more than 10 years and USD 2 billion to bring a new drug to market [1]. Fewer than 500 effective drug targets had been discovered by 2022, which is a negligible portion of the estimated druggable targets in humans [2][3][4]. Target identification is one of the most important processes to determine the biological cause of a disease and provide effective therapies [5]. It is the process of selecting appropriate biological molecules or cellular pathways that can be altered by drugs to achieve therapeutic benefits. The availability of biomedical data has increased in recent years, ranging from basic research into the causes of disease to clinical studies. However, this large amount of information poses challenges for data analysis in terms of scalability, data integration, data quality, noise, computational complexity, interpretability, and validation. AI can manage and analyse such complex networks of biological data. Recently, a promising method for target identification has been developed that combines multi-omics data with AI algorithms [6]. Puna et al. combined various bioinformatics and DL-based models trained using disease-specific text and multi-omics data to prioritize treatable genes and identify potential therapeutic targets in amyotrophic lateral sclerosis (ALS), yielding 18 potential therapeutic targets for ALS. Leveraging more than 20 AI and bioinformatics models, PandaOmics assesses targets by considering their associations with specific diseases, along with information on druggability, developmental stage, and tissue specificity. By combining omics AI scores, text-based AI scores, finance scores, and key opinion leaders (KOL) ratings, PandaOmics could forecast the target genes linked to a particular disease through the use of sophisticated DL models and AI techniques [7]. Validation of proposed AI therapeutic targets for ALS in an ALS-mimicking drosophila model revealed eight unreported targets whose elimination significantly reverses ocular neurodegeneration. Zhang et al. also developed a ML-based technique to recognize KANK1 as a novel ALS-related gene in the same therapeutic area and confirmed the neurotoxic consequences of KANK1 mutations replicated by CRISPR-Cas9 in human neurons [8]. DeepDTnet was developed by Zeng et al. to aid in the in silico discovery of molecular targets for already approved drugs. It is based on 15 different types of chemicals, genomic, phenotypic, and cellular networks. In a mouse model of multiple sclerosis, one of the discovered drugs targeting human ROR-γt demonstrated specific therapeutic benefit [9].
AI has received a lot of attention, and ML-based algorithms, especially AI techniques, have produced remarkable results in the pharmaceutical industry [10]. The modern DL designs such as generative adversarial networks (GANs), recurrent neural networks (RNNs), and transfer learning approaches are attracting more attention and are used in many more healthcare applications than older ML techniques [11]. AI-powered Large Modules (LMs) help to speed up target identification and simplify searches. In addition to using a multitask learning neural network with shared character and word layers (MTM-CW), automatic biomedical named entity recognition (BioNER) is a useful method for identifying chemicals, diseases, and genes that are embedded in free-text documents [12][13]. AI-driven LMs offer potential advantages, including the capability to analyse data and assist in the identification and prioritization of targets [12].
Furthermore, Fabris et al.—using genetic or protein traits such as gene ontology terms, protein–protein interactions, and biological pathways—developed a DL approach with a new modular architecture to identify human genes associated with several age-related diseases [14] (Figure 1).
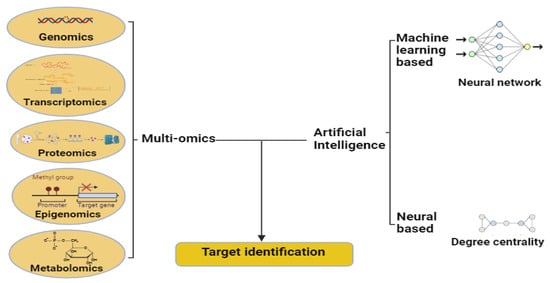
Figure 1. Artificial intelligence employs machine learning and neural network techniques to effectively identify targets. Figure created with BioRender.com.
Synthetic data are data that have been intentionally created to resemble real-world patterns and characteristics. AI algorithms can be used to generate synthetic data to replicate different biological circumstances, allowing scientists to explore and test a wider range of possible outcomes [15][16]. Additionally, the predictions of AI systems may be verified using synthetic data that provide higher assurance for the target discovery process. There are two groups of AI algorithms; i.e., ML-based bioanalysis, and network-based bioanalysis algorithms. These are commonly used for predictive cancer target identification and drug development. However, even after the use of ML algorithm, there are still three significant obstacles to target identification and drug development for cancer [17]. The difficulties are the lack of reliable data for validation, integration of disparate information. and DL models that are difficult to understand [18][19][20]. Although there has been significant improvement in this field, a lot more progress is required.
2. AI for Insightful MD Data Analysis
Molecular dynamics (MD) are extensively employed in the field of drug discovery, playing a crucial role in understanding molecular interactions and predicting drug behaviour. Its research applications include lipid membrane permeability, protein–ligand binding, protein–protein interactions, and partition coefficients. The advantage of MD is the ability to determine thermodynamic characteristics such as the free energy of binding in atomistic detail [21]. Based on a general model of physics that governs interatomic interactions, MD predict how each atom in a protein or other molecular system will move over time [22]. The fundamental concept of MD is to determine the force that all other atoms in a biomolecular system exert on each atom by knowing the position of all. To determine the forces in an MD simulation, a model called a molecular mechanical force field is used, which is often adjusted to the results of some experimental observations and the results of quantum mechanical calculations. Additionally, these simulations can predict how biomolecules respond to changes such as mutation, phosphorylation, protonation, or the addition or removal of a ligand at the atomic level [23]. Traditional MD tools such as GROMACS 2021, Amber 20, NAMD 2.14, and GROMOS 56a7 are well established and used to study interactions between disease targets and drugs [24][25][26][27]. With increased computing power and advanced software, MD is an effective method for studying systems of biomolecules and ligands; however, it has to deal with enormous amounts of data and extremely complex simulated systems [28]. The massive amounts of data generated by MD can be processed quickly and efficiently using DL techniques. To increase the efficiency and speed of analysing MD data, DL can transform the high-dimensional structure space into a low-dimensional feature space. Surface extraction and free energy kinetics, force fields, coarse-grained molecular dynamics, thermodynamics, and other applications are just a few of the many applications of DL in molecular simulations. ML models are important tools in the field of MD. With its exceptional ability to predict chemical qualities such as binding affinity and solubility, RF models are a useful tool for managing intricate, high-dimensional data. MD trajectories are analysed using RNNs, which capture temporal relationships and provide insights into dynamic processes. Because they are good at evaluating chemical structures, graph neural networks (GNNs) are a powerful tool for tasks such as drug-target binding and protein–protein interaction prediction. Meanwhile, the creation of new molecular structures is facilitated by GANs, which accelerates progress in materials research and drug development. These ML models including GANs, GNNs, RNNs, and RFs have transformed the field of molecular dynamics and offer invaluable resources for comprehending and investigating molecular behaviour [29]. Plante et al. used a DL technique to process large MD trajectories and classify GPCR ligands into full, partial, or reverse agonists with good accuracy. The red, green, and blue (RGB) pixels represent the X, Y, and Z coordinates of the protein atoms in the MD trajectory. The confusion matrix was used to train the neural network to predict the classification class labels of GPCR ligands.
In molecular docking, the scoring factor or scoring function (SF), is a mathematical function used to evaluate the binding affinity between a ligand and a receptor. Van der Waals forces, electrostatic interactions, hydrogen bonds, and hydrophobic interactions are only a few of the molecular interactions that are taken into account by the SF factor. Through its guidance of lead identification, virtual screening, and ligand optimization activities, it plays a critical part in the drug development process. When comparing scoring criteria between ML and traditional methodologies, ML approaches have several benefits over conventional methods. These techniques utilize sophisticated algorithms, such as neural networks or RF, to identify intricate patterns and correlations within the data. More accurate predictions are made possible by their ability to manage high-dimensional feature spaces and capture non-linear correlations. Conventional methods, on the other hand, rely on pre-established mathematical formulas that are derived from empirical or physical principles. Conventional methods may not be able to fully capture the intricacy of molecular interactions, despite their widespread usage and insightful contributions. In 2017, Ragoza et al. developed a technique using CNN scoring algorithms to automatically learn the basic binding-related properties of the PLI, given the full 3D description of the PLI as input. Their CNN scoring functions were optimized to distinguish known binders from non-ligands, and between valid and invalid binder positions [30]. A ML-based grading mechanism was created by Nguyen et al. in 2018 to choose the postures produced by GOLD, GLIDE, and Autodock Vina. Using a targeting ligand, they created a complex training dataset through WPB. They then docked the ligands again to the proteins of the selected complexes and used CNN to collect topological features, and implemented RF to understand biomolecular structure. The final predictions for this strategy were determined by combining the energy values predicted by these two machine learning algorithms [31].
3. Compound Screening with AI
The aim of drug discovery is to identify small molecules that can alter the function of the target protein and disease phenotypic characteristics. At the same time, there is also a need to search for small molecules with good pharmacokinetic properties and minimal toxicity. The identification of drug candidates, their validation, pharmacokinetics, and preclinical toxicity assessment are difficult, time-consuming, and expensive processes. It takes an average of 10 to 12 years for a drug to reach the market, and between USD 800 and USD 1.8 billion is spent on each successfully discovered drug [32][33]. The application of AI in drug screening began a revolutionary era in pharmaceutical research, significantly reducing R&D costs by 50% while increasing efficiency and accuracy [34]. AI addresses various challenges related to drug screening, including predicting physicochemical properties and assessing bioactivity and toxicity.
3.1. Primary Drug Screening: Enhancing Cell Classification and Sorting
AI can help to ease the load of repetitive and challenging tasks, making the drug development process faster. These tasks involve various activities such as sorting cells, calculating properties of small molecules, using computers to create organic compounds, designing new compounds, developing tests, and predicting the 3D shapes of target molecules [35]. Accurate identification of cell type is possible using AI-based approaches, particularly the least square vector support method (LS-SVM). With a classification accuracy of 95.34%, this method makes a significant contribution to high-throughput automated cell sorting techniques [35][36][37]. In addition, the interpretation of computerized electrocardiography (ECG), which is a fundamental step in the clinical processes of diagnosis and treatment, has shown the promise of AI [38].
Based on a CNN architecture, Nitta et al. developed the Intelligent Image-activated Cell Sorting system (iIACS), which can sort cells in real time based on cell pictures. The system combines a double membrane pump, a two-stage microfluidic chip with 3D hydraulic focusing, and a high-speed fluorescence microscope to enable automatic liquid focusing, cell sorting, and real-time detection. The intercellular contacts and intracellular localization of proteins allows sorting of blood cells and microalgae in real time. To obtain higher-quality cell images and reduce processing time, researchers modified the iIACS-based cell imaging approach with an image sensor-based optomechanical flow imaging method and improved the system hardware [36].
3.2. Secondary Drug Screening
3.2.1. Predicting Physicochemical Properties
AI is ideal for secondary drug screening, where predicting physicochemical properties is crucial for drug development. Deep neural networks (DNNs) algorithms that use chemical descriptors such as SMILES strings and potential energy measurements are used to generate viable compounds [39][40]. To predict chemical properties, these networks are divided into generation and prediction phases. Each stage is trained independently through supervised learning [39]. The study highlights the importance of considering physiochemical parameters such as melting point and logP when selecting the best drug candidates. Using data from the Environmental Protection Agency (EPA) and the Estimation Program Interface (EPI) package, Yang et al. developed a quantitative structure–property relationship (QSPR) method to determine six physicochemical properties of environmental pollutants. These include the bioconcentration factor (BCF), log P, log S, melting point (MP), boiling point (BP), and vapour pressure (log VP). The lipophilicity and solubility of various substances were predicted using neural networks based on ALGOPS software (version 2.1) and ADMET predictor. It has been shown that it is possible to predict the solubility of compounds using DL techniques such as graph-based convolutional neural networks (CVNN) and recurrent neural networks on undirected graphs [41]. Panapitiya et al. evaluated various molecular representation techniques (such as molecular descriptors, SMILES, molecular graphs, and 3D atomic coordinates) and deep learning techniques (such as fully connected neural networks, RNNs, graph neural networks, and SchNet) for solubility prediction. According to the authors’ results, which were based on the same test dataset, the fully connected neural network outperformed other neural networks in predicting solubility with chemical descriptors. In addition, the researchers examined the importance of different variables in prediction and found that two-dimensional molecular descriptors made the greatest contribution [42].
3.2.2. Predicting Bioactivity: Optimizing Compound Activity
ML techniques, including matched molecular pairs (MMPs), RF, gradient boosting machines (GBMs), and DNNs, are used to predict chemical bioactivity. The performance of RF and GBMs is outperformed particularly well by the combination of MMP with DNN [43][44][45]. This method can predict multiple bioactivity characteristics including oral exposure, intrinsic clearance, ADME, and mode of action, which helps in decision-making in drug development [46][47][48]. Koutsoukas et al. used a multilayer perceptron model based on molecular fingerprint representation of the compounds to predict various bioactivity (pKi and pIC50) against seven targets, including two GPCRs, cannabinoid receptor 1 and dopamine receptor D4. Additionally, the researchers found that MLP performs better than traditional ML techniques on large datasets. However, they pointed out that DL models require much finer tuning of hyperparameters to achieve high predictive performance [49]. Stokes et al. proposed a direct message neural network capable of predicting antimicrobial activity. They generated a feature vector for each molecule by first building a molecular graph based on its SMILES and then combining bonding information (e.g., bond type and stereochemistry) and atomic features (e.g., the number of bonds for each atom and the atom number) used. The optimized feature vector was fed into a feedforward neural network, which acquired the antibacterial properties of the chemical by repeatedly performing the message transmission process [50].
3.2.3. Toxicity Prediction: Mitigating Risk through AI
AI-based toxicity prediction plays a key role in evaluating drug safety. The DeepTox algorithm is effective in correctly assessing the toxicity of a substance [51]. This capacity to forecast could prevent probable side effects during drug development. The application of open-source technologies such as TargeTox and PrOCTOR enhances researchers’ proficiency in the identification and resolution of toxicological issues, thereby advancing the field of toxicity prediction [52][53]. However, the literature lacks essential information, even for basic molecular structures. This absence hinders the ability to conduct a fundamental environmental assessment for a therapeutic molecule’s synthesis path. To address this, simple algorithms use substance interpolation based on known toxicity and similar structures. By leveraging AI expertise, researchers can efficiently discover target-specific compounds, predict physicochemical properties, assess bioactivity, and predict compound toxicity.
4. Drug Design and Optimization
The aim of drug design is to obtain small molecules that could meet various criteria, including efficacy for pharmacological purposes, an appropriate safety profile, appropriate chemical and biological properties, and sufficient innovation to secure intellectual property rights for commercial success, etc. [54]. The computational tools have been crucial to drug discovery and have completely changed the way drugs are designed. There are still several problems associated with traditional computational techniques, including input time, computational cost, and reliability [55][56]. AI could overcome all the obstacles associated with computational drug development, thereby increasing the usefulness of computational techniques in drug development.
4.1. AI in Molecular Design
The process of automatically suggesting novel chemical structures that align most effectively with a specified molecular profile is referred to as “de novo molecular design” [57]. A virtual chemical library is created for computational testing followed by synthesis, and characterization. The variational autoencoder and GANs are two significant technological advances in deep generative modelling [58][59]. “Molecular Autoencoder”, studied by Gómez-Bombarelli et al., was the first to demonstrate detailed generative modelling of molecules. An interesting method is the Variational Autoencoder, which connects an encoder network with a decoder network consisting of two neural networks [60]. The encoder network transforms the chemical structures defined in SMILES into a latent space represented by a real-valued continuous vector. The decoder component can convert latent spatial vectors into chemical structures. De novo design has also been successfully applied through RNNs. They were originally developed in the field of natural language processing [61]. Sequential information is fed into the RNN. The most commonly used models for modelling and creating sequences are RNNs [62][63]. A GAN was successfully employed by Kadurin et al. to recommend potential anticancer drugs [64]. Assmann et al. discussed the practical difficulties in using de novo design to facilitate the identification of new CDK9 inhibitors. The use of the molecules suggested by the molecular generator as seeds in the similarity search of the Enamine REAL library is characterized by an improved variation of the vs. approach. Of the 69 compounds tested, seven were active against CDK9 [65]. Perron et al. have published another useful example of applying generative methods to find optimum solutions to multiparameter objectives using an RNN-based generative model [66]. Li et al. looked at the potential of RNN-based de novo design techniques to produce new molecule inhibitors in chemical space that has been thoroughly researched. Scientists announced their search for new inhibitors of CDK4 kinases and the well-studied proto-oncogene protein serine/threonine kinase 1 (PIM1). After testing four different drugs, they identified a potent PIM1 inhibitor and two key CDK4 inhibitor compounds [67].
4.2. Predicting Pharmacokinetics and Pharmacodynamics
The key concepts of pharmacology include pharmacokinetics and pharmacodynamics. While pharmacodynamics focuses on how a drug works in the body and how it affects other systems in the body, pharmacokinetics deals with the study of drug absorption, distribution, metabolism, and elimination (ADME) [68]. The application of AI techniques in pharmacokinetics and pharmacodynamics has created new opportunities to improve drug development and personalized treatments. It can analyse complex datasets, identify trends and make predictions that could improve patient outcomes, improve drug delivery and minimize side effects [69][70]. ML and DL techniques are widely used to predict pharmacokinetic parameters. Numerous ML techniques—including Bayesian model, RF, SVM, ANN, and decision tree—have been used to predict the ADME of drugs. To predict various pharmacokinetic parameters such as drug absorption, bioavailability, clearance, volume of distribution, and half-life, DL algorithms such as Convolutional Neural Networks (CNN), Short-Term Memory (LSTM), and RNN are often used. A computational method called quantitative structure–activity relationship (QSAR) uses the chemical structure of a molecule to predict its biological activity [71][72][73]. With improved training data, a 47th version of admetSAR 2.0 is now available. This program also includes a module called ADMETopt, which is used to optimize lead activity based on expected ADMET attributes [74]. AI techniques facilitate the modelling of drug–receptor interactions and the prediction of drug efficacy and toxicity in the field of pharmacodynamics. The use of AI in pharmacokinetics and pharmacodynamics can significantly accelerate the drug discovery process and improve precision medicine [70][75]. Obrezanova et al. used conventional ML techniques and multitask convolutional neural networks to calculate time-dependent pharmacokinetic profiles and nine in vivo pharmacokinetic parameters in rats (oral and intravenous administration) based on in vitro measured ADME properties and molecular chemical structures of 3000 different compounds [76]. Ye et al. used transfer learning and multitasked learning to pre-train the model on over 30 million bioactivity data points. The model was then used to estimate four human pharmacokinetic parameters: oral bioavailability, plasma protein binding, Vd, and half-life, for 1104 FDA-approved small-molecule drugs. Compared to other traditional ML techniques, their DL model showed the highest performance (although not always by a significant margin) and generalization ability, achieving a mean absolute error or MAE = 0.31 for oral bioavailability and MAE = 0.17 for Vd [77]. Interestingly, Lou et al. created a model that predicts the bioavailability of mAbs administered through subcutaneous preparation in humans. A dataset of 45 clinical mAbs—with sequence and structure-based features including isoelectric point, total charge, aggregation propensity, solubility score, surface hydrophobicity spots, positive charge, and negative charge (with a threshold of 70% bioavailability)—were used to build a classification model. The study used a range of traditional Scikit-Learn ML techniques such as Adaptive Boost, Multilayer Perceptron, RF, and SVM. Among them, the tree approach showed the highest accuracy, reaching 78% [78].
Two areas that benefit greatly from the implementation of AI algorithms are drug design and optimization. De novo design, virtual screening, and structure-based drug design are just a few examples of these algorithms. The application of AI to drug development and optimization has a transformative impact on the discipline, enabling the rapid discovery of new therapeutic candidates and the more targeted and effective exploration of chemical space.
References
- Hinkson, I.V.; Madej, B.; Stahlberg, E.A. Accelerating therapeutics for opportunities in medicine: A paradigm shift in drug discovery. Front. Pharmacol. 2020, 11, 770.
- Zhou, Y.; Zhang, Y.; Lian, X.; Li, F.; Wang, C.; Zhu, F.; Qiu, Y.; Chen, Y. Therapeutic target database update 2022: Facilitating drug discovery with enriched comparative data of targeted agents. Nucleic Acids Res. 2022, 50, D1398–D1407.
- Kana, O.; Brylinski, M. Elucidating the druggability of the human proteome with e findsite. J. Comput.-Aided Mol. Des. 2019, 33, 509–519.
- Finan, C.; Gaulton, A.; Kruger, F.A.; Lumbers, R.T.; Shah, T.; Engmann, J.; Galver, L.; Kelley, R.; Karlsson, A.; Santos, R. The druggable genome and support for target identification and validation in drug development. Sci. Transl. Med. 2017, 9, eaag1166.
- Schenone, M.; Dančík, V.; Wagner, B.K.; Clemons, P.A. Target identification and mechanism of action in chemical biology and drug discovery. Nat. Chem. Biol. 2013, 9, 232–240.
- Pun, F.W.; Ozerov, I.V.; Zhavoronkov, A. AI-powered therapeutic target discovery. Trends Pharmacol. Sci. 2023, 44, 561–572.
- Pun, F.W.; Liu, B.H.M.; Long, X.; Leung, H.W.; Leung, G.H.D.; Mewborne, Q.T.; Gao, J.; Shneyderman, A.; Ozerov, I.V.; Wang, J. Identification of therapeutic targets for amyotrophic lateral sclerosis using PandaOmics–An AI-Enabled Biological Target Discovery Platform. Front. Aging Neurosci. 2022, 14, 638.
- Zhang, S.; Cooper-Knock, J.; Weimer, A.K.; Shi, M.; Moll, T.; Marshall, J.N.; Harvey, C.; Nezhad, H.G.; Franklin, J.; dos Santos Souza, C. Genome-wide identification of the genetic basis of amyotrophic lateral sclerosis. Neuron 2022, 110, 992–1008.e1011.
- Zeng, X.; Zhu, S.; Lu, W.; Liu, Z.; Huang, J.; Zhou, Y.; Fang, J.; Huang, Y.; Guo, H.; Li, L. Target identification among known drugs by deep learning from heterogeneous networks. Chem. Sci. 2020, 11, 1775–1797.
- McCulloch, W.S.; Pitts, W. A logical calculus of the ideas immanent in nervous activity. Bull. Math. Biophys. 1943, 5, 115–133.
- Zhavoronkov, A.; Mamoshina, P.; Vanhaelen, Q.; Scheibye-Knudsen, M.; Moskalev, A.; Aliper, A. Artificial intelligence for aging and longevity research: Recent advances and perspectives. Ageing Res. Rev. 2019, 49, 49–66.
- Giorgi, J.M.; Bader, G.D. Towards reliable named entity recognition in the biomedical domain. Bioinformatics 2020, 36, 280–286.
- Wang, X.; Zhang, Y.; Ren, X.; Zhang, Y.; Zitnik, M.; Shang, J.; Langlotz, C.; Han, J. Cross-type biomedical named entity recognition with deep multi-task learning. Bioinformatics 2019, 35, 1745–1752.
- Fabris, F.; Palmer, D.; Salama, K.M.; de Magalhaes, J.P.; Freitas, A.A. Using deep learning to associate human genes with age-related diseases. Bioinformatics 2020, 36, 2202–2208.
- Shayakhmetov, R.; Kuznetsov, M.; Zhebrak, A.; Kadurin, A.; Nikolenko, S.; Aliper, A.; Polykovskiy, D. Molecular generation for desired transcriptome changes with adversarial autoencoders. Front. Pharmacol. 2020, 11, 269.
- Viñas, R.; Andrés-Terré, H.; Liò, P.; Bryson, K. Adversarial generation of gene expression data. Bioinformatics 2022, 38, 730–737.
- Li, G.; Luo, J.; Xiao, Q.; Liang, C.; Ding, P.; Cao, B. Predicting microrna-disease associations using network topological similarity based on deepwalk. IEEE Access 2017, 5, 24032–24039.
- Camacho, D.M.; Collins, K.M.; Powers, R.K.; Costello, J.C.; Collins, J.J. Next-generation machine learning for biological networks. Cell 2018, 173, 1581–1592.
- Li, Z.; An, L.; Li, H.; Wang, S.; Zhou, Y.; Yuan, F.; Li, L. Identifying novel genes and chemicals related to nasopharyngeal cancer in a heterogeneous network. Sci. Rep. 2016, 6, 25515.
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M. Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 2019, 18, 463–477.
- Bennett, W.D.; He, S.; Bilodeau, C.L.; Jones, D.; Sun, D.; Kim, H.; Allen, J.E.; Lightstone, F.C.; Ingólfsson, H.I. Predicting small molecule transfer free energies by combining molecular dynamics simulations and deep learning. J. Chem. Inf. Model. 2020, 60, 5375–5381.
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652.
- Hollingsworth, S.A.; Dror, R.O. Molecular dynamics simulation for all. Neuron 2018, 99, 1129–1143.
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25.
- Case, D.A.; Cheatham III, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688.
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130.
- Scott, W.R.; Hünenberger, P.H.; Tironi, I.G.; Mark, A.E.; Billeter, S.R.; Fennen, J.; Torda, A.E.; Huber, T.; Krüger, P.; Van Gunsteren, W.F. The GROMOS biomolecular simulation program package. J. Phys. Chem. A 1999, 103, 3596–3607.
- Wang, Y.; Ribeiro, J.M.L.; Tiwary, P. Machine learning approaches for analyzing and enhancing molecular dynamics simulations. Curr. Opin. Struct. Biol. 2020, 61, 139–145.
- Noé, F.; Tkatchenko, A.; Müller, K.-R.; Clementi, C. Machine learning for molecular simulation. Annu. Rev. Phys. Chem. 2020, 71, 361–390.
- Ragoza, M.; Hochuli, J.; Idrobo, E.; Sunseri, J.; Koes, D.R. Protein–ligand scoring with convolutional neural networks. J. Chem. Inf. Model. 2017, 57, 942–957.
- Nguyen, D.D.; Cang, Z.; Wu, K.; Wang, M.; Cao, Y.; Wei, G.-W. Mathematical deep learning for pose and binding affinity prediction and ranking in D3R Grand Challenges. J. Comput.-Aided Mol. Des. 2019, 33, 71–82.
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214.
- Myers, S.; Baker, A. Drug discovery—An operating model for a new era. Nat. Biotechnol. 2001, 19, 727–730.
- Wang, L.; Ding, J.; Pan, L.; Cao, D.; Jiang, H.; Ding, X. Artificial intelligence facilitates drug design in the big data era. Chemom. Intell. Lab. Syst. 2019, 194, 103850.
- Tripathy, R.K.; Mahanta, S.; Paul, S. Artificial intelligence-based classification of breast cancer using cellular images. Rsc Adv. 2014, 4, 9349–9355.
- Nitta, N.; Sugimura, T.; Isozaki, A.; Mikami, H.; Hiraki, K.; Sakuma, S.; Iino, T.; Arai, F.; Endo, T.; Fujiwaki, Y. Intelligent image-activated cell sorting. Cell 2018, 175, 266–276.e213.
- Samui, P.; Kothari, D. Utilization of a least square support vector machine (LSSVM) for slope stability analysis. Sci. Iran. 2011, 18, 53–58.
- Fernández-Ruiz, I. Artificial intelligence to improve the diagnosis of cardiovascular diseases. Nat. Rev. Cardiol. 2019, 16, 133.
- Popova, M.; Isayev, O.; Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 2018, 4, eaap7885.
- Hessler, G.; Baringhaus, K.-H. Artificial intelligence in drug design. Molecules 2018, 23, 2520.
- Yang, X.; Wang, Y.; Byrne, R.; Schneider, G.; Yang, S. Concepts of artificial intelligence for computer-assisted drug discovery. Chem. Rev. 2019, 119, 10520–10594.
- Panapitiya, G.; Girard, M.; Hollas, A.; Sepulveda, J.; Murugesan, V.; Wang, W.; Saldanha, E. Evaluation of deep learning architectures for aqueous solubility prediction. ACS Omega 2022, 7, 15695–15710.
- Pereira, J.C.; Caffarena, E.R.; Dos Santos, C.N. Boosting docking-based virtual screening with deep learning. J. Chem. Inf. Model. 2016, 56, 2495–2506.
- Sheridan, R.P.; Wang, W.M.; Liaw, A.; Ma, J.; Gifford, E.M. Extreme gradient boosting as a method for quantitative structure–activity relationships. J. Chem. Inf. Model. 2016, 56, 2353–2360.
- Wallach, I.; Dzamba, M.; Heifets, A. AtomNet: A deep convolutional neural network for bioactivity prediction in structure-based drug discovery. arXiv 2015, arXiv:1510.02855.
- Leach, A.G.; Jones, H.D.; Cosgrove, D.A.; Kenny, P.W.; Ruston, L.; MacFaul, P.; Wood, J.M.; Colclough, N.; Law, B. Matched molecular pairs as a guide in the optimization of pharmaceutical properties; a study of aqueous solubility, plasma protein binding and oral exposure. J. Med. Chem. 2006, 49, 6672–6682.
- Paixão, P.; Gouveia, L.F.; Morais, J.A. Prediction of the in vitro intrinsic clearance determined in suspensions of human hepatocytes by using artificial neural networks. Eur. J. Pharm. Sci. 2010, 39, 310–321.
- Keefer, C.E.; Chang, G.; Kauffman, G.W. Extraction of tacit knowledge from large ADME data sets via pairwise analysis. Bioorganic Med. Chem. 2011, 19, 3739–3749.
- Koutsoukas, A.; Monaghan, K.J.; Li, X.; Huan, J. Deep-learning: Investigating deep neural networks hyper-parameters and comparison of performance to shallow methods for modeling bioactivity data. J. Cheminform. 2017, 9, 42.
- Stokes, J.M.; Yang, K.; Swanson, K.; Jin, W.; Cubillos-Ruiz, A.; Donghia, N.M.; MacNair, C.R.; French, S.; Carfrae, L.A.; Bloom-Ackermann, Z. A deep learning approach to antibiotic discovery. Cell 2020, 180, 688–702.e613.
- Jain, S.; Siramshetty, V.B.; Alves, V.M.; Muratov, E.N.; Kleinstreuer, N.; Tropsha, A.; Nicklaus, M.C.; Simeonov, A.; Zakharov, A.V. Large-scale modeling of multispecies acute toxicity end points using consensus of multitask deep learning methods. J. Chem. Inf. Model. 2021, 61, 653–663.
- Lysenko, A.; Sharma, A.; Boroevich, K.A.; Tsunoda, T. An integrative machine learning approach for prediction of toxicity-related drug safety. Life Sci. Alliance 2018, 1.
- Basile, A.O.; Yahi, A.; Tatonetti, N.P. Artificial intelligence for drug toxicity and safety. Trends Pharmacol. Sci. 2019, 40, 624–635.
- Segall, M. Advances in multiparameter optimization methods for de novo drug design. Expert Opin. Drug Discov. 2014, 9, 803–817.
- Baldi, A. Computational approaches for drug design and discovery: An overview. Syst. Rev. Pharm. 2010, 1, 99.
- Lavecchia, A.; Cerchia, C. In silico methods to address polypharmacology: Current status, applications and future perspectives. Drug Discov. Today 2016, 21, 288–298.
- Meyers, J.; Fabian, B.; Brown, N. De novo molecular design and generative models. Drug Discov. Today 2021, 26, 2707–2715.
- Kingma, D.P.; Welling, M. Auto-encoding variational bayes. arXiv 2013, arXiv:1312.6114.
- Goodfellow, I.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative adversarial nets. Adv. Neural Inf. Process. Syst. 2014, 27.
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent. Sci. 2018, 4, 268–276.
- Bengio, Y. Learning deep architectures for AI. Found. Trends® Mach. Learn. 2009, 2, 1–127.
- Géron, A. Hands-On Machine Learning with Scikit-Learn, Keras, and TensorFlow; O’Reilly Media, Inc.: Sebastopol, CA, USA, 2022.
- Heaton, J.; Goodfellow, I.; Bengio, Y.; Courville, A. Deep Learning; The MIT Press: Cambridge, MA, USA, 2016; 800p, ISBN 0262035618.
- Kadurin, A.; Aliper, A.; Kazennov, A.; Mamoshina, P.; Vanhaelen, Q.; Khrabrov, K.; Zhavoronkov, A. The cornucopia of meaningful leads: Applying deep adversarial autoencoders for new molecule development in oncology. Oncotarget 2017, 8, 10883.
- Assmann, M.; Bal, M.; Craig, M.; D’Oyley, J.; Phillips, L.; Triendl, H.; Bates, P.A.; Bashir, U.; Ruprah, P.; Shaker, N. A novel machine learning approach uncovers new and distinctive inhibitors for cyclin-dependent kinase 9. BioRxiv 2020.
- Perron, Q.; Mirguet, O.; Tajmouati, H.; Skiredj, A.; Rojas, A.; Gohier, A.; Ducrot, P.; Bourguignon, M.P.; Sansilvestri-Morel, P.; Do Huu, N. Deep generative models for ligand-based de novo design applied to multi-parametric optimization. J. Comput. Chem. 2022, 43, 692–703.
- Li, X.; Xu, Y.; Yao, H.; Lin, K. Chemical space exploration based on recurrent neural networks: Applications in discovering kinase inhibitors. J. Cheminform. 2020, 12, 42.
- Zhavoronkov, A.; Vanhaelen, Q.; Oprea, T.I. Will artificial intelligence for drug discovery impact clinical pharmacology? Clin. Pharmacol. Ther. 2020, 107, 780–785.
- van Laar, S.A.; Gombert-Handoko, K.B.; Guchelaar, H.J.; Zwaveling, J. An electronic health record text mining tool to collect real-world drug treatment outcomes: A validation study in patients with metastatic renal cell carcinoma. Clin. Pharmacol. Ther. 2020, 108, 644–652.
- Noorbakhsh-Sabet, N.; Zand, R.; Zhang, Y.; Abedi, V. Artificial intelligence transforms the future of health care. Am. J. Med. 2019, 132, 795–801.
- Bhattamisra, S.K.; Banerjee, P.; Gupta, P.; Mayuren, J.; Patra, S.; Candasamy, M. Artificial Intelligence in Pharmaceutical and Healthcare Research. Big Data Cogn. Comput. 2023, 7, 10.
- Westreich, D.; Lessler, J.; Funk, M.J. Propensity score estimation: Neural networks, support vector machines, decision trees (CART), and meta-classifiers as alternatives to logistic regression. J. Clin. Epidemiol. 2010, 63, 826–833.
- Daoui, O.; Elkhattabi, S.; Chtita, S.; Elkhalabi, R.; Zgou, H.; Benjelloun, A.T. QSAR, molecular docking and ADMET properties in silico studies of novel 4, 5, 6, 7-tetrahydrobenzo -thiazol-2-Yl derivatives derived from dimedone as potent anti-tumor agents through inhibition of C-Met receptor tyrosine kinase. Heliyon 2021, 7, e07463.
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069.
- Kim, H.; Kim, E.; Lee, I.; Bae, B.; Park, M.; Nam, H. Artificial intelligence in drug discovery: A comprehensive review of data-driven and machine learning approaches. Biotechnol. Bioprocess Eng. 2020, 25, 895–930.
- Obrezanova, O. Artificial intelligence for compound pharmacokinetics prediction. Curr. Opin. Struct. Biol. 2023, 79, 102546.
- Ye, Z.; Yang, Y.; Li, X.; Cao, D.; Ouyang, D. An integrated transfer learning and multitask learning approach for pharmacokinetic parameter prediction. Mol. Pharm. 2018, 16, 533–541.
- Lou, H.; Hageman, M.J. Machine learning attempts for predicting human subcutaneous bioavailability of monoclonal antibodies. Pharm. Res. 2021, 38, 451–460.
More
Information
Subjects:
Medicine, Research & Experimental
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
23 Feb 2024
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No