 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jinsai Shang | -- | 2214 | 2024-02-19 07:56:22 | | | |
| 2 | Mona Zou | Meta information modification | 2214 | 2024-02-20 08:55:03 | | | | |
| 3 | Mona Zou | + 2 word(s) | 2216 | 2024-02-26 10:32:37 | | |
Video Upload Options
Lung cancer is one of the most lethal malignancies worldwide. Peroxisome proliferator-activated receptor gamma (PPARγ, NR1C3) is a ligand-activated transcriptional factor that governs the expression of genes involved in glucolipid metabolism, energy homeostasis, cell differentiation, and inflammation. Multiple studies have demonstrated that PPARγ activation exerts anti-tumor effects in lung cancer through regulation of lipid metabolism, induction of apoptosis, and cell cycle arrest, as well as inhibition of invasion and migration. Interestingly, PPARγ activation may have pro-tumor effects on cells of the tumor microenvironment, especially myeloid cells. Recent clinical data has substantiated the potential of PPARγ agonists as therapeutic agents for lung cancer. Additionally, PPARγ agonists also show synergistic effects with traditional chemotherapy and radiotherapy. However, the clinical application of PPARγ agonists remains limited due to the presence of adverse side effects.
1. Structure of PPARγ
2. Transcriptional Activities of PPARγ
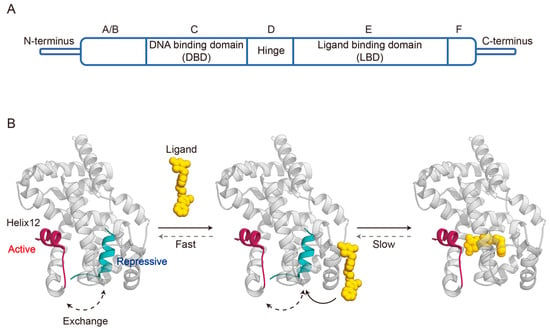
3. Dynamic Mechanisms of Ligand Binding Agonism
4. Role of PPARγ Activation in Lung Cancer
4.1. Regulation of Lipid Metabolism
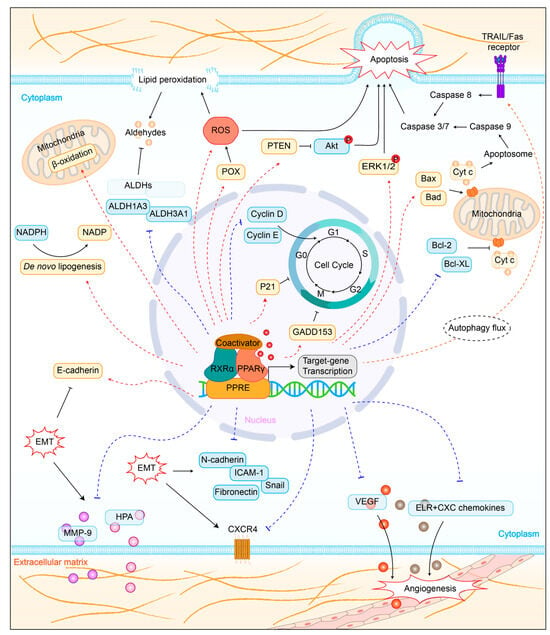
4.2. Promotion of Cell Apoptosis
4.3. Induction of Cell Cycle Arrest
4.4. Inhibition of Tumor Metastasis
4.5. Influence on Tumor Immunity
References
- Fajas, L.; Auboeuf, D.; Raspé, E.; Schoonjans, K.; Lefebvre, A.M.; Saladin, R.; Najib, J.; Laville, M.; Fruchart, J.C.; Deeb, S.; et al. The organization, promoter analysis, and expression of the human PPARgamma gene. J. Biol. Chem. 1997, 272, 18779–18789.
- Fajas, L.; Fruchart, J.C.; Auwerx, J. PPARgamma3 mRNA: A distinct PPARgamma mRNA subtype transcribed from an independent promoter. FEBS Lett. 1998, 438, 55–60.
- Sundvold, H.; Lien, S. Identification of a novel peroxisome proliferator-activated receptor (PPAR) gamma promoter in man and transactivation by the nuclear receptor RORalpha1. Biochem. Biophys. Res. Commun. 2001, 287, 383–390.
- Tontonoz, P.; Hu, E.; Graves, R.A.; Budavari, A.I.; Spiegelman, B.M. mPPAR gamma 2: Tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994, 8, 1224–1234.
- Blanquart, C.; Barbier, O.; Fruchart, J.C.; Staels, B.; Glineur, C. Peroxisome proliferator-activated receptors: Regulation of transcriptional activities and roles in inflammation. J. Steroid Biochem. Mol. Biol. 2003, 85, 267–273.
- Aranda, A.; Pascual, A. Nuclear hormone receptors and gene expression. Physiol. Rev. 2001, 81, 1269–1304.
- Shao, D.; Rangwala, S.M.; Bailey, S.T.; Krakow, S.L.; Reginato, M.J.; Lazar, M.A. Interdomain communication regulating ligand binding by PPAR-gamma. Nature 1998, 396, 377–380.
- Guan, Y.; Breyer, M.D. Peroxisome proliferator-activated receptors (PPARs): Novel therapeutic targets in renal disease. Kidney Int. 2001, 60, 14–30.
- Chandra, V.; Huang, P.; Hamuro, Y.; Raghuram, S.; Wang, Y.; Burris, T.P.; Rastinejad, F. Structure of the intact PPAR-gamma-RXR- nuclear receptor complex on DNA. Nature 2008, 456, 350–356.
- Kroker, A.J.; Bruning, J.B. Review of the Structural and Dynamic Mechanisms of PPARγ Partial Agonism. PPAR Res. 2015, 2015, 816856.
- Danielian, P.S.; White, R.; Lees, J.A.; Parker, M.G. Identification of a conserved region required for hormone dependent transcriptional activation by steroid hormone receptors. EMBO J. 1992, 11, 1025–1033.
- Nolte, R.T.; Wisely, G.B.; Westin, S.; Cobb, J.E.; Lambert, M.H.; Kurokawa, R.; Rosenfeld, M.G.; Willson, T.M.; Glass, C.K.; Milburn, M.V. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 1998, 395, 137–143.
- Frkic, R.L.; Marshall, A.C.; Blayo, A.L.; Pukala, T.L.; Kamenecka, T.M.; Griffin, P.R.; Bruning, J.B. PPARγ in Complex with an Antagonist and Inverse Agonist: A Tumble and Trap Mechanism of the Activation Helix. iScience 2018, 5, 69–79.
- Hernandez-Quiles, M.; Broekema, M.F.; Kalkhoven, E. PPARgamma in Metabolism, Immunity, and Cancer: Unified and Diverse Mechanisms of Action. Front. Endocrinol. 2021, 12, 624112.
- Ricote, M.; Glass, C.K. PPARs and molecular mechanisms of transrepression. Biochim. Biophys. Acta 2007, 1771, 926–935.
- Shang, J.; Mosure, S.A.; Zheng, J.; Brust, R.; Bass, J.; Nichols, A.; Solt, L.A.; Griffin, P.R.; Kojetin, D.J. A molecular switch regulating transcriptional repression and activation of PPARγ. Nat. Commun. 2020, 11, 956.
- Kallenberger, B.C.; Love, J.D.; Chatterjee, V.K.; Schwabe, J.W. A dynamic mechanism of nuclear receptor activation and its perturbation in a human disease. Nat. Struct. Biol. 2003, 10, 136–140.
- Brzozowski, A.M.; Pike, A.C.; Dauter, Z.; Hubbard, R.E.; Bonn, T.; Engström, O.; Ohman, L.; Greene, G.L.; Gustafsson, J.A.; Carlquist, M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997, 389, 753–758.
- Johnson, B.A.; Wilson, E.M.; Li, Y.; Moller, D.E.; Smith, R.G.; Zhou, G. Ligand-induced stabilization of PPARgamma monitored by NMR spectroscopy: Implications for nuclear receptor activation. J. Mol. Biol. 2000, 298, 187–194.
- Bruning, J.B.; Chalmers, M.J.; Prasad, S.; Busby, S.A.; Kamenecka, T.M.; He, Y.; Nettles, K.W.; Griffin, P.R. Partial agonists activate PPARgamma using a helix 12 independent mechanism. Structure 2007, 15, 1258–1271.
- Hughes, T.S.; Chalmers, M.J.; Novick, S.; Kuruvilla, D.S.; Chang, M.R.; Kamenecka, T.M.; Rance, M.; Johnson, B.A.; Burris, T.P.; Griffin, P.R.; et al. Ligand and receptor dynamics contribute to the mechanism of graded PPARγ agonism. Structure 2012, 20, 139–150.
- Brust, R.; Shang, J.; Fuhrmann, J.; Mosure, S.A.; Bass, J.; Cano, A.; Heidari, Z.; Chrisman, I.M.; Nemetchek, M.D.; Blayo, A.L.; et al. A structural mechanism for directing corepressor-selective inverse agonism of PPARγ. Nat. Commun. 2018, 9, 4687.
- Shang, J.; Kojetin, D.J. Structural mechanism underlying ligand binding and activation of PPARγ. Structure 2021, 29, 940–950.e944.
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. CR 2015, 34, 111.
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473.
- Phan, L.M.; Yeung, S.C.; Lee, M.H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1–19.
- Phan, A.N.H.; Vo, V.T.A.; Hua, T.N.M.; Kim, M.K.; Jo, S.Y.; Choi, J.W.; Kim, H.W.; Son, J.; Suh, Y.A.; Jeong, Y. PPARγ sumoylation-mediated lipid accumulation in lung cancer. Oncotarget 2017, 8, 82491–82505.
- Andela, V.B.; Altuwaijri, S.; Wood, J.; Rosier, R.N. Inhibition of beta-oxidative respiration is a therapeutic window associated with the cancer chemo-preventive activity of PPARgamma agonists. FEBS Lett. 2005, 579, 1765–1769.
- Singh, S.; Brocker, C.; Koppaka, V.; Chen, Y.; Jackson, B.C.; Matsumoto, A.; Thompson, D.C.; Vasiliou, V. Aldehyde dehydrogenases in cellular responses to oxidative/electrophilic stress. Free Radic. Biol. Med. 2013, 56, 89–101.
- Pors, K.; Moreb, J.S. Aldehyde dehydrogenases in cancer: An opportunity for biomarker and drug development? Drug Discov. Today 2014, 19, 1953–1963.
- Dinavahi, S.S.; Bazewicz, C.G.; Gowda, R.; Robertson, G.P. Aldehyde Dehydrogenase Inhibitors for Cancer Therapeutics. Trends Pharmacol. Sci. 2019, 40, 774–789.
- Muzio, G.; Trombetta, A.; Maggiora, M.; Martinasso, G.; Vasiliou, V.; Lassen, N.; Canuto, R.A. Arachidonic acid suppresses growth of human lung tumor A549 cells through down-regulation of ALDH3A1 expression. Free. Radic. Biol. Med. 2006, 40, 1929–1938.
- Hua, T.N.M.; Namkung, J.; Phan, A.N.H.; Vo, V.T.A.; Kim, M.K.; Jeong, Y.; Choi, J.W. PPARgamma-mediated ALDH1A3 suppression exerts anti-proliferative effects in lung cancer by inducing lipid peroxidation. J. Recept. Signal Transduct. Res. 2018, 38, 191–197.
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516.
- Sharma, A.; Boise, L.H.; Shanmugam, M. Cancer Metabolism and the Evasion of Apoptotic Cell Death. Cancers 2019, 11, 1144.
- Nicholson, D.W.; Thornberry, N.A. Caspases: Killer proteases. Trends Biochem. Sci. 1997, 22, 299–306.
- Reed, J.C. Apoptosis-targeted therapies for cancer. Cancer Cell 2003, 3, 17–22.
- Ge, L.N.; Yan, L.; Li, C.; Cheng, K. Bavachinin exhibits antitumor activity against non-small cell lung cancer by targeting PPARγ. Mol. Med. Rep. 2019, 20, 2805–2811.
- Li, M.Y.; Hsin, M.K.; Yip, J.; Mok, T.S.; Underwood, M.J.; Chen, G.G. PPARgamma activation extinguishes smoking carcinogen by inhibiting NNK-mediated proliferation. Am. J. Respir. Cell Mol. Biol. 2010, 42, 113–122.
- Liu, Y.; Tian, Y.; Cai, W.; Guo, Y.; Xue, C.; Wang, J. DHA/EPA-Enriched Phosphatidylcholine Suppresses Tumor Growth and Metastasis via Activating Peroxisome Proliferator-Activated Receptor γ in Lewis Lung Cancer Mice. J. Agric. Food Chem. 2021, 69, 676–685.
- Yue, H.; Tian, Y.; Zhao, Z.; Bo, Y.; Guo, Y.; Wang, J. Comparative Study of Docosahexaenoic Acid with Different Molecular Forms for Promoting Apoptosis of the 95D Non-Small-Cell Lung Cancer Cells in a PPARγ-Dependent Manner. Mar. Drugs 2022, 20, 599.
- Kim, T.W.; Hong, D.W.; Kang, C.M.; Hong, S.H. A novel PPARɣ ligand, PPZ023, overcomes radioresistance via ER stress and cell death in human non-small-cell lung cancer cells. Exp. Mol. Med. 2020, 52, 1730–1743.
- Kim, T.W.; Hong, D.W.; Hong, S.H. CB13, a novel PPARγ ligand, overcomes radio-resistance via ROS generation and ER stress in human non-small cell lung cancer. Cell Death Dis. 2020, 11, 848.
- Nazim, U.M.; Moon, J.H.; Lee, Y.J.; Seol, J.W.; Park, S.Y. PPARγ activation by troglitazone enhances human lung cancer cells to TRAIL-induced apoptosis via autophagy flux. Oncotarget 2017, 8, 26819–26831.
- Kim, K.Y.; Ahn, J.H.; Cheon, H.G. Apoptotic action of peroxisome proliferator-activated receptor-gamma activation in human non small-cell lung cancer is mediated via proline oxidase-induced reactive oxygen species formation. Mol. Pharmacol. 2007, 72, 674–685.
- Li, M.; Lee, T.W.; Yim, A.P.; Mok, T.S.; Chen, G.G. Apoptosis induced by troglitazone is both peroxisome proliferator-activated receptor-gamma- and ERK-dependent in human non-small lung cancer cells. J. Cell. Physiol. 2006, 209, 428–438.
- Satoh, T.; Toyoda, M.; Hoshino, H.; Monden, T.; Yamada, M.; Shimizu, H.; Miyamoto, K.; Mori, M. Activation of peroxisome proliferator-activated receptor-gamma stimulates the growth arrest and DNA-damage inducible 153 gene in non-small cell lung carcinoma cells. Oncogene 2002, 21, 2171–2180.
- Ni, J.; Zhou, L.L.; Ding, L.; Zhao, X.; Cao, H.; Fan, F.; Li, H.; Lou, R.; Du, Y.; Dong, S.; et al. PPARγ agonist efatutazone and gefitinib synergistically inhibit the proliferation of EGFR-TKI-resistant lung adenocarcinoma cells via the PPARγ/PTEN/Akt pathway. Exp. Cell Res. 2017, 361, 246–256.
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol. 2022, 23, 74–88.
- Keshamouni, V.G.; Reddy, R.C.; Arenberg, D.A.; Joel, B.; Thannickal, V.J.; Kalemkerian, G.P.; Standiford, T.J. Peroxisome proliferator-activated receptor-gamma activation inhibits tumor progression in non-small-cell lung cancer. Oncogene 2004, 23, 100–108.
- Han, S.; Sidell, N.; Fisher, P.B.; Roman, J. Up-regulation of p21 gene expression by peroxisome proliferator-activated receptor gamma in human lung carcinoma cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 1911–1919.
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458.
- Castaneda, M.; den Hollander, P.; Kuburich, N.A.; Rosen, J.M.; Mani, S.A. Mechanisms of cancer metastasis. Semin. Cancer Biol. 2022, 87, 17–31.
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28.
- Reka, A.K.; Kurapati, H.; Narala, V.R.; Bommer, G.; Chen, J.; Standiford, T.J.; Keshamouni, V.G. Peroxisome proliferator-activated receptor-gamma activation inhibits tumor metastasis by antagonizing Smad3-mediated epithelial-mesenchymal transition. Mol. Cancer Ther. 2010, 9, 3221–3232.
- Li, J.; Chen, L.; Yu, P.; Liu, B.; Zhu, J.; Yang, Y. Telmisartan exerts anti-tumor effects by activating peroxisome proliferator-activated receptor-γ in human lung adenocarcinoma A549 cells. Molecules 2014, 19, 2862–2876.
- Yin, H.; Liu, Y.; Yue, H.; Tian, Y.; Dong, P.; Xue, C.; Zhao, Y.T.; Zhao, Z.; Wang, J. DHA- and EPA-Enriched Phosphatidylcholine Suppress Human Lung Carcinoma 95D Cells Metastasis via Activating the Peroxisome Proliferator-Activated Receptor γ. Nutrients 2022, 14, 4675.
- Choudhary, R.; Li, H.; Winn, R.A.; Sorenson, A.L.; Weiser-Evans, M.C.; Nemenoff, R.A. Peroxisome proliferator-activated receptor-gamma inhibits transformed growth of non-small cell lung cancer cells through selective suppression of Snail. Neoplasia 2010, 12, 224–234.
- Han, S.; Ritzenthaler, J.D.; Rivera, H.N.; Roman, J. Peroxisome proliferator-activated receptor-gamma ligands suppress fibronectin gene expression in human lung carcinoma cells: Involvement of both CRE and Sp1. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L419–L428.
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770.
- Keshamouni, V.G.; Arenberg, D.A.; Reddy, R.C.; Newstead, M.J.; Anthwal, S.; Standiford, T.J. PPAR-gamma activation inhibits angiogenesis by blocking ELR+CXC chemokine production in non-small cell lung cancer. Neoplasia 2005, 7, 294–301.
- Jin, M.Z.; Jin, W.L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 166.
- Li, H.; Sorenson, A.L.; Poczobutt, J.; Amin, J.; Joyal, T.; Sullivan, T.; Crossno, J.T., Jr.; Weiser-Evans, M.C.; Nemenoff, R.A. Activation of PPARγ in myeloid cells promotes lung cancer progression and metastasis. PLoS ONE 2011, 6, e28133.
- Cheng, H.S.; Yip, Y.S.; Lim, E.K.Y.; Wahli, W.; Tan, N.S. PPARs and Tumor Microenvironment: The Emerging Roles of the Metabolic Master Regulators in Tumor Stromal-Epithelial Crosstalk and Carcinogenesis. Cancers 2021, 13, 2153.
- Gou, Q.; Che, S.; Chen, M.; Chen, H.; Shi, J.; Hou, Y. PPARγ inhibited tumor immune escape by inducing PD-L1 autophagic degradation. Cancer Sci. 2023, 114, 2871–2881.


