1. Structure of PPARγ
The
PPARG gene gives rise to four transcripts by differential promoter usage and alternative splicing, which results in the production of PPARγ1 (encoded by
PPARG1,
PPARG3,
PPARG4 mRNAs) and PPARγ2 (encoded by
PPARG2 mRNA) isoforms [
12,
13,
14,
15]. PPARγ2 has the same sequence as PPARγ1, except for an additional 28 amino acids at its N-terminus [
12]. PPARγ1 is ubiquitously abundant in many tissues, whereas PPARγ2 is preferably expressed in adipocytes.
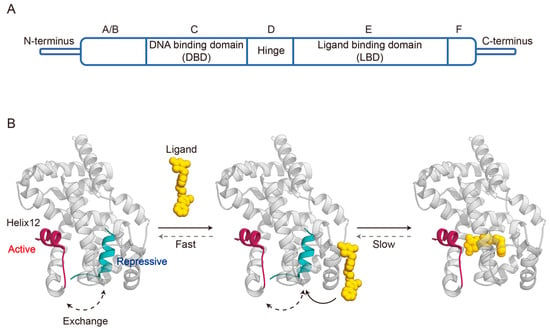
PPARγ shares a typical NR domain structure composed of five domains: A/B, C, D, E, and F domain (
Figure 1A) [
16]. The N-terminal A/B domain is highly variable among the NR family, and harbors a ligand-independent transcriptional activation function region termed AF-1 that regulates PPARγ activation through interdomain coordination and phosphorylation [
17,
18]. The C domain, also known as the DNA-binding domain (DBD), is the most conserved part of NRs and consists of two zinc finger motifs with nine cysteines [
17]. This domain specifically recognizes and binds to the PPAR response elements (PPRE) on the target gene promoter to initiate transcription after forming a heterodimer with the retinoic X receptor α (RXRα) [
19,
20]. The poorly conserved D domain serves as a flexible hinge that allows rotation between the DNA-binding and ligand-binding domains, as well as containing a nuclear localization signal [
17]. The E domain, also named the ligand binding domain (LBD), is the largest region of PPARγ and has four main functions, including a second dimerization interface, the ligand binding pocket, a coregulator binding surface, and ligand-dependent activation function referred to as AF-2 [
21]. The C-terminal F domain is relatively small, and may contribute to interaction with cofactors [
22].
Figure 1. Domain organization of PPARγ (A) and schematic of agonist binding with PPARγ LBD (PDB code: 6ONJ and 6DGL) (B).
Among PPARγ domain structures, the LBD was the first one to be characterized in conformation [
23]. The PPARγ LBD consists of 13 α-helices (termed helix 1–12 and helix 2′) arranged in a three-layered sandwich and a small four-stranded β-sheet. The ligand binding pocket is located in the center of the LBD, and has a large Y- or T-shaped cavity with three branches [
21]. Branch I is composed of helix3, helix5, helix11, and helix12 in the C-terminal AF-2 region; branch II is positioned around helix2′, helix3, helix6, helix7, and the β-sheet region; while branch III is surrounded by the β-sheet, helix2, helix3, and helix5. Adjacent to the ligand-binding pocket, there is an AF-2 coregulator interaction surface formed by the three-dimensional association of helix3, helix4, helix5, and helix12 [
23,
24].
2. Transcriptional Activities of PPARγ
PPARγ regulates gene expression through transactivation and transrepression. In the absence of ligands, PPARγ-RXRα heterodimer binds to PPREs and subsequently recruits corepressors and associated chromatin-modifying enzymes to silence target gene transcription, a process known as ligand-independent repression [
25]. Once the ligand binds to PPARγ, the PPARγ-RXRα heterodimer undergoes a conformational change that releases corepressors in exchange for coactivators, resulting in the transcription of target genes. Furthermore, PPARγ can also negatively regulate gene expression in a ligand-dependent manner by antagonizing other transcription factors, such as nuclear factor-κB and activator protein-1 [
26].
3. Dynamic Mechanisms of Ligand Binding Agonism
In the absence of ligands, helix 12 acts as a highly flexible switching element in equilibrium between many different conformations, ranging from active to repressive (
Figure 1B) [
34,
42]. Agonists stabilize helix 12 in a conformation that exposes the AF-2 surface for the binding of coregulators that control target genes transcription [
43]. Full PPARγ agonists stabilize an active AF-2 surface conformation via forming a critical hydrogen bond with Y473 residues on helix 12, facilitating the recruitment and binding of coactivators [
44]. Partial agonists generally do not form hydrogen bonds with the key residues of the AF-2 regions, including Y473, but mildly stabilize helix 12 through interactions with other regions of the ligand-binding pocket, resulting in differential coactivator recruitment profiles and weak transcriptional activation compared to full agonists [
21,
45,
46]. In contrast, inverse agonists exert transcriptional repression via stabilizing in a conformation that favors the recruitment of corepressors. However, the structural mechanism underlying the ligand-induced repression state is limited. Brust et al. identified R288 as the critical corepressor-selective inverse agonist (T0070907) switch residue, and found that T0070907-bound PPARγ exchanges between two long-lived conformations, one similar to the coactivator-bound state and the other similar to the corepressor-bound state [
33]. Subsequently, our study further verified the structurally diverse mechanism of the inverse agonist-bound state and revealed the mechanism of action of T0070907 [
34]. Briefly, T0070907 can stabilize helix 12 within the orthosteric pocket by pointing the pyridyl group toward the AF-2 surface, thereby increasing corepressor binding affinity.
The preceding section has provided a description of the binding modes demonstrated by various agonists. Subsequently, the dynamic changes in agonist binding with PPARγ LBD will be outlined below. Prior studies remain controversial as to whether ligand binding proceeds through induced fitting or conformational selection mechanisms. Notably, our recent study supported the existence of the induced fit mechanism involving a two-step process of an initial ligand encounter complex followed by a conformational change (
Figure 1B) [
47]. In the absence of ligand, helix 12 in apo-PPARγ LBD exchanges between transcriptionally repressive and a solvent-exposed active conformation through entering and exiting the orthosteric ligand-binding pocket. Agonist binds to the ligand entry site via an initial fast step to form an encounter complex, and this process can occur to either of these conformations. Subsequently, the agonist slowly enters the orthosteric ligand-binding pocket and forms the final ligand-binding pose. In this step, agonist binding to the repressive LBD conformation (helix 12 within the orthosteric pocket) would push helix 12 into an active conformation, while agonist binding to the active LBD conformation (helix 12 outside the orthosteric pocket) would facilitate transition into the final ligand-binding pose.
4. Role of PPARγ Activation in Lung Cancer
4.1. Regulation of Lipid Metabolism
Metabolic reprogramming is a crucial hallmark of malignancy, allowing tumor cells to meet demands for growth, proliferation, and metastasis, as well as be robust against unfavorable environments [
48,
49]. Thus, targeting abnormal tumor metabolic activities, including lipid metabolism, is a rapidly emerging direction for anti-cancer therapy [
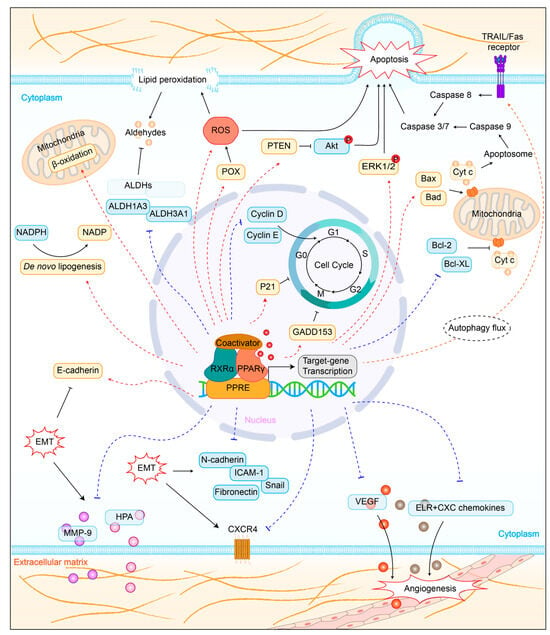
50]. PPARγ is a central regulator of lipid metabolism. Several studies have shown that PPARγ upregulates fatty acid synthesis and β-oxidation in lung cancer (
Figure 2). For example, Phan et al. found that pioglitazone-mediated PPARγ activation induced de novo fatty acid synthesis and β-oxidation in lung cancer [
51]. Importantly, dramatic lipid synthesis could deplete nicotinamide adenine dinucleotide phosphate (NADPH), a major reducing agent important for cellular anti-oxidation systems, leading to disrupted redox balance which, in turn, suppresses lung cancer. Moreover, Andela et al. reported a shift in cellular energy metabolism towards fatty acid oxidation in the lung alveolar carcinoma cell line via treatment with PPARγ agonist troglitazone [
52].
Figure 2. Role of PPARγ activation in lung cancer cells. PPARγ, peroxisome proliferator-activated receptor gamma; RXRα, retinoic X receptor α; PPRE, PPAR response elements; GADD153, DNA-damage inducible gene 153; NADPH, nicotinamide adenine dinucleotide phosphate; ALDH, aldehyde dehydrogenases; ROS, reactive oxygen species; POX, proline oxidase; PTEN, phosphatase and tensin homolog; ERK1/2, extracellular signal-regulated protein kinases 1 and 2; cyt-c, mitochondrial cytochrome c; EMT, epithelial-mesenchymal transition; MMP-9, matrix metalloproteinase 9; HPA, heparanase; ICAM-1, intercellular adhesion molecule-1; VEGF, vascular endothelial growth factor.
Aldehyde dehydrogenases (ALDHs) act as an ‘aldehyde scavenger’ during lipid peroxidation and exhibit high activity in lung cancer [
53,
54]. Inhibition of ALDHs can expose cancer cells to highly reactive and toxic aldehydes, resulting in cell damage and apoptosis [
55]. Notably, PPARγ has been reported to downregulate certain members of the ALDH family to function as a lung cancer inhibitor. For instance, arachidonic acid-induced PPARγ activation suppressed the growth of A549 cells through increasing lipid peroxidation and decreasing ALDH3A1 expression [
56]. Additionally, TZD-mediated PPARγ activation inhibited ALDH1A3 expression to exert anti-proliferative functions in H1993 cells [
57].
4.2. Promotion of Cell Apoptosis
Apoptosis is a homeostatic mechanism to maintain cell populations in normal tissues, whereas tumor cells engage various mechanisms to evade apoptosis for unrestricted proliferation [
58,
59]. Classical pathways of apoptosis include the intrinsic mitochondrial pathway and the extrinsic pathway that induces via the activation of death receptors on the cell surface, both of which result in the activation of cysteine aspartyl-specific proteases (also known as Caspases) to cleave several proteins leading to cell death [
60,
61]. PPARγ promotes apoptosis in lung cancer through dysregulating critical factors in these pathways (
Figure 2). Specifically, PPARγ activation could increase the expression of pro-apoptotic factors Bax and Bad, decrease the expression of anti-apoptotic factors Bcl-2 and Bcl-XL, enhance caspase3 and caspase9 activity, and trigger mitochondrial cytochrome c release [
62,
63,
64,
65,
66,
67]. Furthermore, PPARγ activation enhanced TRAIL-induced apoptosis in human lung adenocarcinoma cells via autophagy flux [
68]. Notably, PPARγ is also able to regulate the upstream signaling pathways to provoke apoptosis. For instance, KR-62980 or rosiglitazone-mediated PPARγ activation promoted the generation of reactive oxygen species (ROS) via proline oxidase (POX) induction, leading to apoptotic cell death in NSCLC [
69]. Troglitazone-mediated PPARγ activation induced the phosphorylation of extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) and subsequently caused apoptosis in NCI-H23 lung cancer cells via a mitochondrial pathway [
70]. Another study showed that troglitazone-mediated PPARγ activation stimulated the expression of DNA-damage inducible gene 153 (GADD153) to trigger growth arrest and endoplasmic reticulum stress-induced apoptosis of NSCLC cells [
71]. PPARγ agonist efatutazone could induce the cell cycle arrest and apoptosis of EGFR-TKI-resistant lung adenocarcinoma cells via PPARγ/phosphatase and tensin homolog (PTEN)/Akt pathway [
72].
4.3. Induction of Cell Cycle Arrest
Cell division is tightly regulated by multiple conserved cell cycle control mechanisms, such as cyclins and cyclin-dependent kinases (CDKs), G1-S transcriptional regulation, DNA damage checkpoint, DNA replication stress checkpoint, and spindle assembly checkpoint [
73]. The eukaryotic cell cycle can be divided into G0, G1, S, G2, and M phases. DNA replicates during the S phase and cell separates during the M phase. Several lines of evidence have shown that PPARγ is involved in cell cycle processes to induce the growth arrest of lung cancer cells (
Figure 2). Troglitazone-mediated PPARγ activation could induce cell cycle arrest in the G0/G1 phase by downregulation of cyclins D and E [
74]. The PPARγ ligands PGJ2, ciglitazone, troglitazone, and GW1929 suppressed human lung carcinoma cell growth by stimulating cyclin-dependent kinase inhibitor p21 expression and reducing cyclin D1 expression [
75].
4.4. Inhibition of Tumor Metastasis
Metastasis is the cause of 90% of cancer-related deaths [
76]. In the process of metastasis, normal cells transform into carcinogenic cells that proliferate uncontrollably, evade the immune system, resist programmed cell death, stimulate angiogenesis, acquire invasive potential, survive in the bloodstream, and establish cancerous growth in distant organs [
77]. Epithelial–mesenchymal transition (EMT), a process through which epithelial cells lose apical–basal polarity, and cell–cell junctions, as well as attain a mesenchymal phenotype with invasive and migratory capabilities, is a critical event in the initiation of metastasis [
77,
78]. The mechanism of PPARγ in lung cancer-related EMT is not yet fully understood. Multiple studies have shown that PPARγ can regulate the expression of EMT-related molecules to exert an inhibitory effect on metastasis (
Figure 2). Specifically, PPARγ activation increased expression of epithelial marker E-cadherin, decreased expression of mesenchymal marker N-cadherin, Snail, and fibronectin, as well as down-regulated expression of matrix metalloproteinase 9 (MMP-9) and heparanase (HPA) [
79,
80,
81,
82,
83]. Moreover, PPARγ activation could also suppress the expression of invasion-related proteins, such as intercellular adhesion molecule-1 (ICAM-1) and C-X-C chemokine receptor type 4 (CXCR4), which function as facilitators in EMT [
80,
81,
83].
Angiogenesis is the major route by which cancer cells spread from the primary tumor to other sites [
77]. Tumor angiogenesis is regulated by both pro- and anti-angiogenic factors, and an imbalance between the two can lead to malformation of the vasculature with excessive branching, hyperpermeability, and leakage [
77,
84]. PPARγ has been reported to be involved in regulating angiogenic factors (
Figure 2). For instance, PPARγ activation could inhibit angiogenesis by blocking the production of ELR + CXC chemokines in NSCLC [
85]. Moreover, vascular endothelial growth factor (VEGF) was drastically downregulated through the PPARγ/NF-κB signaling pathway in human lung carcinoma 95D cells [
64].
4.5. Influence on Tumor Immunity
The role of PPARγ activation in lung cancer immunity remains a controversial issue, which may correlate with the complexity of the immune microenvironment. Notably, the tumor microenvironment encompasses not only malignant cells, but also stromal cells, vascular endothelial cells, as well as various types of immune cells including tumor-associated macrophages and myeloid-derived suppressor cells (MDSCs) [
86]. Interestingly, PPARγ seems to have opposing effects on cancer progression among different cells, with anti-oncogenic effects on cancer cells but pro-oncogenic effects on cancer-associated immune cells [
87,
88]. Gou et al. found that PPARγ inhibits the tumor immune escape by inducing PD-L1 autophagic degradation in NSCLC cells [
89]. Interestingly, this process was independent of the transcriptional activity of PPARγ, but rather formed autophagy receptors through the binding of PPARγ to microtubule-associated protein 1A/1B-light chain 3 (LC3), leading to degradation of PD-L1 in lysosomes. Nevertheless, Li et al. suggested that PPARγ activation in myeloid cells promoted lung cancer progression and metastasis [
87].
This entry is adapted from the peer-reviewed paper 10.3390/biom14020190