Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mehdi Ahangariahangarkolaei | -- | 4110 | 2024-02-19 03:20:18 | | | |
| 2 | Rita Xu | Meta information modification | 4110 | 2024-02-19 03:40:27 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ahangari, M.; Szalai, B.; Lujan, J.; Zhou, M.; Luo, H. High-Capacity Ni-Rich Cathode Materials for Lithium-Ion Batteries. Encyclopedia. Available online: https://encyclopedia.pub/entry/55129 (accessed on 28 July 2026).
Ahangari M, Szalai B, Lujan J, Zhou M, Luo H. High-Capacity Ni-Rich Cathode Materials for Lithium-Ion Batteries. Encyclopedia. Available at: https://encyclopedia.pub/entry/55129. Accessed July 28, 2026.
Ahangari, Mehdi, Benedek Szalai, Josue Lujan, Meng Zhou, Hongmei Luo. "High-Capacity Ni-Rich Cathode Materials for Lithium-Ion Batteries" Encyclopedia, https://encyclopedia.pub/entry/55129 (accessed July 28, 2026).
Ahangari, M., Szalai, B., Lujan, J., Zhou, M., & Luo, H. (2024, February 19). High-Capacity Ni-Rich Cathode Materials for Lithium-Ion Batteries. In Encyclopedia. https://encyclopedia.pub/entry/55129
Ahangari, Mehdi, et al. "High-Capacity Ni-Rich Cathode Materials for Lithium-Ion Batteries." Encyclopedia. Web. 19 February, 2024.
Copy Citation
Lithium-ion batteries are undoubtedly known as the most promising rechargeable batteries. Ternary Ni-rich Li[NixCoyMnz]O2 and Li[NixCoyAlz]O2 cathode materials stand as the ideal candidate for a cathode active material to achieve high capacity and energy density, low manufacturing cost, and high operating voltage.
Ni-rich cathode
surface modification
elemental doping
1. Introduction
In recent decades, the urgent global issues of ever-increasing fossil fuel demand and the alarming threat of global warming have spurred the development of renewable and green energy sources [1]. Within this array of sustainable energy options—for example, hydroelectric, solar, tide, or wind, which all contribute to the global energy needs—rechargeable lithium-ion batteries (LIBs) have prominently emerged as the most sought-after for numerous practical applications, decisively spearheading the electrification of vehicles. This preference arises from their distinctive blend of characteristics, including their lightweight design, impressive power output, substantial energy capacity (ranging from 250 to 693 Wh L−1 and 100 to 265 Wh kg−1), relatively long service life, desirable rate capability, safety features, and minimal environmental impact. Currently, LIBs have achieved widespread success in small-scale portable consumer electronic devices such as cellular phones and laptop computers [2][3]. However, when it comes to utilization of the current state-of-the-art LIBs in more substantial applications, such as passenger electric vehicles (EVs), hybrid electric vehicles, and large-scale energy storage systems, there are formidable challenges and requirements that must be meticulously addressed to compete with traditional internal combustion engine vehicles and receive wider consumer acceptance. These challenges encompass the imperative to meet demanding criteria, such as fast charging and rating capabilities, elevated energy, power density for starting, accelerating, and uphill driving, cost-effectiveness, battery safety under harsh conditions at elevated temperatures, and prolonged lifespan [4]. One way to reduce costs is to utilize high gravimetric and volumetric energy density materials, thereby requiring a smaller number of needed materials. In addition to driving range, charging time is also central to customer experience [5][6][7][8][9][10].
The electrochemical characteristics of LIBs are primarily governed by the properties of their cathode materials. While every component of the battery system plays a vital role in achieving optimal performance and continues to be the focus of extensive research, a notable technical bottleneck in LIBs technology pertains to the limited capacity of cathode materials, typically falling below 270 mAh g−1. This inferior capacity stands in contrast to the more robust capacity of graphite, SnO2, Sn, and Si anode materials, which can reach up to 350, 782, 994, and 4200 mAh g−1, respectively [11]. In order to resolve the capacity shortfall exhibited by cathode materials, immense efforts have been directed on research and development endeavors aimed at creating new materials and refining existing materials that combine high-capacity and high-voltage. Cathode materials are typically categorized into the three most prominent classes: (1) Layered oxide LiMO2 (M = Mn, Co, and Ni) structure, (2) Spinel-framework LiM2O4 (M = Mn, Co, and Ni) structure, and (3) Olivine LiMPO4 (M = Fe, Mn, Co, and Ni) structure [12]. These categories exhibit varying actual capacities, typically falling within the range of 120 mAh g−1 to 210 mAh g−1 [13][14][15]. Among the various materials presently available explored as potential candidates for battery cathodes, layered transition metal oxides (LTMOs) have emerged as particularly promising options. Layered oxide materials (for example LiCoO2 and LiNixCoyMnzO2) with a layered lattice α-NaFeO2 hexagonal structure and R-3m space group play a crucial role in facilitating the reversible (de)intercalation of lithium ions. In this structure, lithium ions and TM cations are placed within octahedral 3a and 3b sites, respectively, while oxygen anions occupy the octahedral 6c sites, showing a cubic close-packed arrangement (as depicted in Figure 1) [16].
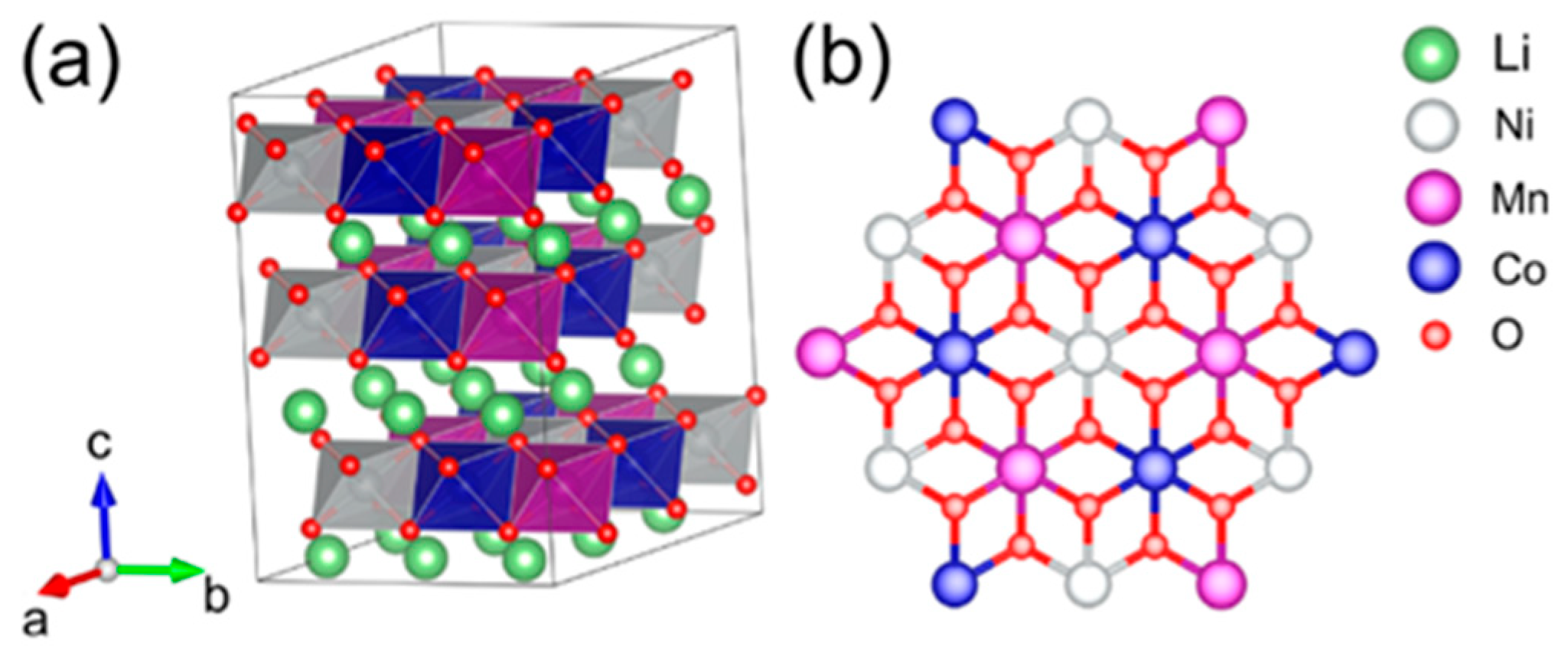
Figure 1. (a) Schematic view of the bulk structure of NCM111 with R-3m space group, and (b) illustration depicting the ion-ordering in NCM111 TM layers.
The evolution of Ni-rich cathode materials traces its roots to the constrained development of LiCoO2 electrodes. Despite the excellent performance of LiCoO2, limitations such as practical delivery of only 50% of the theoretical capacity (140 mAh g−1), high cost, and the material’s toxic property, hinder its application in EVs. As a response, rigorous research endeavors have been undertaken to replace Co with alternative elements such as Ni and Mn in cathode hosts. However, the absence of Co in the layered oxide structure often results in structural instability at low-temperature operations and lower electronic conductivity [17][18]. N. Zhang et al. [19] reported that Co helps Ni and Li ordering, and in Co absence, more Ni and Li ion-exchange sites are possible. Also, Mn provides structural stability, as its oxidation state is +4 throughout electrochemical cycling [20]. Consequently, by taking partial advantage of the high capacity of LiNiO2, the layered character of LiCoO2, and the low cost and thermal stability of LiMnO2, LiNixCoyMnzO2 (NCM) cathode materials have appeared as viable contenders to substitute the traditional LiCoO2 cathode, addressing some of the inherent challenges associated with Co-based formulations [21][22][23]. Ni-rich layered oxide cathode materials (x > 0.5) have attracted great interest as the leading candidates in this domain. This enthusiasm is primarily fueled by their exceptional attributes, including a superior actual capacity exceeding 170 mAh g−1, a volume change by less than 2% during Li insertion/extraction, and a higher average operating cutoff potential surpassing 3.4 V [24][25][26][27]. While Ni-rich cathode materials have received great attention for their remarkable attributes, such as high capacity and energy density (exceeding 600 Wh kg−1), they are susceptible to a range of significant drawbacks. These issues include premature capacity degradation, severe structural and thermal instability, limited calendar life, electrolyte decomposition, intrinsic low-rate capability, rapid voltage decay leading to sluggish Li+ ion transport dynamics, and safety concerns. These intrinsic defects have posed significant obstacles to their widespread commercialization, particularly in the context of EVs [28][29][30][31][32].
2. Strategies and Countermeasures
Extensive research has been undertaken to enhance Ni-rich cathode materials performance in response to the growing demand for batteries with higher energy density. Common strategies employed for this purpose include elemental substitution, surface treatment, compositional adjustments, as well as the utilization of single crystals and concentration gradient structures. These methods are widely adopted to address the associated challenges and requirements effectively.
2.1. Elemental Doping
The incorporation of cation and anion doping has emerged as a prominent strategy to enhance the structural stability of Ni-enriched layered oxides [33][34]. This approach proves pivotal in improving the cycling performance of cathode materials by substituting unstable elements with inactive materials. It effectively hinders microcrack propagation within primary particles through the pillaring effect of cationic species, while stabilizing the oxygen octahedral site through anionic doping [35][36]. As an example, Mg emerged as a promising cation element with the potential to serve as an inert stabilizer, leading to enhanced performance in NCM622 cathode materials by mitigating structural deterioration. The introduction of Mg at concentrations of 1%, 3%, and 5% resulted in a reduction of Li/Ni cation intermixing by 1.58%, 1.77%, and 3.20%, respectively, compared to the 5.18% observed in undoped NCM. When Mg ions partially substituted for Ni in Mg-doped NCM, the unit cell parameters a and c decreased in magnitude, while the c/a ratios increased. This change typically signified the development of a layered material structure. The reduction in unit cell dimensions a and c was attributed to the incorporation of Mg2+ ions into both the Li layer and the transition metal layer. This incorporation led to a decrease in the overall content of Ni2+ ions residing within the Li layer. This phenomenon is comprehensible because the diminished presence of Ni2+ ions in the structure of Mg-doped powders resulted in a slight reduction in lattice parameters. Furthermore, it is noteworthy that the bond dissociation energy of Mg-O (∆Hf298 = 394 kJ mol−1) surpasses that of Ni-O (∆Hf298 = 391 kJ mol−1). Therefore, the introduction of Mg2+ ions into the host structure contributed to the enhancement of structural stability [37]. Anionic dopants, like F, have the capacity to occupy oxygen sites without compromising the capacity and energy density of the active materials. Among these anionic dopants, fluorine exhibits the highest electronegativity and establishes a remarkably stable structure due to the formidable bonds formed between transition metals and fluorine. Furthermore, the enhanced electrochemical performance of cathode materials doped with fluorine may be attributed to their shielding effect against HF production, which can arise during the decomposition of the liquid electrolyte [38][39][40]. Sung-Beom Kim et al. [41] employed a solid-state reaction method to synthesize F-doped NCM811. The pristine material exhibited a discharge capacity of 97.9 mAh g−1 after 100 cycles at a rate of 100 mA g−1, within the voltage range of 2.8–4.3 V. In contrast, the discharge capacities of LiNi0.8Co0.1Mn0.1O2−xFx, with x values of 0.02, 0.04, 0.06, and 0.08, were measured at 107.8, 133.2, 169.6, and 154.5 mAh g−1, respectively. Furthermore, assessing the capacity retention of various samples, including NCM-bare, NCMF-2, NCMF-4, NCMF-6, and NCMF-8 after 100 cycles, revealed retention percentages of 60.7%, 66.2%, 78.8%, 96.8%, and 89.5%, respectively. A diverse array of elements has been successfully employed as cation and anion dopants for transition metals and oxygen sites, aiming to enhance the electrochemical performance of Ni-rich cathode materials. Notable examples include Mo [42][43], Al [44][45], B [46], Nb [47][48][49], K [50], Na [51], Mg [37], Ti [52][53][54][55][56][57][58], PO43- [59], and F [41][60][61]. It is worth mentioning that doping Ni-rich cathode materials with cations of varying valence states engenders distinct mechanisms that contribute to performance enhancement. Figure 2a–d provides a visual representation of the electrochemical effects resulting from a 1% molar elemental doping with diverse valence states on LiNi0.91Co0.09O2 active material [62]. Notably, all doped materials, irrespective of low valence (Mg2+ and Al3+) or high valence (Ti4+, Ta5+, and Mo6+) states, exhibited superior electrochemical performance compared to the undoped counterpart in initial cycles, regardless of cycling rate and temperature. While samples doped with low valence states displayed satisfactory performance over extended cycles, such as 1000 cycles, capacity retention significantly decreased to 54.2% and 46.9% for Al and Mg-doped samples, respectively. In contrast, Ta- and Mo-doped samples maintained capacity retention even after 3000 cycles. Structural evaluations of samples, depicted in cross-sectional SEM images (Figure 2e) of cathodes cycled up to 1000 cycles, revealed that undoped NC90 exhibited severe intergranular cracks and disintegration into individual grains. Conversely, Mg and Al-doped samples showed incipient microcrack propagation, while Ta and Mo-doped samples retained particle coherency with minimal signs of microcrack nucleation and propagation, aligning with their exceptional cycling stabilities. These observations suggest diverse mechanisms contributing to the enhancement of doped samples.

Figure 2. Performance evaluation of cathodes (NC90, Mg-NC90, Al-NC90, Ti-NC90, Ta-NC90, and Mo-NC90) in pouch-type full cells, (a) showcasing first charge–discharge cycle curves at 0.1C and 30 °C, (b,c) cycling at 0.5C over 100 cycles at both 30 °C and 60 °C, and (d) extended cycling at 1C and 25 °C within the voltage range of 3.0–4.2 V vs. graphite; (e) presents cross-sectional images of recovered cathodes after 1000 cycles at 100% DOD, discharged state of 2.7 V. (f) Distinctions in the mechanisms and impacts of low-valence (Al) and high-valence (Nb, Ta, and Mo) dopants, encompassing their incorporation into the bulk material and the coating of grain boundaries. Reprinted from Ref. [63] with permission from John Wiley and Sons.
Park et al. [63] have proposed mechanisms for improvement through cation doping with different valence states (Figure 2f). The authors investigated the effects of a 1% molar doping of Al3+, Nb5+, Ta5+, and Mo6+ in LiNiO2. They suggested that cations with low valence states, such as Al3+, participated in the bulk and acted as pillars to improve structural stability and electrochemical performance. Regarding high valence cations, such as Mo6+, Nb5+, and Ta5+, these elements not only participated in the bulk structure, but also reacted with Li resources and Li-X-O compounds forming and segregating at grain boundaries, inhibiting grain boundary migration, and preserving the size of primary particles. For instance, Al 1% molar doping in LiNiO2 resulted in LiAlO2 formation at 650 °C, disappearing at 680 °C. In contrast, compounds such as LiNbO3 appeared at low temperatures, and at higher calcination temperatures, the insoluble Li3NbO4 phase were segregated in the grain boundaries. Consequently, if doping elements persisted as Li-X-O compounds during high-temperature calcination, they coated the grain boundary, inhibiting boundary migration and suppressing primary particle coarsening. For elements like Al and Mg with low valence states, it was suggested that higher doping molar concentrations (3%) could be beneficial for grain boundary reinforcement. In a study by Hüger et al. [64], it was revealed that compounds like LiNbO3 and LiTaO3 exhibit higher Li diffusivity, or in other words, had a higher Li insertion/extraction rate (about 1 × 10−18 and 8 × 10−19 m2 s−1, respectively) compared to other compounds such as LiAlO2 and LiGaO2 (about 4 × 10−21 and 1 × 10−21 m2 s−1, respectively). Furthermore, the bond dissociation energy of Nb-O and Ta-O is nearly twice (∆Hf298 = 753 and 805 kJ mol−1, respectively) that of Mg-O and Ni-O, thereby enhancing structural stability.
2.2. Surface Coating
Surface treatment represents a foremost strategy to mitigate interfacial side reactions between cathode materials surface and electrolytes, with the aim of enhancing the retention of oxide ion vacancies within the crystal lattice post-initial charge. This approach concurrently addresses challenges such as the suppression of electrolyte decomposition, the mitigation of cathode-electrolyte interphase formation, the preservation of low microstrain for improved structural integrity and crystallinity during cycling, and the sequestration of HF from the electrolyte [65][66][67][68][69]. In the context of Ni-rich materials, a coating which is generally composed of nanoparticles and typically ranging from 5–20 nm can be broadly classified into three categories: oxides, phosphates, and fluorides. Among these, metal oxide coatings, including Al2O3 [70], Li4Ti5O12 [71], ZrO2 [72][73], Li2WO4 [74], and Li2ZrO3 [75] have emerged as widely adopted choices to address the limitations of Ni-rich materials. Shim et al. [76] specifically applied an acidic WO3 coating to NCM811 particles, leveraging its intercalation host properties attributed to its well-defined cubic ReO3-type structure and high resistance against HF attack. As depicted in Figure 3a,b, the WO3 coating significantly enhanced the cycling performance of NCM811 within the 3–4.3 V range, and notably, under challenging conditions spanning a voltage range of 3–4.6 V. The capacity retention of the pristine active material at upper cutoff voltages of 4.3 V and 4.6V was 82% and 64.44%, respectively, while for the WO3-coated NCM811, the corresponding values were 85.8% and 76.46%, respectively. In terms of rate capability performance (Figure 3c), the coated sample exhibited superior performance attributed to expedited Li ion and electron kinetics, stemming from enhanced structural stability and reduced resistance. This improvement was further attributed to the suppression of side reactions between the cathode material and electrolyte. Moreover, Figure 3d,e illustrates the absence of discernible microcracks along grain boundaries in NCM protected by a 2.14 nm thick WO3 coating. In contrast, the untreated NCM, subjected to cycling-induced expansion and shrinkage, exhibited electrolyte infiltration into the interior, leading to structural collapse. This observation underscored the effectiveness of the WO3 coating in preserving the structural integrity of NCM during cycling. The incorporation of a La2O3 coating as a protective layer has proven highly effective in augmenting the electrochemical performance of the Ni0.91Co0.06Mn0.03 electrode in LIB [77]. Notably, the capacity retention of the cathode after 100 cycles at 0.5C demonstrated a substantial improvement, with a retention rate of 68.1% for the pristine NCM, whereas the La2O3-coated material exhibited superior retention at 87.2%. The cyclic voltammetry plots (Figure 3f) of both pristine and coated samples revealed significant improvements by coating deposition on NCM material. First, the intensity peak related to phase transformation H2 → H3 that is in charge of particle pulverization and capacity fading decreased significantly. Secondly, the oxidation-reduction gap (ΔE = Eoxidation − Ereduction), which serves as a determinant of electrode polarization, reduced from 0.111 V to 0.094 V [76]. A TiO2 coating was employed to provide comprehensive protection for NCM811 cathode active materials, capitalizing on its structural stability and chemical inertness against the electrolyte at high voltages [78]. Q. Fan et al. [78] reported that the application of a continuous TiO2 nano-coating layer on NCM811 not only resulted in a higher specific discharge capacity, but also significantly improved cycling performance, with 72.2% capacity retention for the coated sample and 45.7% for the bare sample. This improvement was attributed to the reduction in side reactions at the electrode and electrolyte interface, particularly at high upper cut-off voltages. The exceptional barrier properties of the TiO2 coating led to a reduction in electrolyte consumption and the formation of a thick passivation solid electrolyte film. This reduction helped alleviate polarization at the material interface, preventing the inevitable damage resulting from direct contact between the active material and the electrolyte. Figure 3g illustrated a schematic representation of the protective mechanism of TiO2, emphasizing the significance of an intact coating for active materials. This depiction underscores that the development of a partial coating may result in the isolation failure of particles, potentially leading to the collapse of secondary particles. Phosphate coatings have emerged as highly protective coatings for matrix materials, acclaimed for their exceptional thermal and electrochemical stability. This stability arises from the robust P=O bond, with a bond energy of 5.64 eV and the formation of strong covalent bonds between polyanions and metal ions, rendering them resistant to chemical attacks [79][80][81][82]. Among phosphate coatings, the Li3PO4 coating stands out as a prominent choice for Ni-rich active materials due to its rapid Li+ conductivity and high chemical stability [83]. The wet-coating method, utilizing an aqueous solution of NH4H2PO4 or phosphoric acid, has been widely employed to extensively create Li3PO4 coatings on active materials, enabling a substantial reduction of lithium-based surface impurities (LiOH and Li2CO3) and the suppression of detrimental side reactions during both storage and cycling [84][85]. T. Sattar et al. [86] coated Li3PO4 onto layered oxide Ni0.91Co0.06Mn0.03 active material by transforming Li residual compounds into the coating (Figure 3h). The superior electrochemical rating and cycling performance of the coated samples were attributed to the removal of lithium residual compounds and the isolation of particles, preventing direct contact with the electrolyte and excessive growth of the solid electrolyte interface layer. Additionally, the higher dissociation energy bond of P=O compared to transition metal–oxygen bonds contributed to better electrochemical performance, with capacity retention rates of 68.1% for pristine NCM and 82.4% for the surface-modified sample. In the context of EV industries, where thermal stability is crucial, Ni0.8Co0.15Al0.05 cathode materials, employed in Tesla’s Model S and X, have gained prominence [87]. A Li3PO4 coating developed on Ni0.815Co0.15Al0.035 active material has been shown to decrease heat generation during cycling. A study conducted by Tang et al. [88] confirmed through DSC profiles that the Li3PO4-modified samples generated less heat (525 J g−1) compared to the pristine sample (757 J g−1), highlighting the efficacy of the coating in suppressing surface side reactions with the organic electrolyte. The entire process of reaction between Li residual compounds and phosphoric acid can be expressed through the following Reactions (10)–(12) [89][90]:
3Li2O + 2H3PO4 = 2Li3PO4 + 3H2O,
3LiOH + H3PO4 = Li3PO4 + 3H2O,
3Li2CO3 + 2H3PO4 = 2Li3PO4 + 3H2O + 3CO2

Figure 3. Cyclabilities of pristine and WO3-coated NCM at a voltage of (a) 3.0–4.3 V and (b) 3.0–4.6 V at 0.5C, (c) rate capabilities of pristine and WO3-coated NCM. FE-SEM images after 100 cycling for (d) WO3 coated NCM and (e) pristine NCM. (f) Cyclic voltammetry curves for pristine and LaO-0.2. (g) Mechanism for the protective action of TiO2 nano-coating on Ni-rich cathode materials. (h) Schematic illustration depicting the transformation of residual lithium into a Li3PO4 coating.
Fluorides exhibit lower Gibbs energy compared to oxides, indicating their heightened stability in comparison to oxide counterparts. The utilization of metal fluorides as protective coatings has been extensively investigated to address challenges associated with electrode side parasitic reactions with the electrolyte and HF etching. Among the various fluorine coatings, LiF is frequently employed for Ni-rich active materials. A thermodynamically stable lithium fluoride coating, characterized by superior Li ion conductivity compared to other fluorides, has been typically achieved through the mixing of NH4F and NH4HF2 with the original cathode materials [91][92]. H. Kim et al. [93] treated Ni0.85Co0.12Al0.03 cathode material with NH4HF2 and reported that this treatment helped to remove Li residual compounds from the particles’ surface and additionally, both cycling performance and gas evolution behavior were enhanced significantly in the modified sample.
2.3. Structural Engineering
The innovative approach to developing functional layered oxide materials involves creating a comprehensive concentration gradient structure. This strategy utilizes a high-capacity Ni-rich core in conjunction with a chemically stable Co, Mn-rich surface to prevent undesired Ni4+ reduction on the material’s surface. In a typical cathode with a full concentration gradient, the concentration of Ni gradually decreases from the particle’s center to its outer surface, while the concentrations of Co and Mn increase [94]. Hou et al. [95] synthesized a full concentration gradient (FCG) NCM622 with LiNi0.8Co0.2O2 for the inner composition, and LiNi0.4Co0.2Mn0.4O2 for the outer composition. Reports indicated that the FCG-NCM622 cathode exhibited superior capacity retention at both room temperature and 55 °C. Moreover, DSC analysis confirmed the enhanced thermal stability of the structurally modified NCM622 compared to the normal sample. The exothermic peak observed at 282.7 °C for FCG-NCM622 and 266.2 °C for NCM622 indicated better thermal stability for the former. The generated heat was 692 J g−1 for FCG-NCM622 and 969 J g−1 for NCM622, attributed to the presence of Mn on the outer surface of the active material. Recently, a combination of FCG and coating-doping modifications has been employed to improve the thermal and electrochemical performance of Ni-enriched cathode materials [96][97]. Liao et al. [98] utilized dual modification for NCM material, incorporating both a concentration gradient and a protective Al2O3 coating. Figure 4a–c demonstrates that this dual modification significantly enhanced electrochemical performance, resulting in lower voltage decay over cycling compared to a normal cathode. This improvement was attributed to the reduced reaction of the cathode material with the electrolyte, and higher structural stability. The capacity retention for coated CG-NCM after 100 cycles was approximately 88%, outperforming CG (80%) and normal NCM (62.6%) as shown in Figure 4d. The impact of dual modification became more evident in cycling at higher temperatures (55 °C), as illustrated in Figure 4e. In terms of thermal stability, DSC profiles (Figure 4f) confirmed that both coated CG and CG samples exhibit a higher onset temperature for the exothermic reaction compared to the normal sample, indicating the effectiveness of both modifications. Park et al. [99] explored the electrochemical and thermal characteristics of a concentration gradient Ni0.865Co0.120Al0.015 cathode material. Although conventional NCA demonstrated a slightly higher initial discharge capacity at 0.1C in the operating potential range of 2.7–4.3 V compared to CG NCA (225 and 222 mAh g−1, respectively), the capacity retention of the CG electrode at 0.5C, both at room temperature and 60 °C, was notably superior to that of the standard sample. Moreover, differential scanning calorimetry (DSC) measurements on charged samples at 4.3 V revealed an exothermic peak at 192 °C with a heat generation of 1789 J g−1 for conventionally synthesized NCA, whereas for CG NCA, it was observed at 202 °C with a lower heat generation of 1409 J g−1, indicating enhanced thermal stability of the structurally modified NCA. Xu et al. [100] successfully synthesized a concentration gradient cathode material comprising Ni0.7Co0.13Mn0.17 with NCM811 composition at the core and NCM523 at the outer surface (Figure 5a–e). The initial charge–discharge profile (Figure 5f) of NCM523, NCM811, and the CG NCM indicated that the CG sample exhibited a lower discharge capacity compared to NCM811, attributed to its lower Ni content on the surface. Additionally, the plateau at around 4.2 V was shorter for NCM523 and CG compared to NCM811. Moreover, the H2 → H3 phase transition, recognized as the primary reason for volume change and subsequent capacity fading in Ni-rich electrodes, was pronounced for NCM811 compared to the CG cathode material (Figure 5g). The rate capability of the CG electrode outperformed that of NCM811 and NCM523 counterparts, delivering much higher capacity at high discharge rates (Figure 5h). Cycling profiles of the samples at 1C (Figure 5i) revealed that NCM811 experienced catastrophic capacity retention, while the performance of CG was highly similar to that of NCM523. These results suggest that utilizing the CG cathode offers benefits for both NCM523 in terms of capacity retention, and for NCM811 in terms of high capacity delivery.

Figure 4. Charge discharge curves for (a) normal, (b) CG, and (c) CG-Al2O3 electrodes within the voltage range of 2.7 to 4.5 V at room temperature, comparative cycling performances (C/5) for the three cathodes at (d) room temperature and (e) 55 °C, and (f) DSC traces illustrating the heat flow during the reaction of the electrolyte with CC, CG, and CG-Al2O3 electrodes when charged to 4 V.

Figure 5. (a) Illustration depicting the distribution of transition metals within the concentration CG NCM particle, (b) profiles showing the calculated transition metal compositions over time, and (c) from the particle’s core to its surface. (d) EDS line scanning analysis conducted across the precursor particle (the yellow line denotes the scanning path), (e) comparative analysis of the measured composition profiles with the calculated outcomes. Electrochemical performance of CG NCM, NCM523, and NCM811 cells within the voltage range of 3.0–4.3 V: (f) initial charge–discharge profiles at a 0.1C rate, (g) differential capacity curves for the initial charge–discharge cycle, (h) rate capability assessed at different charge-discharge current densities, and (i) cycling performance evaluated at a 1C rate.
References
- Koo, J.K.; Choi, H.; Seo, J.K.; Hwang, S.M.; Lee, J.; Kim, Y.J. Microstructure engineering of nickel-rich oxide/carbon composite cathodes for fast charging of lithium-ion batteries. Ceram. Int. 2022, 48, 31859–31865.
- Rudnicka, E.; Jakobczyk, P.; Lewandowski, A. Thermodynamic and kinetic limits of Li-ion battery operation. J. Energy Storage 2022, 55, 105747.
- Entwistle, T.; Sanchez-Perez, E.; Murray, G.J. Co-precipitation synthesis of nickel-rich cathodes for Li-ion batteries. Energy Rep. 2022, 8, 67–73.
- Chen, Z.; Danilov, D.L.; Rajimakers, L.H.J. Overpotential analysis of graphite-based Li-ion batteries seen from a porous electrode modeling perspective. J. Power Sources 2021, 509, 230345.
- Zheng, H.; Li, J.; Song, X.; Liu, L.; Battaglia, V.S. A comprehensive understanding of electrode thickness effects on the electrochemical performances of Li-ion battery cathodes. Electrochim. Acta 2012, 71, 258–265.
- Xie, Y.; Jin, Y.; Xiang, L. Li-rich layered oxides: Structure, capacity and voltage fading mechanisms and solving strategies. Particuology 2022, 61, 1–10.
- Gamble, F.; Osiecki, J.H.; Cais, M.; Pisharody, R.; Disalvo, F.J.; Geballe, T.H. Intercalation complexes of Lewis bases and layered sulfides: A large class of new superconductors. J. Sci. 1971, 174, 493–497.
- Tang, Z.; Wang, S.; Liao, J.; Wang, S.; He, X.; Pan, B.; He, H.; Chen, C. Facilitating lithium-ion diffusion in layered cathode materials by introducing Li+/Ni2+ antisite defects for high-rate Li-ion batteries. Res. J. 2019, 2019, 2198906.
- Huang, D.; Engtrakul, C.; Nanayakkara, S.; Mulder, D.W.; Han, S.D.; Zhou, M.; Luo, H.; Tenent, R.C. Understanding degradation at the lithium-ion battery cathode/electrolyte interface: Connecting transition-metal dissolution mechanisms to electrolyte composition. ACS Appl. Mater. Interfaces 2021, 13, 11930–11939.
- Deng, J.; Bae, C.; Denlinger, A.; Miller, T. Electric vehicles batteries: Requirements and challenges. Joule 2020, 4, 511–515.
- Jiang, M.; Qian Zhang, Q.; Wu, X.; Chen, Z.; Danilov, D.L.; Eichel, R.; Notte, P.H.L. Synthesis of Ni-rich layered-oxide nanomaterials with enhanced Li-ion diffusion pathways as high-rate cathodes for Li-ion batteries. ACS Appl. Energy Mater. 2020, 3, 6583–6590.
- Manthiram, A. An outlook on lithium ion battery technology. ACS Cent. Sci. 2017, 3, 1063–1069.
- Zaghib, K.; Mauger, A.; Julien, C.M. Olivine-based cathode materials In Rechargeable Batteries. Green Energy Technol. 2015, 25–65.
- Julien, C.M.; Mauger, A.; Zaghib, K.; Groult, H. Comparative issues of cathode materials for Li-ion batteries. Inorganics 2014, 2, 132–154.
- Shen, Y.; Wang, L.; Jizhou Jiang, J.; Duo Wang, D.; Zhang, D.; Yin, D.; Wang, D.L.; Zhang, X.; Huang, G.; Cheng, Y. Stabilization of high-voltage layered oxide cathode by multi-electron rare earth oxide. Chem. Eng. J. 2023, 454, 140249.
- Yang, W. Oxygen release and oxygen redox. Nat. Energy. 2018, 3, 619–620.
- Ryu, H.H.; Sun, H.H.; Myung, S.T.; Chong, S.; Yoon, C.S.; Sun, Y.K. Reducing cobalt from lithium-ion batteries for the electric vehicle era. Energy Environ. Sci. 2021, 14, 844–852.
- Kim, M.H.; Shin, H.S.; Shin, D.; Sun, Y.K. Synthesis and electrochemical properties of LiO2 and LiO2 via co-precipitation. J. Power Sources 2006, 159, 1328–1333.
- Zhang, N.; Li, J.; Li, H.; Liu, A.; Huang, Q.; Ma, L.; Li, Y.; Dahn, J.R. Structural, Electrochemical, and Thermal Properties of Nickel-Rich LiNixMnyCozO2 Materials. Chem. Mater. 2018, 30, 8852–8860.
- Xiao, J.; Chernova, N.A.; Whittingham, M.S. Layered mixed transition metal oxide cathodes with reduced cobalt content for lithium ion batteries. Chem. Mater. 2008, 20, 7454–7464.
- Tian, C.; Lin, F.; Doeff, M.M. Electrochemical characteristics of layered transition metal oxide cathode materials for lithium ion batteries: Surface, bulk behavior, and thermal properties. Acc. Chem. Res. 2017, 51, 89–96.
- Aryal, S.; Durham, J.L.; Lipson, A.L.; Pupek, K.Z.; Kahvecioglu, O. Roles of Mn and Co in Ni-rich layered oxide cathodes synthesized utilizing a Taylor Vortex Reactor. Electrochim. Acta. 2021, 391, 138929.
- Liang, C.; Kong, F.; Longo, R.C.; Santosh, K.C.; Kim, J.S.; Jeon, S.H.; Choi, S.A.; Cho, K. Unraveling the origin of instability in Ni-rich LiNi1-2xCoxMnxO2 (NCM) cathode materials. J. Phys. Chem. C 2016, 120, 6383–6393.
- Whittingham, M.S. Lithium batteries and cathode materials. Chem. Rev. 2004, 104, 4271–4302.
- Belharouak, I.; Sun, Y.K.; Liu, J.; Amine, K. Li(Ni1/3Co1/3Mn1/3)O2 as a suitable cathode for high power applications. J. Power Sources 2003, 123, 247–252.
- Choi, J.; Manthiram, A. Role of chemical and structural stabilities on the electrochemical properties of layered LiNi1/3Mn1/3Co1/3O2 cathodes. J. Electrochem. Soc. 2005, 152, A1714.
- Li, F.; Liu, Z.; Shen, J.; Xu, X.; Zeng, L.; Zhang, B.; Zhu, H.; Liu, Q.; Liu, J.; Zhu, M. A nanorod-like Ni-rich layered cathode with enhanced Li+ diffusion pathways for high-performance lithium-ion batteries. J. Mater. Chem. A 2021, 9, 2830–2839.
- Zhuang, D.; Bazant, M.Z. Theory of layered-oxide cathode degradation in Li-ion batteries by oxidation-induced cation disorder. J. Electrochem. Soc. 2022, 169, 100536.
- Evertz, M.; Horsthemke, F.; Kasnatscheew, J.; Börner, M.; Winter, M.; Nowak, S. Unraveling transition metal dissolution of Li1.04Ni1/3Co1/3Mn1/3O2 (NCM 111) in lithium ion full cells by using the total reflection X-ray fluorescence technique. J. Power Sources 2016, 329, 364–371.
- Kim, T.; Ono, L.K.; Fleck, N.; Raga, S.R.; Qi, Y. Transition metal speciation as a degradation mechanism with the formation of a solid-electrolyte interphase (SEI) in Ni-rich transition metal oxide cathodes. J. Mater. Chem. A. 2018, 6, 14449–14463.
- Kim, H.R.; Woo, S.W.; Kim, J.H.; Cho, W.; Kim, Y.K. Capacity fading behavior of Ni-rich layered cathode materials in Li-ion full cells. J. Electroanal. Chem. 2016, 782, 168–173.
- Cui, J.; Ding, X.; Luo, D.; Xie, H.; Zhang, Z.; Zhang, B.; Tan, F.; Liu, C.; Lin, Z. Effect of cationic uniformity in precursors on Li/Ni mixing of Ni-rich layered cathodes. Energy Fuels 2021, 35, 1842–1850.
- Levartovsky, Y.; Wu, X.; Erk, C.; Maiti, S.; Grinblat, J.; Talianker, M.; Aurbach, D. Enhancement of structural, electrochemical, and thermal properties of Ni-rich LiNi0.85Co0.1Mn0.05O2 cathode materials for Li-ion batteries by Al and Ti doping. Batter. Supercaps 2021, 4, 221–231.
- Hua, W.; Zhang, J.; Zheng, Z.; Liu, W.; Peng, X.; Guo, X.D.; Zhong, B.; Wang, Y.J.; Wang, X. Na-doped Ni-rich LiNi0.5Co0.2Mn0.3O2 cathode material with both high rate capability and high tap density for lithium-ion batteries. Dalton Trans. 2014, 43, 14824–14832.
- Kang, K.; Meng, Y.S.; Bréger, J.; Grey, C.P.; Ceder, G. Electrodes with high power and high capacity for rechargeable lithium batteries. Science 2006, 311, 977–980.
- Rajkamal, A.; Kim, H. Formation of pillar-ions in the Li layer decreasing the Li/Ni disorder and improving the structural stability of cation-doped Ni-rich LiNi0.8Co0.1Mn0.1O2: A first-principles verification. ACS Appl. Energy Mater. 2021, 4, 14068–14079.
- Huang, Z.; Wang, Z.; Zheng, X.; Guo, H.; Li, X.; Jing, Q.; Yang, Z. Effect of Mg doping on the structural and electrochemical performance of LiNi0.6Co0.2Mn0.2O2 cathode materials. Electrochim. Acta 2015, 182, 795–802.
- Liu, K.; Zhang, Q.; Dai, S.; Li, W.; Liu, X.; Ding, F.; Zhang, J. Synergistic effect of F–doping and LiF coating on improving the high-voltage cycling stability and rate capacity of LiNi0.5Co0.2Mn0.3O2 cathode materials for lithium-ion batteries. ACS Appl. Mater. Interfaces 2018, 10, 34153–34162.
- Zhou, S.; Wang, G.; Tang, W.; Xiao, Y.; Yan, K. Enhanced rate performance and high potential as well as decreased strain of LiNi0.6Co0.2Mn0.2O2 by facile fluorine modification. Electrochim. Acta 2018, 261, 565–577.
- Chen, Z.; Xu, M.; Zhu, H.; Xie, T.; Wang, W.; Zhao, Q. Enhanced electrochemical performance of polyacene coated LiMn2O3.95F0.05 for lithium-ion batteries. Appl. Surf. Sci. 2013, 286, 177–183.
- Kim, S.B.; Kim, H.; Park, D.H.; Kim, J.H.; Shin, J.H.; Jang, J.S.; Moon, S.H.; Choi, J.H.; Park, K.W. Li-ion diffusivity and electrochemical performance of Ni-rich cathode material doped with fluoride ions. J. Power Sources 2021, 506, 230219.
- Su, Y.; Yang, Y.; Chen, L.; Lu, Y.; Bao, L.; Chen, G.; Yang, Z.; Zhang, Q.; Wang, J.; Chen, R.; et al. Improving the cycling stability of Ni-rich cathode materials by fabricating surface rock salt phase. Electrochim. Acta 2018, 292, 217–226.
- Susai, F.A.; Bano, A.; Maiti, S.; Grinblat, J.; Chakraborty, A.; Sclar, H.; Kravchuk, T.; Kondrakov, A.; Tkachev, M.; Talianker, M.; et al. Stabilizing Ni-rich high energy cathodes for advanced lithium-ion batteries: The case of LiNi0.9Co0.1O2. J. Mater. Chem. A 2023, 11, 12958–12972.
- Zhao, W.; Zou, L.; Jia, H.; Zheng, J.; Wang, D.; Song, J.; Hong, C.; Liu, R.; Xu, W.; Yang, Y.; et al. Optimized Al doping improves both interphase stability and bulk structural integrity of Ni-rich NMC cathode materials. ACS Appl. Energy Mater. 2020, 3, 3369–3377.
- Jeong, M.; Kim, H.; Lee, W.; Ahn, S.J.; Lee, E.; Yoon, W.S. Stabilizing effects of Al-doping on Ni-rich LiNi0.80Co0.15Mn0.05O2 cathode for Li rechargeable batteries. J. Power Sources 2020, 474, 228592.
- Liu, Y.; Fan, X.; Luo, B.; Zhao, Z.; Shen, J.; Liu, Z.; Xiao, Z.; Zhang, B.; Zhang, J.; Ming, L.; et al. Understanding the enhancement effect of boron doping on the electrochemical performance of single-crystalline Ni-rich cathode materials. J. Colloid. Interface Sci. 2021, 604, 776–784.
- Chu, M.; Huang, Z.; Zhang, T.; Wang, R.; Shao, T.; Wang, C.; Zhu, W.; He, L.; Chen, J.; Zhao, W.; et al. Enhancing the electrochemical performance and structural stability of Ni-rich layered cathode materials via dual-site doping. ACS Appl. Mater. Interfaces 2021, 13, 19950–19958.
- Xin, F.; Goel, A.; Chen, X.; Zhou, H.; Bai, J.; Liu, S.; Wang, F.; Zhou, G.; Whittingham, M.S. Electrochemical characterization and microstructure evolution of Ni-rich layered cathode materials by niobium coating/substitution. Chem. Mater. 2022, 34, 7858–7866.
- Li, J.; Zhang, M.; Zhang, D.; Yan, Y.; Li, Z. An effective doping strategy to improve the cyclic stability and rate capability of Ni-rich LiNi0.8Co0.1Mn0.1O2 cathode. Chem. Eng. J. 2020, 402, 126195.
- Zhang, H.; Wang, X.; Naveed, A.; Zeng, T.; Zhang, X.; Shi, H.; Su, M.; Dou, A.; Zhou, Y.; Liu, Y. Comparison of structural and electrochemical properties of LiNi0.8Co0.15Al0.05O2 with Li site doping by different cations. Appl. Surf. Sci. 2022, 599, 153933.
- Huang, Z.; Wang, Z.; Jing, Q.; Guo, H.; Li, X.; Yang, Z. Investigation on the effect of Na doping on structure and Li-ion kinetics of layered LiNi0.6Co0.2Mn0.2O2 cathode material. Electrochim. Acta 2016, 192, 120–126.
- Liu, D.; Liu, S.; Zhang, C.; You, L.; Huang, T.; Yu, A. Revealing the effect of Ti doping on significantly enhancing cyclic performance at a high cutoff voltage for Ni-rich LiNi0.8Co0.15Al0.05O2 cathode. ACS Sustain. Chem. Eng. 2019, 7, 10661–10669.
- Zhang, H.; Wu, K.; Li, N.; Deng, X.; Jiao, J.; Zhao, E.; Yin, W.; Wang, B.; Zhao, J.; Xiao, X. Enhancing Thermal and High-Voltage Cycling Stability of Ni-Rich Layered Cathodes through a Ti-Doping-Induced Surface-Disordered Structure. ACS Appl. Energy Mater. 2022, 5, 12673–12681.
- Sun, H.; Cao, Z.; Wang, T.; Lin, R.; Li, Y.; Liu, X.; Zhang, L.; Lin, F.; Huang, Y.; Luo, W. Enabling high rate performance of Ni-rich layered oxide cathode by uniform titanium doping. Mater. Today Energy 2019, 13, 145–151.
- Kim, J.H.; Kim, H.; Kim, W.J.; Kim, Y.C.; Jung, J.Y.; Rhee, D.Y.; Song, J.H.; Cho, W.; Park, M.S. Incorporation of titanium into Ni-rich layered cathode materials for lithium-ion batteries. ACS Appl. Energy Mater. 2020, 3, 12204–12211.
- Wu, F.; Liu, N.; Chen, L.; Su, Y.; Tan, G.; Bao, L.; Zhang, Q.; Lu, Y.; Wang, J.; Chen, S.; et al. Improving the reversibility of the H2-H3 phase transitions for layered Ni-rich oxide cathode towards retarded structural transition and enhanced cycle stability. Nano Energy 2019, 59, 50–57.
- Steiner, J.D.; Cheng, H.; Walsh, J.; Zhang, Y.; Zydlewski, B.; Mu, L.; Xu, Z.; Rahman, M.M.; Sun, H.; Michel, F.M.; et al. Targeted surface doping with reversible local environment improves oxygen stability at the electrochemical interfaces of nickel-rich cathode materials. ACS Appl. Mater. Interfaces 2019, 11, 37885–37891.
- Kasim, M.F.; Wan Azizan, W.A.H.; Elong, K.A.; Kamarudin, N.; Yaakob, M.K.; Badar, N. Enhancing the structural stability and capacity retention of Ni-rich LiNi0.7Co0.3O2 cathode materials via Ti doping for rechargeable Li-ion batteries: Experimental and computational approaches. J. Alloys Compd. 2021, 888, 161559.
- Qiu, L.; Xiang, W.; Tian, W.; Xu, C.L.; Li, Y.C.; Wu, Z.G.; Chen, T.R.; Jia, K.; Wang, D.; He, F.R.; et al. Polyanion and cation co-doping stabilized Ni-rich Ni–Co–Al material as cathode with enhanced electrochemical performance for Li-ion battery. Nano Energy 2019, 63, 103818.
- Li, C.; Kan, W.H.; Xie, H.; Jiang, Y.; Zhao, Z.; Zhu, C.; Xia, Y.; Zhang, J.; Xu, K.; Mu, D.; et al. Inducing Favorable Cation Antisite by Doping Halogen in Ni-Rich Layered Cathode with Ultrahigh Stability. Adv. Sci. 2019, 6, 1801406.
- Kim, U.H.; Park, G.T.; Conlin, P.; Ashburn, N.; Cho, K.; Yu, Y.S.; Shapiro, D.A.; Maglia, F.; Kim, S.J.; Lamp, P.; et al. Cation ordered Ni-rich layered cathode for ultra-long battery life. Energy Environ. Sci. 2021, 14, 1573–1583.
- Sun, H.H.; Kim, U.H.; Park, J.H.; Park, S.W.; Seo, D.H.; Heller, A.; Mullins, C.B.; Yoon, C.S.; Sun, Y.K. Transition metal-doped Ni-rich layered cathode materials for durable Li-ion batteries. Nat. Commun. 2021, 12, 6552.
- Park, N.Y.; Kim, S.B.; Kim, M.C.; Han, S.M.; Kim, D.H.; Kim, M.S.; Sun, Y.K. Mechanism of Doping with High-Valence Elements for Developing Ni-Rich Cathode Materials. Adv. Energy Mater. 2023, 13, 2301530.
- Hüger, E.; Riedel, L.; Zhu, J.; Stahn, J.; Heitjans, P.; Schmidt, H. Lithium Niobate for Fast Cycling in Li-ion Batteries: Review and New Experimental Results. Batteries 2023, 9, 244.
- Wu, Y.; Manthiram, A. Impact of surface modifications on the layered solid solution cathodes in (1− z) LiO2−(z) LiO2. Solid. State Ion. 2009, 180, 50–56.
- Ying, J.; Wan, C.; Jiang, C. Surface treatment of LiNi0.8Co0.2O2 cathode material for lithium secondary batteries. J. Power Sources 2001, 102, 162–166.
- Liu, J.; Manthiram, A. Improved electrochemical performance of the 5 V spinel cathode LiMn1.5Ni0.42Zn0.08O4 by surface modification. J. Electrochem. Soc. 2008, 156, A66.
- Kannan, A.; Manthiram, A. Surface/chemically modified LiMn2O4 cathodes for lithium-ion batteries. Electrochem. Solid-State Lett. 2002, 5, A167.
- Sun, Y.K.; Hong, K.J.; Prakash, J.; Amine, K. Electrochemical performance of nano-sized ZnO-coated LiNi0.5Mn1.5O4 spinel as 5 V materials at elevated temperatures. Electrochem. Commun. 2002, 4, 344–348.
- Karayaylali, P.; Tatara, R.; Zhang, Y.; Chan, K.L.; Yu, Y.; Giordano, L.; Maglia, F.; Jung, R.; Lund, I.; Shao-Horn, Y. Coating-dependent electrode-electrolyte interface for Ni-rich positive electrodes in Li-Ion batteries. J. Electrochem. Soc. 2019, 166, A1022.
- Xu, Y.D.; Xiang, W.; Wu, Z.G.; Xu, C.L.; Li, Y.C.; Guo, X.D.; Lv, G.P.; Peng, X.; Zhong, B.H. Improving cycling performance and rate capability of Ni-rich LiNi0.8Co0.1Mn0.1O2 cathode materials by Li4Ti5O12 coating. Electrochim. Acta 2018, 268, 358–365.
- Ho, V.C.; Jeong, S.; Yim, T.; Mun, J. Crucial role of thioacetamide for ZrO2 coating on the fragile surface of Ni-rich layered cathode in lithium ion batteries. J. Power Sources 2020, 450, 227625.
- Schipper, F.; Bouzaglo, H.; Dixit, M.; Erickson, E.M.; Weigel, T.; Talianker, M.; Grinblat, J.; Burstein, L.; Schmidt, M.; Lampert, J.; et al. From surface ZrO2 coating to bulk Zr doping by high temperature annealing of nickel-rich lithiated oxides and their enhanced electrochemical performance in lithium ion batteries. Adv. Energy Mater. 2018, 8, 1701682.
- Sim, S.-J.; Lee, S.-H.; Jin, B.-S.; Kim, H.-S. Effects of lithium tungsten oxide coating on LiNi0.90Co0.05Mn0.05O2 cathode material for lithium-ion batteries. J. Power Sources 2021, 481, 229037.
- Song, B.; Li, W.; Oh, S.-M.; Manthiram, A. Long-life nickel-rich layered oxide cathodes with a uniform Li2ZrO3 surface coating for lithium-ion batteries. ACS Appl. Mater. Interfaces 2017, 9, 9718–9725.
- Shim, T.Y.; Yoo, Y.W.; Hwang, D.Y.; Lee, S.H. Highly improved structural stability and electrochemical properties of Ni-rich NCM cathode materials. Ceram. Int. 2023, 49, 12138–12143.
- Sattar, T.; Sim, S.J.; Jin, B.S.; Kim, H.S. Improving the cycle stability and rate performance of LiNi0.91Co0.06Mn0.03O2 Ni-rich cathode material by La2O3 coating for lithium-ion batteries. Curr. Appl. Phys. 2022, 36, 176–182.
- Fan, Q.; Lin, K.; Yang, S.; Guan, S.; Chen, J.; Feng, S.; Liu, J.; Liu, L.; Li, J.; Shi, Z. Constructing effective TiO2 nano-coating for high-voltage Ni-rich cathode materials for lithium-ion batteries by precise kinetic control. J. Power Sources 2020, 477, 228745.
- Cho, J.; Kim, Y.W.; Kim, B.; Lee, J.G.; Park, B. A breakthrough in the safety of lithium secondary batteries by coating the cathode material with AlPO4 nanoparticles. Angew. Chem. 2003, 115, 1656–1659.
- Rastogi, P.K.; Kim, Y.W. Effects of an inorganic coating on the structure and magnetic properties of a 1 wt.% silicon steel. J. Appl. Phys. 1985, 57, 4223–4225.
- Wang, X.; Wu, Q.; Li, S.; Tong, Z.; Wang, D.; Zhuang, H.L.; Wang, X.; Lu, Y. Lithium-Aluminum-Phosphate coating enables stable 4.6 V cycling performance of LiCoO2 at room temperature and beyond. Energy Storage Mater. 2021, 37, 67–76.
- Cho, S.W.; Kim, G.O.; Ryu, K.S. Sulfur anion doping and surface modification with LiNiPO4 of a LiO2 cathode material for Li-ion batteries. Solid. State Ion. 2012, 206, 84–90.
- Zhang, W.; Liang, L.; Zhao, F.; Liu, Y.; Hou, L.; Yuan, C. Ni-rich LiNi0.8Co0.1Mn0.1O2 coated with Li-ion conductive Li3PO4 as competitive cathodes for high-energy-density lithium-ion batteries. Electrochim. Acta 2020, 340, 135871.
- Jo, C.H.; Cho, D.H.; Noh, H.J.; Yashiro, H.; Sun, Y.K.; Myung, S.T. An effective method to reduce residual lithium compounds on Ni-rich LiO2 active material using a phosphoric acid-derived Li3PO4 nanolayer. Nano Res. 2015, 8, 1464–1479.
- Park, K.; Park, J.H.; Choi, B.; Kim, J.H.; Hong, S.G.; Han, H.N. Metal phosphate-coated Ni-rich layered oxide positive electrode materials for Li-ion batteries: Improved electrochemical performance and decreased Li residuals content. Electrochim. Acta 2017, 257, 217–223.
- Sattar, T.; Sim, S.J.; Jin, B.S.; Kim, H.S. Dual function Li-reactive coating from residual lithium on Ni-rich NCM cathode material for Lithium-ion batteries. Sci. Rep. 2021, 11, 18590.
- Hua, W.; Zhang, J.; Wang, S.; Cheng, Y.; Li, H.; Tseng, J.; Wu, Z.; Shen, C.H.; Dolotko, O.; Liu, H.; et al. Long-Range Cationic Disordering Induces two Distinct Degradation Pathways in Co-Free Ni-Rich Layered Cathodes. Angew. Chem. Int. Ed. 2023, 62, e202214880.
- Tang, Z.F.; Wu, R.; Huang, P.F.; Wang, Q.S.; Chen, C.H. Improving the electrochemical performance of Ni-rich cathode material LiNi0.815Co0.15Al0.035O2 by removing the lithium residues and forming Li3PO4 coating layer. J. Alloys Compd. 2017, 693, 1157–1163.
- Xiong, X.; Ding, D.; Bu, Y.; Wang, Z.; Huang, B.; Guo, H.; Li, X. Enhanced electrochemical properties of a LiNiO2-based cathode material by removing lithium residues with (NH4)2HPO4. J. Mater. Chem. A 2014, 2, 11691–11696.
- Zou, P.; Lin, Z.; Fan, M.; Wang, F.; Liu, Y.; Xiong, X. Facile and efficient fabrication of Li3PO4-coated Ni-rich cathode for high-performance lithium-ion battery. Appl. Surf. Sci. 2020, 504, 144506.
- Huang, J.; Du, K.; Peng, Z.; Cao, Y.; Xue, Z.; Duan, J.; Wang, F.; Liu, Y.; Hu, G. Enhanced High-Temperature Electrochemical Performance of Layered Nickel-Rich Cathodes for Lithium-Ion Batteries after LiF Surface Modification. ChemElectroChem 2019, 6, 5428–5432.
- Sun, X.G.; Jafta, C.J.; Tan, S.; Borisevich, A.; Gupta, R.B.; Paranthaman, M.P. Facile surface coatings for performance improvement of NMC811 battery cathode material. J. Electrochem. Soc. 2022, 169, 020565.
- Kim, H.; Lee, K.; Kim, S.; Kim, Y. Fluorination of free lithium residues on the surface of lithium nickel cobalt aluminum oxide cathode materials for lithium-ion batteries. Mater. Des. 2016, 100, 175–179.
- Lim, B.B.; Yoon, S.J.; Park, K.J.; Yoon, C.S.; Kim, S.J.; Lee, J.J.; Sun, Y.K. Advanced concentration gradient cathode material with two-slope for high-energy and safe lithium batteries. Adv. Funct. Mater. 2015, 25, 4673–4680.
- Hou, P.; Zhang, L.; Gao, X. A high-energy, full concentration-gradient cathode material with excellent cycle and thermal stability for lithium ion batteries. J. Mater. Chem. A 2014, 2, 17130–17138.
- Mohanty, D.; Dahlberg, K.; King, D.M.; David, L.A.; Sefat, A.S.; Wood, D.L.; Daniel, C.; Dhar, S.; Mahajan, V.; Lee, M.; et al. Modification of Ni-rich FCG NMC and NCA cathodes by atomic layer deposition: Preventing surface phase transitions for high-voltage lithium-ion batteries. Sci. Rep. 2016, 6, 26532.
- Kim, U.H.; Myung, S.T.; Yoon, C.S.; Sun, Y.K. Extending the battery life using an Al-doped LiO2 cathode with concentration gradients for lithium-ion batteries. ACS Energy Lett. 2017, 2, 1848–1854.
- Liao, J.Y.; Manthiram, A. Surface-modified concentration-gradient Ni-rich layered oxide cathodes for high-energy lithium-ion batteries. J. Power Sources 2015, 282, 429–436.
- Park, K.J.; Choi, M.J.; Maglia, F.; Kim, S.J.; Kim, K.H.; Yoon, C.S.; Sun, Y.K. High-Capacity Concentration Gradient LiO2 Cathode for Lithium-Ion Batteries. Adv. Energy Mater. 2018, 8, 1703612.
- Xu, X.; Xiang, L.; Wang, L.; Jian, J.; Du, C.; He, X.; Huo, H.; Cheng, X.; Yin, G. Progressive concentration gradient nickel-rich oxide cathode material for high-energy and long-life lithium-ion batteries. J. Mater. Chem. A 2019, 7, 7728–7735.
More
Information
Subjects:
Electrochemistry
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.3K
Revisions:
2 times
(View History)
Update Date:
19 Feb 2024
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No