Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Satoru Matsuda | -- | 1857 | 2024-02-18 13:46:39 | | | |
| 2 | Rita Xu | Meta information modification | 1857 | 2024-02-19 02:32:12 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Nakashima, M.; Suga, N.; Ikeda, Y.; Yoshikawa, S.; Matsuda, S. Innovative Treatment against Polycystic Kidney Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/55119 (accessed on 24 June 2026).
Nakashima M, Suga N, Ikeda Y, Yoshikawa S, Matsuda S. Innovative Treatment against Polycystic Kidney Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/55119. Accessed June 24, 2026.
Nakashima, Moeka, Naoko Suga, Yuka Ikeda, Sayuri Yoshikawa, Satoru Matsuda. "Innovative Treatment against Polycystic Kidney Disease" Encyclopedia, https://encyclopedia.pub/entry/55119 (accessed June 24, 2026).
Nakashima, M., Suga, N., Ikeda, Y., Yoshikawa, S., & Matsuda, S. (2024, February 18). Innovative Treatment against Polycystic Kidney Disease. In Encyclopedia. https://encyclopedia.pub/entry/55119
Nakashima, Moeka, et al. "Innovative Treatment against Polycystic Kidney Disease." Encyclopedia. Web. 18 February, 2024.
Copy Citation
Polycystic kidney disease (PKD) is the most common genetic form of chronic kidney disease (CKD), and it involves the development of multiple kidney cysts.
polycystic kidney disease
chronic kidney disease
autophagy
mitophagy
mitochondria
1. Introduction
Polycystic kidney disease (PKD) is the most familiar genetic type of CKD. The condition is characterized by the development of multiple kidney cysts, and it could subsequently instigate kidney damage and/or renal failure [1]. This disease may be triggered by mutations in PKD1 or PKD2 genes, which encode the integral membrane proteins polycystin-1 and polycystin-2, respectively [2]. Interestingly, these PKD proteins are associated with decreased autophagy [2][3]. Autophagy has been found to be impaired in the epithelial cells of the kidneys in animal models of PKD, as well as in patients with PKD, suggesting that this impairment might contribute to the development and/or progression of PKD [4][5]. In addition, several agents, such as rapamycin, could protect against PKD, which might restore the autophagy in animal models [6]. Mechanistically, aberrant activation of the mammalian target of rapamycin (mTOR), a target molecule of rapamycin, has been shown to be linked to the impaired autophagy as well as the pathology of PKD [7]. Cyst enlargement in PKD kidneys may result in restricted areas of hypoxia [8]. Hypoxic stimuli may then increase the hypoxia-inducible factor-1α (HIF-1α) protein by preventing its degradation by the proteasome. Under hypoxic conditions, the phosphatidylinositol 3-kinase (PI3K) and the mTOR pathway might activate the expression of HIF-1α [9]. (Figure 1) Hypoxia-related events have been revealed to be linked with cyst formation [10][11]. HIF-1α has been found to be also highly expressed in dendritic cells, and its expression is relatively higher in radicular cysts than in odontogenic tumors [12]. In PKD, HIF-1α may not disturb initial cyst formation, but it is important for cyst progression and expansion in later stages of the disease [13]. Conversely, it has also been shown that HIF-1α inhibition could reduce cystic growth [14]. Signaling pathways being related to the activation of HIF-1α during hypoxia could be contributing to cyst expansion in PKD [15]. Reactive oxygen species (ROS) have been also shown to stimulate cyst development in PKD. In addition, several tissues of PKD may exhibit elevated ROS levels that are positively interrelated with disease severity [15][16]. Peroxidation of phospholipids in animal and human kidneys may be caused by high amounts of ROS [17][18]. Cultured renal cysts and MDCK cell cysts in a three-dimensional setup have confirmed a relationship between lipid peroxidation and increased cyst size [14][19]. It is well-known that damaged mitochondria could induce the generation of ROS and may bring about an increase in membrane lipid peroxidation. Therefore, autophagy, mitochondria, hypoxia and/or ROS might be important administrators in the progression in PKD. Undoubtedly, these hypotheses need further investigation. In addition, despite remarkable efforts to clarify all features of PKD through wide-ranging translational research, there is still an unmet clinical requirement for biomarkers and/or prognosticators that may possibly predict the speed of disease progression [20][21][22]. A better comprehending of the pathophysiology of cystic expansion may lead to the advancement of potential therapies to slow cyst development and/or expansion. The development of innovative treatments that may act synergistically or have fewer side effects might considerably improve the treatment consequences.
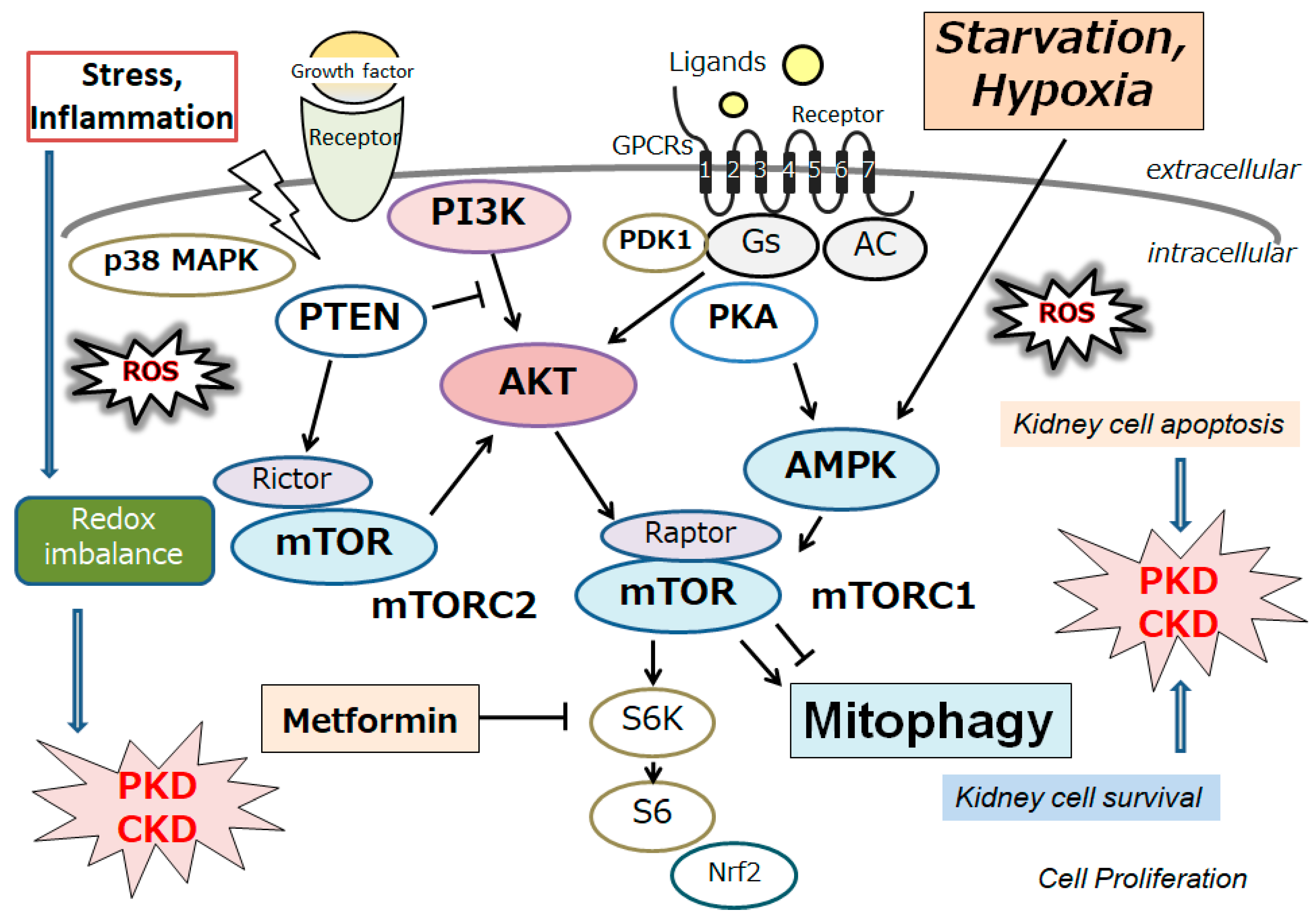
Figure 1. Schematic representation of the relevant signaling pathway potentially being involved in the pathogenesis of polycystic kidney disease (PKD) and/or chronic kidney disease (CKD). Several modulator molecules linked to the PI3K/AKT/mTOR/mTORC1 signaling pathway are demonstrated. Examples of compound metformin, as well as hypoxia and/or starvation, known to act on the AMPK/mTOR and/or mitophagy signaling, are also shown. Arrowhead indicates stimulation, whereas hammerhead shows inhibition. Note that several important activities, such as cytokine-induction and/or inflammatory reactions, have been omitted for clarity. Abbreviation: mTOR, mammalian/mechanistic target of rapamycin; PI3K, phosphoinositide-3 kinase; ROS, reactive oxygen species.
2. Autophagy/Mitophagy and Redox Imbalance in the Homeostasis of Kidney Cells
Substantial evidence has supported an imperative role for autophagy in kidney pathophysiology. Autophagy is firmly regulated to support cells to get used to and/or decrease cellular stress. Some studies have emphasized an intricate signaling system that could detect alterations in energy and/or nutrient condition to either activate or prevent autophagy. Intracellular stresses, which can be brought from ROS, hypoxia, stress of endoplasmic reticulum, several DNA damages and/or inflammatory immune signaling have been revealed as potential stimulators of autophagy [23]. Interestingly, autophagy has been defined as a HIF-1α-dependent response [24]. Autophagy describes the process by which cytoplasmic materials, including organelles, access the lysosomes for hydrolytic degeneration [25], which is also a course of cell repair that may frequently convey the apoptosis termed “self-killing” of cells [26]. Dying cells may frequently exhibit an accumulation of autophagosomes and hence adopt a morphology known as autophagic cell death [26]. Consequently, autophagic cell death might cause cell death with autophagy rather than cell death by autophagy. Hypoxia can regulate the mTOR complex 1 (mTORC1) [27]. Therefore, hypoxia and/or mTOR signaling may be modulators of autophagy [27]. (Figure 1) Mitochondria are particularly sensitive to hypoxa, which might result in both functional and morphological impairments. Mitophagy is an arrangement of autophagy that eliminates surplus mitochondria, facilitates reconstruction of mitochondria, and prevents the accumulation of impaired mitochondria [28]. Therefore, mitophagy might be a key mechanism for preserving the quality of mitochondria by eliminating damaged mitochondria. In response to hypoxia, the PTEN-induced putative kinase 1 (PINK1) may be activated as a regulator of mitophagy, confirming the suitable functioning of the total mitochondrial network [29]. Various stressors, such as hypoxia, ischemia, ageing, and oxidative stress, may lead to an increase in ROS and damages to mitochondria, which may trigger the PINK1 mediated mitophagy [30]. (Figure 2) There is evidence for weakened mitophagy in the renal cells of diabetic mice with reduced expressions of mitochondrial PINK1 [31]. A working mitophagy system may act as a scavenger of damaged mitochondria, and thereby maintain a decent mitochondrial homeostasis.
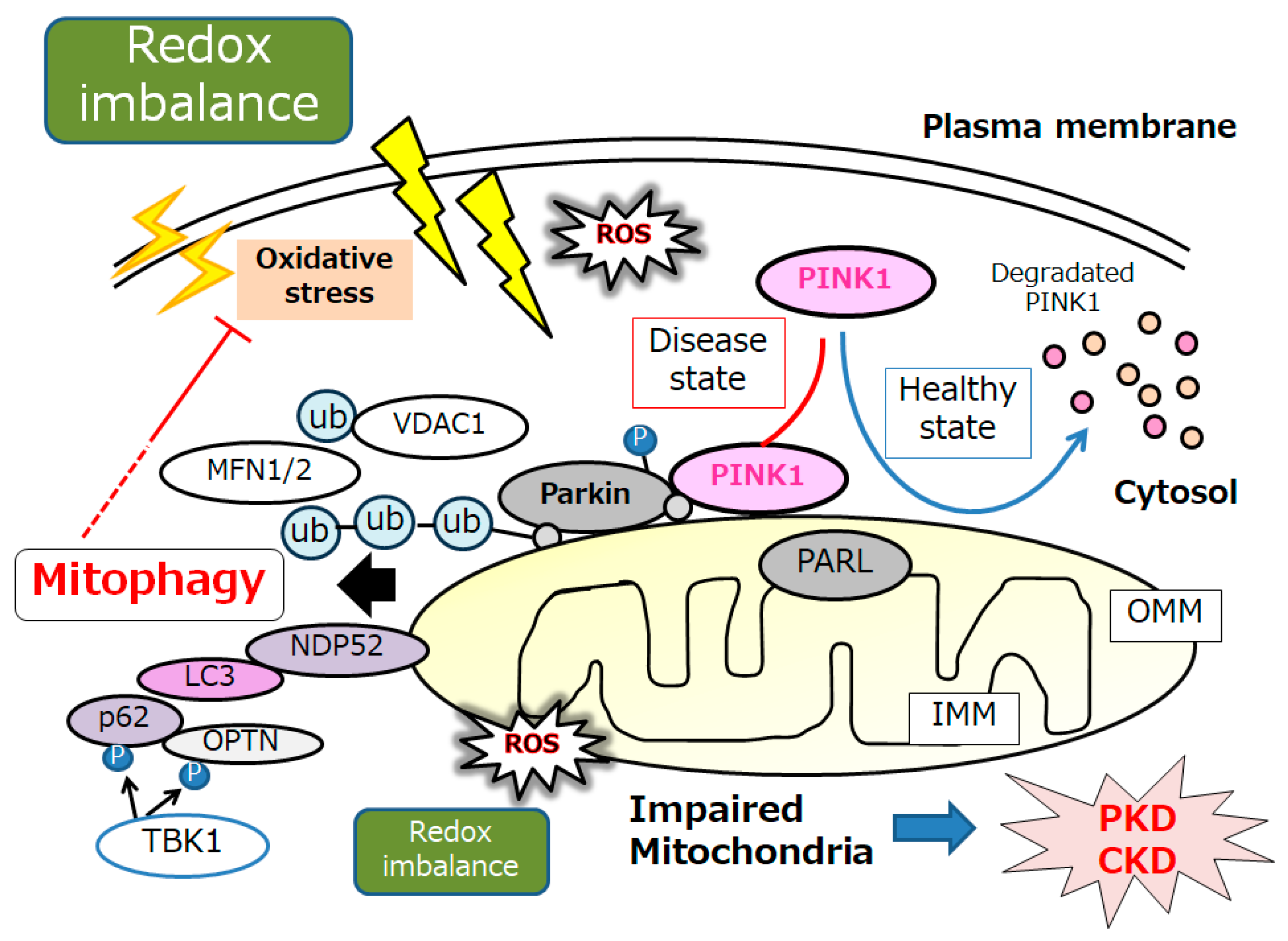
Figure 2. An illustrative representation and overview of PINK1, Parkin, and related molecules in the regulatory pathway for mitophagy. Under the healthy and steady state of cells, PINK1 is despoiled within the surface of mitochondria, which may be reduced by mitochondrial damage due to oxidative stress and/or redox imbalance, resulting in PINK1 and Parkin increases in the outer membrane of mitochondria. Mainly, the PINK1 could phosphorylate ubiquitin to activate the ubiquitin ligase activity for Parkin, where the Parkin is expected to be phosphorylated and ubiquitinated, resulting in the induction of mitophagy. OMM, outer mitochondrial membrane; IMM, inner mitochondrial membrane; MARK2, microtubule affinity regulating kinase 2; MFN1, mitofusin 1; MFN2, mitofusin 2; NDP52, nuclear dot protein 52; PARL, presenilin-associated rhomboid-like; OPTN, optineurin; PINK1, PTEN-induced kinase 1; ROS, reactive oxygen species; VDAC1, voltage-dependent anion channel 1; Ub, ubiquitin.
Mitophagy is principally facilitated by microtubule-associated protein 1 light chain 3 (LC3)-linked receptors. Ubiquitin-dependent mitophagy may include the mitochondrial serine/threonine protein kinase PINK1 and E3 ubiquitin protein ligase Parkin; it also may include the Parkin/PINK1 pathways [32]. Conclusions of the experiment in primary human renal epithelial cells have demonstrated that mitochondrial quality control could be disturbed by mitophagy mediated via PINK/Parkin signaling [33]. PINK1 accumulates on the outer mitochondrial membrane (OMM) after loss of mitochondrial membrane potential, where it recruits and then phosphorylates Parkin to add phosphor-ubiquitin chains on OMM proteins. Interestingly, mitophagy could inhibit oxidative stress via the upregulation of the PINK1-parkin pathway, which could delay kidney senescence in mice [34]. Autophagy receptors, including optineurin, calcium-binding and coiled-coil domain-containing protein 2, also called nuclear dot protein 52 kDa (NDP52), which comprise both ubiquitin binding domains and LC3-interacting regions, could link the ubiquitylated mitochondria to LC3-associated membranes for appropriation [35]. PINK1-mediated phosphorylation of ubiquitin can employ optineurin and/or NDP52 to induce mitophagy without Parkin. By attaching to LC3 at their cytosolic N-terminus, mitophagy receptors could connect impaired mitochondria directly to autophagosomes. (Figure 2) After ubiquitination, impaired mitochondria might be consequently recognized by adapter proteins to be eaten by autophagosomes. Too much mitophagy might result in cellular energy depletion. Therefore, mitophagy may positively or negatively regulate apoptosis, which is a double-edged sword in the pathogenesis of several diseases. For example, a high or low level of mitophagy activity may occasionally induce podocyte apoptosis, which is the collective pathological base for the progression of several kidney diseases [36]. In general, mitophagy may be induced as a protection mechanism for keeping a population of well mitochondria and thus safeguarding cell survival. Although mitophagy may be dispensable for kidney development [37], mitophagy seems to be essential for maintaining kidney integrity and normal physiology in adult kidney cells [38]. Clearance of damaged mitochondria via mitophagy is valuable to the protective effect of impaired kidney cells [39].
3. Autophagy/Mitophagy Involved in the Pathogenesis of Several Kidney Diseases, including Polycystic Kidney
Collecting evidence relates the impaired mitophagy with disease pathogenesis/progression in several pathological situations, including kidney diseases [40]. Acute kidney injury (AKI) may be categorized by a rapid weakening of kidney function, which typically results from renal ischemia, sepsis, and nephrotoxic agents [41]. Mitophagy induction might act as a mutual mechanism to kidney tubular cell protection in many models of AKI [42]. The mitophagy-mediated removal of injured mitochondria might inhibit excessive ROS accumulation, as well as prevent the release of damage-associated molecular arrays which might indorse inflammation during AKI. As renal tissue has massive mitochondrial content, mitophagy and/or mitochondrial biogenesis may be critical to overwhelming stressful illnesses, including AKI [42][43]. Mitophagy might enable compromised cells to persist during kidney interstitial fibrosis, which is a feature of maladaptive restoration in the transition from AKI to chronic kidney disease (CKD) [44]. Mitophagy being induced in distal tubules and pericytes could protect against renal interstitial fibrosis by suppressing the inflammasome of the tumor growth factor β (TGFβ) and the NLR family pyrin domain containing 3 (NLRP3) signaling [45]. Therefore, mitophagy might be a pharmacological target for the management of interstitial fibrosis in kidneys, particularly in regard to offering new concepts for more efficient anti-fibrosis and delaying the development of CKD [46]. It has been shown that mitophagy activation may protect against renal fibrosis via the downregulation of TGF-β1/Smad signaling, improving mitochondrial fitness and alleviating inflammatory infiltration in kidneys [47]. Focal segmental glomerulosclerosis may be one of the fibrotic diseases in kidneys that is characterized by glomerular lesions with podocytes [48]. It has been revealed that podocyte mitophagy could have an imperative role in the development of the focal segmental glomerulosclerosis [48]. In addition, modifications of the apolipoprotein L1 (APOL1) gene have known links with the focal segmental glomerulosclerosis, which may affect endosomal trafficking and/or block mitophagic flux, eventually leading to podocyte injury [49]. Therefore, podocyte mitophagy could counteract the development of the focal segmental glomerulosclerosis [50]. A decrease in podocyte mitophagy may underlie the conceivable progression of podocytopathies, including the focal segmental glomerulosclerosis [51]. Interestingly, activation of mTORC1 has been detected in glomeruli from patients with the focal segmental glomerulosclerosis [52]. Hyperglycemia may inhibit mitophagy in kidney tubules of diabetic patients with diabetes mellitus [53]. It seems that mitophagy has been impaired in the diabetic kidneys of patients with diabetic kidney disease [53]. Defective mitophagy induced by high glucose levels may accelerate the senescence of tubular cells [54]. Treatment with the mitochondria-targeted antioxidant may ameliorate tubular injury in diabetic mice by restoring mitophagy, which might be mediated by an elevation in PINK1 expression provoked by nuclear factor erythroid 2-related factor 2 [53][54]. Therefore, mitophagy in kidney tubules might be helpful for diabetic kidney disease.
References
- Torres, V.E.; Harris, P.C. Progress in the understanding of polycystic kidney disease. Nat. Rev. Nephrol. 2019, 15, 70–72.
- Harris, P.C.; Rossetti, S. Molecular diagnostics for autosomal dominant polycystic kidney disease. Nat. Rev. Nephrol. 2010, 6, 197–206.
- Anvarian, Z.; Mykytyn, K.; Mukhopadhyay, S.; Pedersen, L.B.; Christensen, S.T. Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol. 2019, 15, 199–219.
- Belibi, F.; Zafar, I.; Ravichandran, K.; Segvic, A.B.; Jani, A.; Ljubanovic, D.G.; Edelstein, C.L. Hypoxia-inducible factor-1α (HIF-1α) and autophagy in polycystic kidney disease (PKD). Am. J. Physiol. Renal Physiol. 2011, 300, F1235–F1243.
- Nowak, K.L.; Edelstein, C.L. Apoptosis and autophagy in polycystic kidney disease (PKD). Cell. Signal. 2020, 68, 109518.
- Li, A.; Fan, S.; Xu, Y.; Meng, J.; Shen, X.; Mao, J.; Zhang, L.; Zhang, X.; Moeckel, G.; Wu, D.; et al. Rapamycin treatment dose-dependently improves the cystic kidney in a new ADPKD mouse model via the mTORC1 and cell-cycle-associated CDK1/cyclin axis. J. Cell. Mol. Med. 2017, 21, 1619–1635.
- Kou, P.; Wei, S.; Xiong, F. Recent advances of mTOR inhibitors use in autosomal dominant polycystic kidney disease: Is the road still open? Curr. Med. Chem. 2019, 26, 2962–2973.
- Zeier, M.; Jones, E.; Ritz, E. Autosomal dominant polycystic kidney disease—The patient on renal replacement therapy. Nephrol. Dial. Transplant. 1996, 11 (Suppl. S6), 18–20.
- Dery, M.C.; Michaud, M.D.; Richard, D.E. Hypoxia-inducible factor 1: Regulation by hypoxic and non-hypoxic activators. Int. J. Biochem. Cell Biol. 2005, 37, 535–540.
- De Mendonça, R.P.; Balbinot, K.M.; Martins, B.V.; da Silva Kataoka, M.S.; Mesquita, R.A.; de Jesus Viana Pinheiro, J.; de Melo Alves Júnior, S. Hypoxia and proangiogenic proteins in human ameloblastoma. Sci. Rep. 2020, 10, 17567.
- Pereira-Prado, V.; Vigil-Bastitta, G.; Sánchez-Romero, C.; Arocena, M.; Molina-Frechero, N.; González-González, R.; Meleti, M.; Bologna-Molina, R. Immunoexpression of galectin-3 and its potential relation to hypoxia-inducible factor-1α in ameloblastomas. Biotech. Histochem. 2021, 96, 296–301.
- Da Costa, N.M.M.; de Siqueira, A.S.; Ribeiro, A.L.R.; da Silva Kataoka, M.S.; Jaeger, R.G.; de Alves-Júnior, S.M.; Smith, A.M.; de Jesus Viana Pinheiro, J. Role of HIF-1α and CASPASE-3 in cystogenesis of odontogenic cysts and tumors. Clin. Oral. Investig. 2018, 22, 141–149.
- Buchholz, B.; Eckardt, K.U. Role of oxygen and the HIF-pathway in polycystic kidney disease. Cell. Signal. 2020, 69, 109524.
- Buchholz, B.; Schley, G.; Faria, D.; Kroening, S.; Willam, C.; Schreiber, R.; Klanke, B.; Burzlaff, N.; Jantsch, J.; Kunzelmann, K.; et al. Hypoxia-inducible factor-1α causes renal cyst expansion through calcium-activated chloride secretion. J. Am. Soc. Nephrol. 2014, 25, 465–474.
- Li, Z.L.; Ding, L.; Ma, R.X.; Zhang, Y.; Zhang, Y.L.; Ni, W.J.; Tang, T.T.; Wang, G.H.; Wang, B.; Lv, L.L.; et al. Activation of HIF-1α C-terminal transactivation domain protects against hypoxia-induced kidney injury through hexokinase 2-mediated mitophagy. Cell Death Dis. 2023, 14, 339.
- Schreiber, R.; Buchholz, B.; Kraus, A.; Schley, G.; Scholz, J.; Ousingsawat, J.; Kunzelmann, K. Lipid peroxidation drives renal cyst growth in vitro through activation of TMEM16A. J. Am. Soc. Nephrol. 2019, 30, 228–242.
- Jiang, W.; Liu, J.; Cui, J.; Su, J.; Xu, W.; Zhang, F.; Ding, Y. Ferroptosis plays a crucial role in lung cell damage caused by ventilation stretch. Free Radic. Biol. Med. 2023, 209 Pt 1, 84–95.
- Zhang, X.; Li, L.X.; Ding, H.; Torres, V.E.; Yu, C.; Li, X. Ferroptosis Promotes Cyst Growth in Autosomal Dominant Polycystic Kidney Disease Mouse Models. J. Am. Soc. Nephrol. 2021, 32, 2759–2776.
- Falchook, G.S.; Wheler, J.J.; Naing, A.; Jackson, E.F.; Janku, F.; Hong, D.; Ng, C.S.; Tannir, N.M.; Lawhorn, K.N.; Huang, M.; et al. Targeting hypoxia-inducible factor-1α (HIF-1α) in combination with antiangiogenic therapy: A phase I trial of bortezomib plus bevacizumab. Oncotarget 2014, 5, 10280–10292.
- Gregory, A.V.; Chebib, F.T.; Poudyal, B.; Holmes, H.L.; Yu, A.S.L.; Landsittel, D.P.; Bae, K.T.; Chapman, A.B.; Frederic, R.O.; Mrug, M.; et al. Utility of new image-derived biomarkers for autosomal dominant polycystic kidney disease prognosis using automated instance cyst segmentation. Kidney Int. 2023, 104, 334–342.
- Kim, Y.; Tao, C.; Kim, H.; Oh, G.Y.; Ko, J.; Bae, K.T. A Deep Learning Approach for Automated Segmentation of Kidneys and Exophytic Cysts in Individuals with Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2022, 33, 1581–1589.
- Raj, A.; Tollens, F.; Hansen, L.; Golla, A.K.; Schad, L.R.; Nörenberg, D.; Zöllner, F.G. Deep Learning-Based Total Kidney Volume Segmentation in Autosomal Dominant Polycystic Kidney Disease Using Attention, Cosine Loss, and Sharpness Aware Minimization. Diagnostics 2022, 12, 1159.
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293.
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1α-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903.
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326.
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010.
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295.
- Li, A.; Gao, M.; Liu, B.; Qin, Y.; Chen, L.; Liu, H.; Wu, H.; Gong, G. Mitochondrial autophagy: Molecular mechanisms and implications for cardiovascular disease. Cell Death Dis. 2022, 13, 444.
- Chen, M.; Wang, W.; Fu, X.; Yi, Y.; Wang, K.; Wang, M. Role of Pink1-mediated mitophagy in adenomyosis. PeerJ 2023, 11, e16497.
- Ji, Y.; Leng, Y.; Lei, S.; Qiu, Z.; Ming, H.; Zhang, Y.; Zhang, A.; Wu, Y.; Xia, Z. The mitochondria-targeted antioxidant MitoQ ameliorates myocardial ischemia-reperfusion injury by enhancing PINK1/Parkin-mediated mitophagy in type 2 diabetic rats. Cell Stress Chaperones 2022, 27, 353–367.
- Feng, J.; Lu, C.; Dai, Q.; Sheng, J.; Xu, M. SIRT3 Facilitates Amniotic Fluid Stem Cells to Repair Diabetic Nephropathy through Protecting Mitochondrial Homeostasis by Modulation of Mitophagy. Cell. Physiol. Biochem. 2018, 46, 1508–1524.
- Tang, C.; Livingston, M.J.; Liu, Z.; Dong, Z. Autophagy in kidney homeostasis and disease. Nat. Rev. Nephrol. 2020, 16, 489–508.
- Chen, K.; Chen, J.; Wang, L.; Yang, J.; Xiao, F.; Wang, X.; Yuan, J.; Wang, L.; He, Y. Parkin Ubiquitinates GATA4 and Attenuates the GATA4/GAS1 Signaling and Detrimental Effects on Diabetic Nephropathy. FASEB J. 2020, 34, 8858–8875.
- E, Y.; Lin, Y.; Yan, G.; Yang, J.; Jiao, L.; Wu, R.; Yan, Q.; Chen, Y.; Chen, Y.; Yan, X.; et al. Exogenous H2S alleviates senescence of glomerular mesangial cells through up-regulating mitophagy by activation of AMPK-ULK1-PINK1-parkin pathway in mice. Biochim. Biophys. Acta Mol. Cell Res. 2023, 1870, 119568.
- Saha, B.; Olsvik, H.; Williams, G.L.; Oh, S.; Evjen, G.; Sjøttem, E.; Mandell, M.A. TBK1 is ubiquitinated by TRIM5α to assemble mitophagy machinery. bioRxiv 2023.
- Gao, X.; Liu, Y.; Wang, L.; Sai, N.; Liu, Y.; Ni, J. Morroniside Inhibits H2O2-Induced Podocyte Apoptosis by Down-Regulating NOX4 Expression Controlled by Autophagy In Vitro. Front. Pharmacol. 2020, 11, 533809.
- Liu, S.; Hartleben, B.; Kretz, O.; Wiech, T.; Igarashi, P.; Mizushima, N.; Walz, G.; Huber, T.B. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy 2012, 8, 826–837.
- Forbes, M.S.; Thornhill, B.A.; Chevalier, R.L. Proximal tubular injury and rapid formation of atubular glomeruli in mice with unilateral ureteral obstruction: A new look at an old model. Am. J. Physiol. Renal Physiol. 2011, 301, F110–F117.
- Livingston, M.J.; Wang, J.; Zhou, J.; Wu, G.; Ganley, I.G.; Hill, J.A.; Yin, X.M.; Dong, Z. Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys. Autophagy 2019, 15, 2142–2162.
- Su, L.; Zhang, J.; Gomez, H.; Kellum, J.A.; Peng, Z. Mitochondria ROS and mitophagy in acute kidney injury. Autophagy 2023, 19, 401–414.
- Su, L.; Zhang, J.; Wang, J.; Wang, X.; Cao, E.; Yang, C.; Sun, Q.; Sivakumar, R.; Peng, Z. Pannexin 1 targets mitophagy to mediate renal ischemia/reperfusion injury. Commun. Biol. 2023, 6, 889.
- Wang, Y.; Cai, J.; Tang, C.; Dong, Z. Mitophagy in acute kidney injury and kidney repair. Cells 2020, 9, 338.
- Bhatia, D.; Choi, M.E. The emerging role of mitophagy in kidney diseases. J. Life Sci. 2019, 1, 13–22.
- Baisantry, A.; Bhayana, S.; Rong, S.; Ermeling, E.; Wrede, C.; Hegermann, J.; Pennekamp, P.; Sörensen-Zender, I.; Haller, H.; Melk, A.; et al. Autophagy induces prosenescent changes in proximal tubular S3 segments. J. Am. Soc. Nephrol. 2016, 27, 1609–1616.
- Nam, S.A.; Kim, W.Y.; Kim, J.W.; Kang, M.G.; Park, S.H.; Lee, M.S.; Kim, H.W.; Yang, C.W.; Kim, J.; Kim, Y.K. Autophagy in FOXD1 stroma-derived cells regulates renal fibrosis through TGF-beta and NLRP3 inflammasome pathway. Biochem. Biophys. Res. Commun. 2019, 508, 965–972.
- Sun, J.; Liu, C.; Liu, Y.Y.; Guo, Z.A. Mitophagy in renal interstitial fibrosis. Int. Urol. Nephrol. 2023, 56, 167–179.
- Jin, L.; Yu, B.; Liu, G.; Nie, W.; Wang, J.; Chen, J.; Xiao, L.; Xia, H.; Han, F.; Yang, Y. Mitophagy induced by UMI-77 preserves mitochondrial fitness in renal tubular epithelial cells and alleviates renal fibrosis. FASEB J. 2022, 36, e22342.
- 48 Rosenberg, A.Z.; Kopp, J.B. Focal Segmental Glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2017, 12, 502–517.
- Kumar, V.; Ayasolla, K.; Jha, A.; Mishra, A.; Vashistha, H.; Lan, X.; Qayyum, M.; Chinnapaka, S.; Purohit, R.; Mikulak, J.; et al. Disrupted apolipoprotein L1-miR193a axis dedifferentiates podocytes through autophagy blockade in an APOL1 risk milieu. Am. J. Physiol. Cell Physiol. 2019, 317, C209–C225.
- Asanuma, K.; Tanida, I.; Shirato, I.; Ueno, T.; Takahara, H.; Nishitani, T.; Kominami, E.; Tomino, Y. MAP-LC3, a promising autophagosomal marker, is processed during the differentiation and recovery of podocytes from PAN nephrosis. FASEB J. 2003, 17, 1165–1167.
- Zeng, C.; Fan, Y.; Wu, J.; Shi, S.; Chen, Z.; Zhong, Y.; Zhang, C.; Zen, K.; Liu, Z. Podocyte autophagic activity plays a protective role in renal injury and delays the progression of podocytopathies. J. Pathol. 2014, 234, 203–213.
- Zschiedrich, S.; Bork, T.; Liang, W.; Wanner, N.; Eulenbruch, K.; Munder, S.; Hartleben, B.; Kretz, O.; Gerber, S.; Simons, M.; et al. Targeting mTOR signaling can prevent the progression of FSGS. J. Am. Soc. Nephrol. 2017, 28, 2144–2157.
- Ma, Z.; Li, L.; Livingston, M.J.; Zhang, D.; Mi, Q.; Zhang, M.; Ding, H.F.; Huo, Y.; Mei, C.; Dong, Z. p53/microRNA-214/ULK1 axis impairs renal tubular autophagy in diabetic kidney disease. J. Clin. Investig. 2020, 130, 5011–5026.
- Chen, K.; Dai, H.; Yuan, J.; Chen, J.; Lin, L.; Zhang, W.; Wang, L.; Zhang, J.; Li, K.; He, Y. Optineurin-mediated mitophagy protects renal tubular epithelial cells against accelerated senescence in diabetic nephropathy. Cell Death Dis. 2018, 9, 105.
More
Information
Subjects:
Food Science & Technology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
527
Revisions:
2 times
(View History)
Update Date:
19 Feb 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No