 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Anne VEJUX | + 2478 word(s) | 2478 | 2020-04-08 08:54:00 | | | |
| 2 | Anne VEJUX | Meta information modification | 2478 | 2020-04-10 16:31:39 | | | | |
| 3 | Anne VEJUX | + 124 word(s) | 2602 | 2020-04-10 16:53:45 | | | | |
| 4 | Rita Xu | -151 word(s) | 2451 | 2020-10-29 09:40:45 | | | | |
| 5 | Jamir Pitton Rissardo | + 104 word(s) | 2555 | 2023-09-15 04:40:48 | | |
Video Upload Options
Neurodegenerative diseases are affecting more and more people around the world. Current therapies only treat the symptoms and not the causes of the disease. However, the pathophysiology of these diseases is now better known. In the case of Alzheimer's disease and Parkinson's disease, some common mechanisms have been identified. One of the first known mechanisms is the accumulation of proteins: α-synuclein (Parkinson's disease), Tau (Alzheimer's disease) and β-amyloid (Alzheimer's disease and Parkinson's disease) proteins. Protein accumulation is related to a disruption of mitochondrial activity associated with cell death and oxidative stress. Inflammation is also another important mechanism, which is disrupted in these pathologies.
1. Mechanisms Common to Neurodegenerative Diseases: Pakinson's and Alzheimer's disease
Protein aggregation connected with dysfunctional protein degradation systems, mitochondrial perturbation related with cell death and oxidative stress were identified as mechanisms common to the two most-frequent neurodegenerative diseases in humans : Parkinson’s and Alzheimer’s disease. In the following paragraphs we will detail these mechanisms present in common in these two pathologies.
2. Protein Aggregation and Dysfunctions of Protein Degradation Systems
In Parkinson’s and Alzheimer’s disease, protein aggregation concerns α-synuclein, Tau and β-amyloid proteins, respectively, although β-amyloid proteins are equally present in Parkinson’s disease.
In Parkinson’s disease, insoluble α-synuclein fibrils compose the Lewy bodies and Lewy neurites. These are mainly detected in the pigmented neurons of the substantia nigra and in other neuronal populations at the peripheral and central levels [1][2][3]. Lewy bodies are existent in the dopaminergic neurons of substantia nigra pars compacta. These Lewy bodies are round intraneuronal and positive round inclusions of α-synuclein and ubiquitin [3]. Lewy neurites are atypical neurites made up of α-synuclein filaments and granular material [4]. These Lewy neurites pile up in the amygdala and striatum of Parkinsonian patients [4]. Lewy neurites can restrain neuronal transport and, thus, compromise neuronal activity and survival. In Alzheimer’s disease, the modifications identified are amyloid plaques and neurofibrillary degeneration. Two proteins have been identified as participating in these modifications: Tau protein and Amyloid precursor protein (APP). The APP protein can generate the Aβ peptide after proteolysis. Amyloid plaques are formed by aggregation of Aβ peptide[5]. Neurofibrillary degeneration is composed of argentophilic neurofilaments found in the pericaryon of some cortical neurons but is also detected within myelinated axons, dendrites and synapses [6]. The intensity of these neurofibrillary accumulations in the neocortex is directly connected with the severity of the disease [7]. Neurofibrillary tangles are composed of hyper-phosphorylated Tau proteins arranged in paired helical filaments. Extracellular deposits of Aβ peptide compose amyloid plaques. Glial cells containing phagocytic lysosomes, and amyelinic neurites enclose amyloid plaques. These plaques are in preference detected in certain areas of the brain: cortex, striatum and cerebellum. The less mature plaques are described as diffuse plaques, composed of amorphous, low-density aggregates of Aβ peptides. Mature plaques consist of very dense aggregates of the Aβ peptide and are correlated with neurodegeneration and astroglial reactivity.
Two systems are involved in the degradation of α-synuclein: the ubiquitin-proteasome system (UPS) and the autophagy-lysosome pathway (ALP)[8][9]. The UPS system recognizes short-lived soluble proteins and the ALP pathway identify long-lived macromolecules, cytosolic components and dysfunctional organelles. In the case of Parkinson’s disease, chaperone-mediated autophagy (CMA), pathway belonging to ALP system, is implicated. Indeed, CMA activity is connected to levels of LAMP-2A, this receptor of the CMA pathway attaches to chaperones who have identified proteins having the KFERQ motif. In the substantia nigra of patient brains, a diminution of the expression of LAMP-2A is particularly observed[10][11][12]. This diminution is connected to the accumulation of α-synuclein and nigral cell death [13]. Modifications in enzyme content and acidification of the lysosomes were observed as well as autophagosomes accumulation[14][15][16]. In Alzheimer’s disease, the accumulation of Aβ inside cells in multivesicular bodies can be explained by proteasome inhibition[17]. Both in vitro and in vivo, peptide levels Aβ can increase due to proteasome inhibition [18]. Extracellular Aβ can enter the cell and inhibit proteasome activity in cultured neurons, which causes it to accumulate in the cytosol[18].
3. Mitochondrial Dysfunction and Cell Death
In the case of Parkinson’s disease, a deficiency in the complex I of the mitochondrial respiratory chain is detected at the level of the patient’s black matter in the neurons and in the glia, as well as at the level of skeletal muscle and platelets[19][20][21][22]. This change in the activity of the mitochondrial respiratory chain favors the augmentation of mitochondria-dependent apoptotic processes[23]. On experimental models and in patients, modifications in mitochondrial dynamics, at the bioenergetic level and the decrease of complex I activity of the respiratory chain, have been detected[24][25]. Moreover, a relationship exists between the genes involved in Parkinson’s disease and mitochondrial function. Indeed, mutations in the LRRK2 gene are connected with dysfunctions in the mitochondria [26]. The proteins encoded by the PARK2 and PINK1 genes are implicated in the elimination of dysfunctional mitochondria by the process of mitophagy[26]. In patients with Parkinson’s disease, it has been observed a reduction of degradation of the MIRO protein. This outer mitochondrial membrane protein allows the mitochondria to bind to the microtubules. This diminution leads to loss of function in the mitochondria, which can lead to an increase in oxidative stress[27].
In addition to what has just been described, α-synuclein can interact with the mitochondria and change their function. α-synuclein can be found in the outer membrane of the mitochondria. It can interfere with members of the TOM complex (translocase/receptor system) and induce inhibition of mitochondrial import of proteins [28]. Excessive production of oxidative stress can be produced during interactions between mitochondria and α-synuclein [29]. α-synuclein can also disrupt the fusion process, and inhibits complex I [30][31]. Histological experiments demonstrate cell death is the process in charge for Parkinson’s disease: detection of fragmented DNA, apoptosis in patients’ brains (TUNEL methods), presence of active forms of caspases -1, -3, -8 and -9 at the level of the black substance[32][33]. The intrinsic and extrinsic pathways of cell death are also highlighted in post-mortem studies: implication of mitochondrial pathway (p53-GAPDH-Bax pathway), and Fas/FADD death receptor pathway, (high levels of Fas, FADD and caspase-8 in the brains of Parkinsonian patients)[34].
Loss of neurons in the cortical II layer of the entorhinal cortex is a major feature of Alzheimer's disease[35]. Neurons, in the entorhinal cortex, synthesize acetylcholine and innervate the hippocampus and neocortex. This type of neuron can also be affected by cell death in Alzheimer’s disease. The two processes responsible for the cell death of these cells are accumulation of amyloid plaques and neurofibrillary degeneration. In various cell cultures and animal models of Alzheimer’s disease, it has been shown exogenous Aβ and pseudo-hyperphosphorylated Tau can induce neuronal death[36], neurons internalizing the extracellular Aβ peptide[37]. The disruption of mitochondrial membranes and of enzymatic activities of the respiratory chain can be induced by Aβ peptide, disturbance that could lead to a production of reactive oxygen species [38]. Induced oxidative stress can activated numerous enzymes, as calpains and caspases, leading to neuronal cell death[39].
4. Oxidative Stress
The exact causes of Parkinson's disease are still being studied, but significant evidence suggests that oxidative stress plays a major role in the degeneration of dopaminergic neurons. Since maintaining a balanced redox potential is crucial for neuron survival, it is unsurprising that any disruption to this balance can interfere with other cellular processes and ultimately lead to cell death. It is also believed that various mechanisms at play can contribute to neurodegeneration in Parkinson's disease, creating a feed-forward scenario where primary insults lead to oxidative stress. This stress can then damage essential pathogenetic proteins and disrupt lipid membranes, producing more ROS and further worsening the condition.[40]
Different free radicals, reactive oxygen species (ROS) and reactive oxygen and nitrogen species (RONS) can induce oxidative stress. Hydrogen peroxide (H2O2), in the presence of iron (in ionic form), provides two hydroxyl radicals (.OH), nitrogen monoxide (NO.) and superoxide (O2.−). Free radicals create damages to proteins, lipids and DNA. This would promote neuronal degeneration.
In postmortem studies in patient’sbrain, damage to proteins, lipids and DNA related to free radicals has been detected at the level of substantia nigra[41]. The dysfunctions of the complex I of the mitochondria lead to an augmentation in the production of free radicals, and inversely, oxidative stress causes dysfunction of the mitochondria[41]. Genetic modifications may also be involved in oxidative stress. The DJ-1 or PARK7 genes code for a protein that might have antioxidant activity. Mutations in these genes are concomitant with the increase of oxidative stress. In DJ-1-deficient mice, increased protein oxidation is present in stressed dopaminergic neurons[42]. Changes in mitochondrial activity and oxidative stress can induce to lysosome depletion and functional impairment of the autophagy/lysosome system. These observations demonstrate the very close relationship between the different processes responsible for the pathology[43].
In Alzheimer’s disease, ROS break up the cytoskeleton, unorganized, particularly at the level of dendrites [39] but, also, the plasma membrane and the membranes of the cell organelles. This process induces activation of the BACE protein and, as a result, drives the metabolism of the APP protein towards the amyloidogenesis pathway with increased production of the toxic Aβ peptide. As previously stated in Parkinson's disease, proteins, nucleic acids and lipids have been damaged by oxidative stress[44][45]. The Aβ peptide may also behave as a pro-oxidant or by impacting NMDA receptor-dependent calcium influxes. These actions can induce mitochondrial dysfunction and in turn ROS overproduction[46][47].
5. Inflammation and Immunity
The central nervous system (CNS) has its own immune system coordinated by immunocompetent cells. Microglial cells, glial cells (astrocytes) and, possibly, neurons are the mains actors implicated in the inflammatory process during Alzheimer’s disease[48][49]. Both microglial cells and astrocytes change rapidly in morphology, antigenicity and function in response to the disease[50]. Microglial cells are cells with support and protection functions for neurons, acting as immunocompetent defense cells. They orchestrate the endogenous immune response of the CNS. Major histocompatibility complex type II (MHCII) can be express in the cells and these microglial cells produce proinflammatory cytokines, chemokines, ROS and complement proteins. These cells play a major role in the cellular immune response to the various lesions encountered in Alzheimer's disease, such as Aβ and senile plaques[51]. Microglial cells can accumulate around amyloid deposits as a result of attraction and activation by the peptide Aβ. The activation of microglial cells lead to increased MHC II cell expression and increased secretion of proinflammatory cytokines (interleukin-1 (IL-1), interleukin-6 (IL-6) and TNF-α (tumor necrosis factor-α)), as well as chemokines (interleukin-8 (IL-8), MIP-1α (macrophage inflammatory protein-1α) and MCP1 (monocyte chemoattractant protein-1))[52][53]. Following chemokine recruitment, the passage of peripheral circulating macrophages across the blood-brain barrier is promoted by Aβ , which may induce an increase in the extent of inflammation. A phagocytic response can be induce in microglial cells by Aβ in a dose- and time-dependent manner[54]. At the same time as the arrival of microglial cells, there is an internalization and co-localization of the lysosome and Aβ associated with damage to neighboring neurons[55][56]. Reactive astrocytes also participate to neurodegeneration by promoting apoptotic cell death[57]. In Alzheimer's disease, astrocytes are present in amyloid deposits, where they secrete several pro-inflammatory molecules such as interleukins, prostaglandins, leukotrienes, thromboxanes and complement factors. These molecules are like those secreted by microglial cells, with which they are co-located. In the brain of non-demented elderly people, diffuse plaques consisting of Aβ granules and associated with astrocytes have been observed. Astrocytes could thus be implicated in the phagocytosis of Aβ[58] and that, probably, the pathology of Alzheimer's disease could have as a possible cause a deficit in the elimination of Aβ by astrocytes. In the brain of Alzheimer’s disease patients[50] and transgenic models of Alzheimer’s disease mice[59], reactive astrocytes are found very close to the plaques, surrounding the Aβ deposits, a mechanism by which the cells could act as a protective barrier between healthy and inflammatory tissues[50]. In addition, the fibrillar form of amyloid peptide operates with complement proteins and in consequence induces the innate immune system[60]. For example, the interelation Aβ/C1q, which implicates the 11 N-terminal residues of Aβ, initiates the classical complement pathway[61]. This phenomenom leads to a cascade of activations that permits the formation of lytic complexes, partly responsible for neuronal death, and inflammatory activation[62]. Neurons can produce certain cytokines such as IL-1[63], IL-6[64], TNF-α[65] and some pentraxins, namely CRP and Ap (amyloid P) and thus participates in the inflammatory process[66]. The mRNA expression levels of proteins of the classical complement pathway are expanded in the brains of Alzheimer’s patients compared to those of controls[67].
Neuroinflammation and, more specifically, microglial cells are believed to be involved in the development and progression of the death of the dopaminergic neurons of the substantia nigra[68]. Using positron emission tomography (PET) imaging, microglial activation was observed in the pons, central gray nuclei, striatum and frontal and temporal cortical regions of patients with Parkinson's disease[69][70]. Post-mortem immunohistological analyses show in the CNS an increase in cell density, branch retraction and hypertrophy of the cell body and expression of MHC II molecules, processes that are synonymous with microglial activation[71][72][73][74], just as increased expression of the lysosomal marker CD68[75][72]. The two main functions attributed to microglial cells are phagocytosis and antigenic presentation. In the 6-hydroxydopamine (6-OHDA) model, these two processes can evolve according to independent kinetics as shown by the analysis of the lysosomal marker CD68, reflecting the activity of phagocytosis, and MHC II molecules, reflecting the role of antigen presentation[76][77][78][79]. Optimal phagocytic activity may occur before or after maximum neuronal death as evidenced by the level of CD68 expression[76][80]. Different hypotheses can be made, given the contradictory results obtained, as to the role of microglial activation. A process of deregulation of phagocytic functions could increase the autonomous mechanisms of neuronal death. The main function of microglial cells could also be the removal of neuronal cell debris. Activated microglia could be have the ability to secrete a set of pro-inflammatory or anti-inflammatory cytokines. Potentially neurotoxic pro-inflammatory proteins have been detected in the brains of patients with Parkinson's disease, including COX and iNOS, TNFα, IL1β and IFNγ[81][82]. T lymphocytes are the main cells that produce IFNγ, which is known to induce activation of macrophages (including microglia). The deleterious role of IFNγ in the pathophysiology of Parkinson's disease has been demonstrated in numerous animal studies[83][84][85][86]. It has been suggested that the dialogue between Th1 cells and microglial cells in the substantia nigra of patients with Parkinson's disease could be a key process in dopaminergic neuronal loss. In the MPTP model, the degeneration of dopaminergic neurons is inhibited by regulatory T lymphocytes (Treg) that modify the molecular behaviour of microglial cells[87][88]. As a result, microglial cells may have distinct or even opposite effect actions depending on the stage of Parkinson's disease, the immunogenetic terrain, and a set of instructional signals delivered by infiltrated T cells, among other factors.
The main signaling pathways implicated in protein aggregation, cell death, oxidative stress and inflammation are summarized in Figure 1.
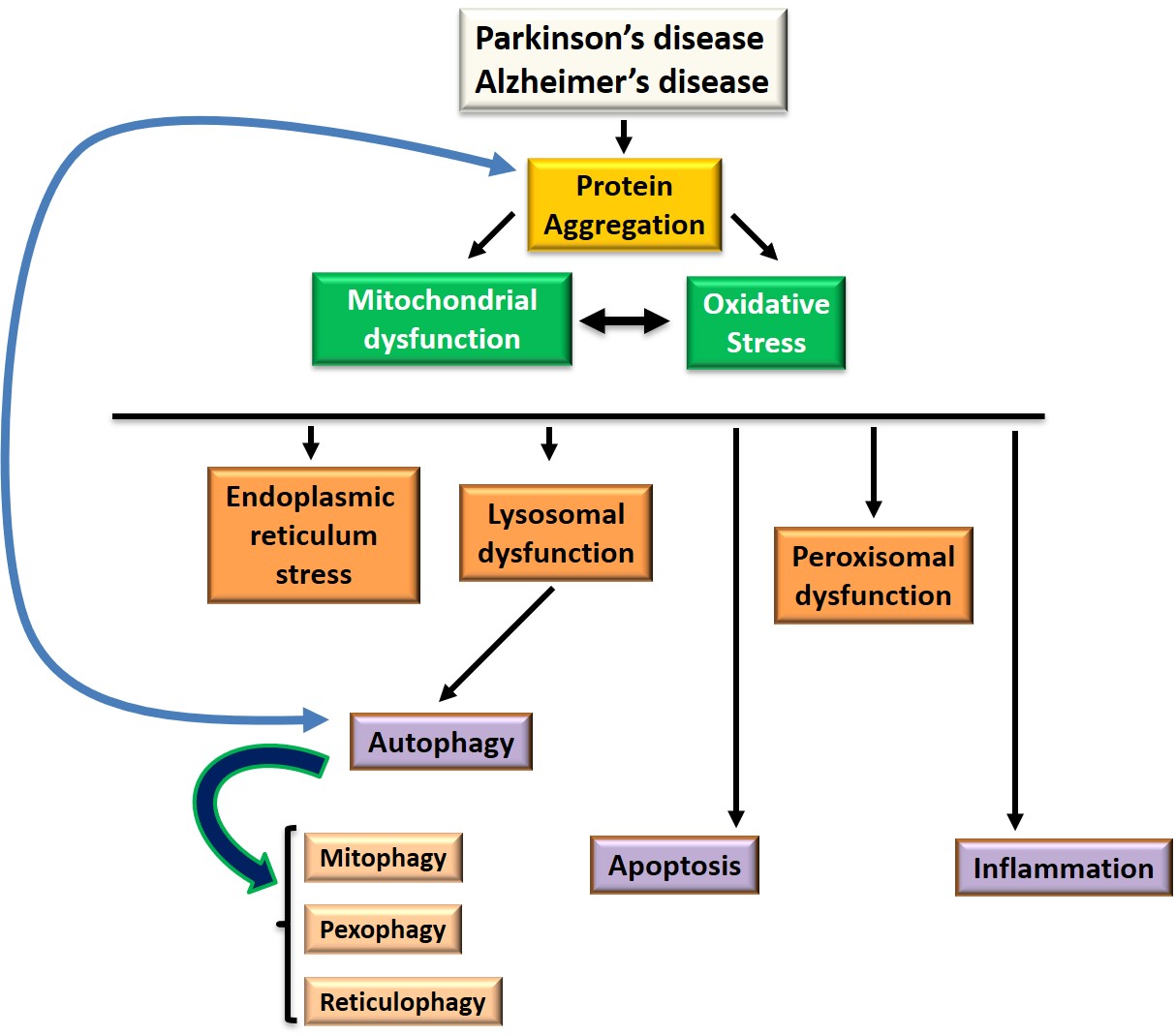
Figure 1: Signaling pathways at the level of target organelles and common processes involved in Parkinson's and Alzheimer's disease: autophagy, apoptosis, oxidative stress and inflammation
The study of these common mechanisms will make it possible to identify natural or synthetic molecules capable of inhibiting them either by using them in natural form or by coupling the compounds of interest to nanoparticles to facilitate their access to the brain.
References
- Maria Grazia Spillantini; Marie Luise Schmidt; Virginia M.-Y. Lee; John Q. Trojanowski; Ross Jakes; Michel Goedert; α-Synuclein in Lewy bodies. Nature 1997, 388, 839-840, 10.1038/42166.
- K Jellinger; Neuropathological substrates of Alzheimer's disease and Parkinson's disease.. Journal of neural transmission. Supplementum 1987, 24, 109-29.
- Hitoshi Takahashi; Koichi Wakabayashi; The cellular pathology of Parkinson's disease.. Neuropathology 2001, 21, 315-322, 10.1046/j.1440-1789.2001.00403.x.
- John E. Duda; Benoit I. Giasson; Meghann E. Mabon; Virginia M.-Y. Lee; John Q. Trojanowski; Novel antibodies to synuclein show abundant striatal pathology in Lewy body diseases. Annals of Neurology 2002, 52, 205-210, 10.1002/ana.10279.
- D J Selkoe; Alzheimer's Disease Is a Synaptic Failure. Science 2002, 298, 789-791, 10.1126/science.1074069.
- K. S. Kosik; L. K. Duffy; M. M. Dowling; Carmela R. Abraham; A. McCluskey; D. J. Selkoe; Microtubule-associated protein 2: monoclonal antibodies demonstrate the selective incorporation of certain epitopes into Alzheimer neurofibrillary tangles.. Proceedings of the National Academy of Sciences 1984, 81, 7941-7945, 10.1073/pnas.81.24.7941.
- A Delacourte; Biochemical and molecular characterization of neurofibrillary degeneration in frontotemporal dementias.. Dementia and Geriatric Cognitive Disorders 1999, 10, 75-79, 10.1159/000051218.
- Alfred L. Goldberg; Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895-899, 10.1038/nature02263.
- Daniel J. Klionsky; Scott D. Emr; Autophagy as a Regulated Pathway of Cellular Degradation. Science 2000, 290, 1717-1721, 10.1126/science.290.5497.1717.
- Lydia Alvarez; María Cruz Rodriguez-Oroz; J M Cooper; Cristina Caballero; Isidro Ferrer; José A Obeso; Anthony H. V. Schapira; Chaperone-Mediated Autophagy Markers in Parkinson Disease Brains. Archives of Neurology 2010, 67, 1464–1472, 10.1001/archneurol.2010.198.
- Ana Maria Cuervo; J. F. Dice; Regulation of lamp2a levels in the lysosomal membrane.. Traffic 2000, 1, 570-583, 10.1034/j.1600-0854.2000.010707.x.
- Ana Maria Cuervo; Aldrin V. Gomes; Junor A. Barnes; J. F. Dice; Selective Degradation of Annexins by Chaperone-mediated Autophagy. Journal of Biological Chemistry 2000, 275, 33329-33335, 10.1074/jbc.m005655200.
- Maria Xilouri; Oeystein Roed Brekk; Alexia Polissidis; Margarita Chrysanthou-Piterou; Ismini Kloukina; Leonidas Stefanis; Impairment of chaperone-mediated autophagy induces dopaminergic neurodegeneration in rats. Autophagy 2016, 12, 2230-2247, 10.1080/15548627.2016.1214777.
- Ana Maria Cuervo; Leonidas Stefanis; Ross Fredenburg; Peter T. Lansbury; David Sulzer; Impaired Degradation of Mutant -Synuclein by Chaperone-Mediated Autophagy. Science 2004, 305, 1292-1295, 10.1126/science.1101738.
- Wai Haung Yu; Beatriz Dorado; Helen Yvette Figueroa; Lili Wang; Emmanuel Planel; Mark R. Cookson; Lorraine N. Clark; Karen E. Duff; Metabolic Activity Determines Efficacy of Macroautophagic Clearance of Pathological Oligomeric α-Synuclein. The American Journal of Pathology 2009, 175, 736-747, 10.2353/ajpath.2009.080928.
- Brian Spencer; Rewati Potkar; Margarita Trejo; Edward Rockenstein; Christina Patrick; Ryan Gindi; Anthony Adame; Tony Wyss-Coray; Eliezer Masliah; Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson's and Lewy body diseases.. The Journal of Neuroscience 2009, 29, 13578-88, 10.1523/JNEUROSCI.4390-09.2009.
- Claudia Guimas Almeida; Reisuke H. Takahashi; Gunnar K. Gouras; β-Amyloid Accumulation Impairs Multivesicular Body Sorting by Inhibiting the Ubiquitin-Proteasome System. The Journal of Neuroscience 2006, 26, 4277-4288, 10.1523/JNEUROSCI.5078-05.2006.
- Sangsoo Oh; Hyun Seok Hong; Enmi Hwang; Hae Jin Sim; Woojin Lee; Su Jeon Shin; Inhee Mook-Jung; Amyloid peptide attenuates the proteasome activity in neuronal cells. Mechanisms of Ageing and Development 2005, 126, 1292-1299, 10.1016/j.mad.2005.07.006.
- Yoshikuni Mizuno; Shigeo Ohta; Masashi Tanaka; Shinzaburo Takamiya; Keiji Suzuki; Takeshi Sato; Hiroshi Oya; Takayuki Ozawa; Yasuo Kagawa; Deficiencies in Complex I subunits of the respiratory chain in Parkinson's disease. Biochemical and Biophysical Research Communications 1989, 163, 1450-1455, 10.1016/0006-291x(89)91141-8.
- A H Schapira; J.M. Cooper; D. Dexter; Peter Jenner; J.B. Clark; C.D. Marsden; MITOCHONDRIAL COMPLEX I DEFICIENCY IN PARKINSON'S DISEASE. The Lancet 1989, 333, 1269, 10.1016/s0140-6736(89)92366-0.
- Laurence A. Bindoff; M. Birch-Machin; N.E.F. Cartlidge; W.D. Parker; D.M. Turnbull; MITOCHONDRIAL FUNCTION IN PARKINSON'S DISEASE. The Lancet 1989, 334, 49, 10.1016/s0140-6736(89)90291-2.
- William Davis Parker; Sally J. Boyson; Janice K. Parks; Abnormalities of the electron transport chain in idiopathic parkinson's disease. Annals of Neurology 1989, 26, 719-723, 10.1002/ana.410260606.
- Celine Perier; Kim Tieu; Christelle Guégan; Casper Caspersen; Vernice Jackson-Lewis; Valerio Carelli Md; Andrea Martinuzzi; Michio Hirano; Serge Przedborski; Miquel Vila; et al. Complex I deficiency primes Bax-dependent neuronal apoptosis through mitochondrial oxidative damage. Proceedings of the National Academy of Sciences 2005, 102, 19126-19131, 10.1073/pnas.0508215102.
- Brent Ryan; Selim Hoek; Edward A. Fon; Richard Wade-Martins; Mitochondrial dysfunction and mitophagy in Parkinson's: from familial to sporadic disease. Trends in Biochemical Sciences 2015, 40, 200-210, 10.1016/j.tibs.2015.02.003.
- Konstanze F. Winklhofer; Christian Haass; Mitochondrial dysfunction in Parkinson's disease. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2010, 1802, 29-44, 10.1016/j.bbadis.2009.08.013.
- Ingrid González-Casacuberta; Diana Luz Juárez-Flores; Constanza Morén; Gloria Garrabou; Bioenergetics and Autophagic Imbalance in Patients-Derived Cell Models of Parkinson Disease Supports Systemic Dysfunction in Neurodegeneration.. Frontiers in Neuroscience 2019, 13, 894, 10.3389/fnins.2019.00894.
- Chung-Han Hsieh; Atossa Shaltouki; Ashley E. Gonzalez; Alexandre Bettencourt Da Cruz; Lena F. Burbulla; Erica St. Lawrence; Birgitt Schüle; Dimitri Krainc; Theo D. Palmer; Xinnan Wang; et al. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson's Disease.. Cell Stem Cell 2016, 19, 709-724, 10.1016/j.stem.2016.08.002.
- Latha Devi; Vijayendran Raghavendran; Badanavalu M. Prabhu; Narayan G. Avadhani; Hindupur K. Anandatheerthavarada; Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain.. Journal of Biological Chemistry 2008, 283, 9089-100, 10.1074/jbc.M710012200.
- Roberto Di Maio; Paul J. Barrett; Eric K. Hoffman; Caitlyn W. Barrett; Alevtina Zharikov; Anupom Borah; Xiaoping Hu; Jennifer McCoy; Charleen Chu; Edward A Burton; et al.Teresa G. HastingsJohn T Greenamyre α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Science Translational Medicine 2016, 8, 342ra78-342ra78, 10.1126/scitranslmed.aaf3634.
- Frits Kamp; Nicole Exner; Anne Kathrin Lutz; Nora Wender; Jan Hegermann; Bettina Brunner; Brigitte Nuscher; Tim Bartels; Armin Giese; Klaus Beyer; et al.Stefan EimerKonstanze F. WinklhoferChristian Haass Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. The EMBO Journal 2010, 29, 3571-3589, 10.1038/emboj.2010.223.
- Amy K. Reeve; Marthe Ludtmann; P R Angelova; E M Simcox; Mathew Horrocks; David Klenerman; S Gandhi; Doug M. Turnbull; Andrey Y. Abramov; Aggregated α-synuclein and complex I deficiency: exploration of their relationship in differentiated neurons.. Cell Death & Disease 2015, 6, e1820-e1820, 10.1038/cddis.2015.166.
- Hideki Mochizuki; Keigo Goto; Hideo Mori; Yoshikuni Mizuno; Histochemical detection of apoptosis in Parkinson's disease. Journal of the Neurological Sciences 1996, 137, 120-123, 10.1016/0022-510x(95)00336-z.
- Julie K. Andersen; Does neuronal loss in Parkinson's disease involve programmed cell death?. BioEssays 2001, 23, 640-646, 10.1002/bies.1089.
- Andreas Hartmann; Jean-Denis Troadec; Stephane Hunot; Kristy Kikly; Baptiste A. Faucheux; Annick Mouatt-Prigent; Merle Ruberg; Yves Agid; Etienne C. Hirsch; Caspase-8 Is an Effector in Apoptotic Death of Dopaminergic Neurons in Parkinson's Disease, But Pathway Inhibition Results in Neuronal Necrosis. The Journal of Neuroscience 2001, 21, 2247-2255, 10.1523/JNEUROSCI.21-07-02247.2001.
- Helen Murray; Molly E. V. Swanson; Birger Dieriks; Clinton Turner; Richard Faull; Maurice A. Curtis; Neurochemical Characterization of PSA-NCAM+ Cells in the Human Brain and Phenotypic Quantification in Alzheimer's Disease Entorhinal Cortex.. Neuroscience 2018, 372, 289-303, 10.1016/j.neuroscience.2017.12.019.
- Neelam Shahani; Srinivasa Subramaniam; Tobias Wolf; Christian Tackenberg; Roland Brandt; Tau Aggregation and Progressive Neuronal Degeneration in the Absence of Changes in Spine Density and Morphology after Targeted Expression of Alzheimer's Disease-Relevant Tau Constructs in Organotypic Hippocampal Slices. The Journal of Neuroscience 2006, 26, 6103-6114, 10.1523/JNEUROSCI.4245-05.2006.
- Peter M. Clifford; Shabnam Zarrabi; Gilbert Siu; Kristin J. Kinsler; Mary C. Kosciuk; Venkateswar Venkataraman; Michael R. D'andrea; Steven Dinsmore; Robert G. Nagele; Aβ peptides can enter the brain through a defective blood–brain barrier and bind selectively to neurons. Brain Research 2007, 1142, 223-236, 10.1016/j.brainres.2007.01.070.
- P. Hemachandra Reddy; Amyloid precursor protein-mediated free radicals and oxidative damage: Implications for the development and progression of Alzheimer's disease. Journal of Neurochemistry 2006, 96, 1-13, 10.1111/j.1471-4159.2005.03530.x.
- Alexandre Fifre; Isabelle Sponne; Violette Koziel; Badreddine Kriem; Frances T. Yen Potin; Bernard E. Bihain; Jean-Luc Olivier; Thierry Oster; Thierry Pillot; Microtubule-associated Protein MAP1A, MAP1B, and MAP2 Proteolysis during Soluble Amyloid β-Peptide-induced Neuronal Apoptosis. Journal of Biological Chemistry 2005, 281, 229-240, 10.1074/jbc.m507378200.
- Kuo-Hsuan Chang; Chiung-Mei Chen; The Role of Oxidative Stress in Parkinson’s Disease. Antioxidants 2020, 9, 597.
- Peter Jenner; Oxidative stress in Parkinson's disease. Annals of Neurology 2003, 53, S26-S38, 10.1002/ana.10483.
- Michela Di Nottia; Marcella Masciullo; Daniela Verrigni; Sara Petrillo; Anna Modoni; Valentina Rizzo; D. Di Giuda; Teresa Rizza; Marcello Niceta; Alessandra Torraco; et al.Marzia BianchiMassimo SantoroAnna Rita BentivoglioEnrico Silvio BertiniFiorella PiemonteRosalba CarrozzoG. Silvestri DJ-1 modulates mitochondrial response to oxidative stress: clues from a novel diagnosis of PARK7. Clinical Genetics 2016, 92, 18-25, 10.1111/cge.12841.
- Benjamin Dehay; Jordi Bove; Natalia Rodriguez-Muela; Celine Perier; Ariadna Recasens; Patricia Boya; Miquel Vila; Pathogenic Lysosomal Depletion in Parkinson's Disease. The Journal of Neuroscience 2010, 30, 12535-12544, 10.1523/JNEUROSCI.1920-10.2010.
- Akihiko Nunomura; Kazuhiro Honda; Atsushi Takeda; Keisuke Hirai; Xiongwei Zhu; Mark A. Smith; George Perry; Oxidative Damage to RNA in Neurodegenerative Diseases. Journal of Biomedicine and Biotechnology 2006, 2006, 1-6, 10.1155/JBB/2006/82323.
- Rukhsana Sultana; D. Allan Butterfield; Role of Oxidative Stress in the Progression of Alzheimer's Disease. Journal of Alzheimer's Disease 2010, 19, 341-353, 10.3233/jad-2010-1222.
- D. Allan Butterfield; Aaron M. Swomley; Rukhsana Sultana; Amyloid β-Peptide (1–42)-Induced Oxidative Stress in Alzheimer Disease: Importance in Disease Pathogenesis and Progression. Antioxidants & Redox Signaling 2013, 19, 823-835, 10.1089/ars.2012.5027.
- Fernanda G. De Felice; Pauline T. Velasco; Mary P. Lambert; Kirsten Viola; Sara J. Fernandez; Sergio T. Ferreira; William L. Klein; Aβ Oligomers Induce Neuronal Oxidative Stress through anN-Methyl-D-aspartate Receptor-dependent Mechanism That Is Blocked by the Alzheimer Drug Memantine. Journal of Biological Chemistry 2007, 282, 11590-11601, 10.1074/jbc.m607483200.
- Haruhiko Akiyama; Steven Barger; Scott Barnum; Bonnie Bradt; Joachim Bauer; Greg M. Cole; Neil R. Cooper; Piet Eikelenboom; Mark Emmerling; Berndt L. Fiebich; et al.Caleb E. FinchSally FrautschyW.S.T. GriffinHarald HampelMichael HullGary LandrethLih–Fen LueRobert MrakIan R. MacKenziePatrick L. McGeerM.Kerry O’BanionJoel PachterGuilio PasinettiCarlos Plata–SalamanJ RogersRussell RydelYong ShenWolfgang StreitRonald StrohmeyerIkuo TooyomaFreek L. Van MuiswinkelRobert VeerhuisUglas WalkerScott WebsterBeatrice WegrzyniakGary WenkTony Wyss–Coray Inflammation and Alzheimer’s disease. Neurobiology of Aging 2000, 21, 383-421, 10.1016/s0197-4580(00)00124-x.
- Haruhiko Akiyama; Tetsuaki Arai; Hiromi Kondo; Eiko Tanno; Chie Haga; Kenji Ikeda; Cell Mediators of Inflammation in the Alzheimer Disease Brain. Alzheimer Disease & Associated Disorders 2000, 14, S47-S53, 10.1097/00002093-200000001-00008.
- Michael V. Sofroniew; Harry V. Vinters; Astrocytes: biology and pathology. Acta Neuropathologica 2009, 119, 7-35, 10.1007/s00401-009-0619-8.
- Rajesh N. Kalaria; Microglia and Alzheimer’s disease. Current Opinion in Hematology 1999, 6, 15, 10.1097/00062752-199901000-00004.
- J Rogers; Lih-Fen Lue; Microglial chemotaxis, activation, and phagocytosis of amyloid β-peptide as linked phenomena in Alzheimer's disease. Neurochemistry International 2001, 39, 333-340, 10.1016/s0197-0186(01)00040-7.
- Dawling A. Dionisio-Santos; John A. Olschowka; M. Kerry O’Banion; Exploiting microglial and peripheral immune cell crosstalk to treat Alzheimer’s disease. Journal of Neuroinflammation 2019, 16, 74, 10.1186/s12974-019-1453-0.
- Karla K. Kopec; Richard T. Carroll; Alzheimer's ?-Amyloid Peptide 1-42 Induces a Phagocytic Response in Murine Microglia. Journal of Neurochemistry 2002, 71, 2123-2131, 10.1046/j.1471-4159.1998.71052123.x.
- Tristan Bolmont; Florent Haiss; Daniel Eicke; Rebecca Radde; Chester A. Mathis; William Klunk; Shinichi Kohsaka; Mathias Jucker; Michael E. Calhoun; Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance.. The Journal of Neuroscience 2008, 28, 4283-92, 10.1523/JNEUROSCI.4814-07.2008.
- Melanie Meyer‐Luehmann; Tara L. Spires-Jones; Claudia Prada; Monica Garcia‐Alloza; Alix De Calignon; Anete Rozkalne; Jessica Koenigsknecht-Talboo; David M. Holtzman; Brian J. Bacskai; Bradley T. Hyman; et al. Rapid appearance and local toxicity of amyloid-β plaques in a mouse model of Alzheimer’s disease. Nature 2008, 451, 720-4, 10.1038/nature06616.
- Fiorella Malchiodi-Albedi; Maria Rosaria Domenici; Silvia Paradisi; Antonietta Bernardo; Maria Antonietta Ajmone-Cat; Luisa Minghetti; Astrocytes contribute to neuronal impairment in ?A toxicity increasing apoptosis in rat hippocampal neurons. Glia 2001, 34, 68-72, 10.1002/glia.1041.
- J.L. Perez; I. Carrero; P. Gonzalo; J. Arévalo-Serrano; José Miguel Sanz Anquela; Francisco Javier Ortega; Manuel J. Rodriguez; Alicia Gonzalo-Ruiz; Soluble oligomeric forms of beta-amyloid (Aβ) peptide stimulate Aβ production via astrogliosis in the rat brain. Experimental Neurology 2010, 223, 410-421, 10.1016/j.expneurol.2009.10.013.
- H. Yamaguchi; Shiro Sugihara; Akira Ogawa; Takaomi C. Saido; Yasuo Ihara; Diffuse plaques associated with astroglial amyloid β protein, possibly showing a disappearing stage of senile plaques. Acta Neuropathologica 1998, 95, 217-222, 10.1007/s004010050790.
- M. R. Emmerling; M.Desiree Watson; Charlotte A. Raby; Katharyn Spiegel; The role of complement in Alzheimer’s disease pathology. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2000, 1502, 158-171, 10.1016/s0925-4439(00)00042-9.
- Michael C Carroll; THE ROLE OF COMPLEMENT AND COMPLEMENT RECEPTORS IN INDUCTION AND REGULATION OF IMMUNITY. Annual Review of Immunology 1998, 16, 545-568, 10.1146/annurev.immunol.16.1.545.
- Andrea J. Tenner; Complement in Alzheimer's disease: opportunities for modulating protective and pathogenic events.. Neurobiology of Aging 2002, 22, 849-861, 10.1016/s0197-4580(01)00301-3.
- W.J. Friedman; Cytokines Regulate Expression of the Type 1 Interleukin-1 Receptor in Rat Hippocampal Neurons and Glia. Experimental Neurology 2001, 168, 23-31, 10.1006/exnr.2000.7595.
- Li Yuekui; Steven Barger; Ling Liu; Robert E. Mrak; W. Sue T. Griffin; Yuekui Li; S100β Induction of the Proinflammatory Cytokine Interleukin-6 in Neurons. Journal of Neurochemistry 2001, 74, 143-150, 10.1046/j.1471-4159.2000.0740143.x.
- A.E. Renauld; Robert N. Spengler; Tumor necrosis factor expressed by primary hippocampal neurons and SH-SY5Y cells is regulated by ?2-adrenergic receptor activation. Journal of Neuroscience Research 2001, 67, 264-274, 10.1002/jnr.10101.
- Koji Yasojima; Claudia Schwab; Edith G. McGeer; P L McGeer; Human neurons generate C-reactive protein and amyloid P: upregulation in Alzheimer's disease.. Brain Research 2000, 887, 80-89, 10.1016/s0006-8993(00)02970-x.
- Yong Shen; Rena Li; Edith G McGeer; Patrick L McGeer; Neuronal expression of mRNAs for complement proteins of the classical pathway in Alzheimer brain.. Brain Research 1997, 769, 391-395, 10.1016/s0006-8993(97)00850-0.
- Malú G. Tansey; Matthew S. Goldberg; Neuroinflammation in Parkinson's disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiology of Disease 2009, 37, 510-8, 10.1016/j.nbd.2009.11.004.
- Yasuomi Ouchi; Etsuji Yoshikawa; Yoshimoto Sekine; Masami Futatsubashi; Toshihiko Kanno; Tomomi Ogusu; Tatsuo Torizuka; Microglial activation and dopamine terminal loss in early Parkinson's disease. Annals of Neurology 2005, 57, 168-175, 10.1002/ana.20338.
- Alexander Gerhard; Nicola Pavese; Gary Hotton; Federico E Turkheimer; Meltem Es; Alexander Hammers; Karla Eggert; Wolfgang Oertel; Richard B. Banati; David J. Brooks; et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson's disease. Neurobiology of Disease 2006, 21, 404-412, 10.1016/j.nbd.2005.08.002.
- Carolyn Orr; Dominic B. Rowe; Yoshikuni Mizuno; Hideo Mori; Glenda M Halliday; A possible role for humoral immunity in the pathogenesis of Parkinson's disease. Brain 2005, 128, 2665-2674, 10.1093/brain/awh625.
- Emilie Croisier; Linda B Moran; David T. Dexter; Ronald K. B. Pearce; Manuel B Graeber; Microglial inflammation in the parkinsonian substantia nigra: relationship to alpha-synuclein deposition. Journal of Neuroinflammation 2005, 2, 14-14, 10.1186/1742-2094-2-14.
- Kazuhiro Imamura; Nozomi Hishikawa; Makoto Sawada; Toshiharu Nagatsu; Mari Yoshida; Yoshio Hashizume; Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson's disease brains. Acta Neuropathologica 2003, 106, 518-526, 10.1007/s00401-003-0766-2.
- P. L. McGeer; S. Itagaki; B. E. Boyes; E. G. McGeer; Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology 1988, 38, 1285-1285, 10.1212/wnl.38.8.1285.
- Richard B. Banati; Susan E. Daniel; Stavia B. Blunt; Glial pathology but absence of apoptotic nigral neurons in long-standing Parkinson's disease. Movement Disorders 1998, 13, 221-227, 10.1002/mds.870130205.
- Lilia Marinova-Mutafchieva; Mona Sadeghian; Lauren Broom; John B. Davis; Andrew D. Medhurst; David T. Dexter; Relationship between microglial activation and dopaminergic neuronal loss in the substantia nigra: a time course study in a 6-hydroxydopamine model of Parkinson’s disease. Journal of Neurochemistry 2009, 110, 966-975, 10.1111/j.1471-4159.2009.06189.x.
- Marianne Vázquez-Claverie; Pablo Garrido-Gil; Waldy San Sebastian; Amaya Izal-Azcárate; Silvia Belzunegui; Irene Marcilla; Berta López; Rosario Luquin; Acute and Chronic 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Administrations Elicit Similar Microglial Activation in the Substantia Nigra of Monkeys. Journal of Neuropathology & Experimental Neurology 2009, 68, 977-984, 10.1097/nen.0b013e3181b35e41.
- Vanesa Sanchez-Guajardo; Fabia Febbraro; Deniz Kirik; Marina Romero-Ramos; Microglia Acquire Distinct Activation Profiles Depending on the Degree of α-Synuclein Neuropathology in a rAAV Based Model of Parkinson's Disease. PLOS ONE 2010, 5, e8784, 10.1371/journal.pone.0008784.
- H Akiyama; P L McGeer; Microglial response to 6-hydroxydopamine-induced substantia nigra lesions. Brain Research 1989, 489, 247-253, 10.1016/0006-8993(89)90857-3.
- Vincent Henry; Vincent Paillé; Faustine Lelan; Philippe Brachet; Philippe Damier; Kinetics of Microglial Activation and Degeneration of Dopamine-Containing Neurons in a Rat Model of Parkinson Disease Induced by 6-Hydroxydopamine. Journal of Neuropathology & Experimental Neurology 2009, 68, 1092-1102, 10.1097/nen.0b013e3181b767b4.
- C. Knott; G. Stern; G.P. Wilkin; Inflammatory Regulators in Parkinson's Disease: iNOS, Lipocortin-1, and Cyclooxygenases-1 and -2. Molecular and Cellular Neuroscience 2000, 16, 724-739, 10.1006/mcne.2000.0914.
- Stephane Hunot; F. Boissière; B. Faucheux; B. Brugg; A. Mouatt-Prigent; Y. Agid; E.C. Hirsch; Nitric oxide synthase and neuronal vulnerability in parkinson's disease. Neuroscience 1996, 72, 355-363, 10.1016/0306-4522(95)00578-1.
- Paramita Chakrabarty; Carolina Ceballos-Diaz; Wen-Lang Lin; Amanda Beccard; Karen Jansen-West; Nikolaus McFarland; Christopher Janus; Dennis W. Dickson; Pritam Das; Todd E. Golde; et al. Interferon-γ induces progressive nigrostriatal degeneration and basal ganglia calcification.. Nature Neuroscience 2011, 14, 694-6, 10.1038/nn.2829.
- Emily N. Mangano; Darcy Litteljohn; Remmick So; Eric Nelson; Sarah Peters; Cheri Bethune; Jessica Bobyn; Shawn Hayley; Interferon-γ plays a role in paraquat-induced neurodegeneration involving oxidative and proinflammatory pathways. Neurobiology of Aging 2012, 33, 1411-1426, 10.1016/j.neurobiolaging.2011.02.016.
- Carlos Barcia; Francisco Ros; Valentina Annese; A Gómez; F Ros-Bernal; D Aguado-Yera; M E Martínez-Pagán; V De Pablos; E Fernandez-Villalba; M. Trinidad Herrero; et al. IFN-γ signaling, with the synergistic contribution of TNF-α, mediates cell specific microglial and astroglial activation in experimental models of Parkinson's disease. Cell Death & Disease 2011, 2, e142-e142, 10.1038/cddis.2011.17.
- D. Litteljohn; E. Mangano; N. Shukla; S. Hayley; Interferon-γ deficiency modifies the motor and co-morbid behavioral pathology and neurochemical changes provoked by the pesticide paraquat. Neuroscience 2009, 164, 1894-1906, 10.1016/j.neuroscience.2009.09.025.
- Ashley D. Reynolds; Rebecca Banerjee; Jianou Liu; Howard E. Gendelman; R. Lee Mosley; Neuroprotective activities of CD4+CD25+ regulatory T cells in an animal model of Parkinson’s disease. Journal of Leukocyte Biology 2007, 82, 1083-1094, 10.1189/jlb.0507296.
- Lisa M. Kosloski; Elizabeth A. Kosmacek; Katherine E. Olson; R. Lee Mosley; Howard E. Gendelman; GM-CSF induces neuroprotective and anti-inflammatory responses in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine intoxicated mice.. Journal of Neuroimmunology 2013, 265, 1-10, 10.1016/j.jneuroim.2013.10.009.

