 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Nompumelelo Anna-Cletta Gumede | -- | 3082 | 2024-02-16 08:45:18 | | | |
| 2 | Sirius Huang | + 12 word(s) | 3094 | 2024-02-17 02:17:14 | | |
Video Upload Options
Myocardial infarction (MI) is a significant contributor to CVD-related mortality. Type 2 diabetes mellitus (T2DM) is a risk factor for MI. Stress activates the hypothalamus–pituitary–adrenal axis (HPA axis), sympathetic nervous system (SNS), and endogenous OPS. These pro-opiomelanocortin (POMC) derivatives increase the blood glucose and cardiovascular response by inhibiting the PI3K/AkT insulin signaling pathway and increasing cardiac contraction. Sustained activation of the POMC derivatives may lead to developing myocardial infarction. Suffering from T2DM and stress increases the risk of developing CVD. T2DM is preceded by prediabetes, which is a state of blood glucose level being above normal but below the level of T2DM diagnosis. Research has shown that T2DM-related complications begin during prediabetes; therefore, there is a possibility of the dysregulation of the POMC derivatives during prediabetes and pathways that could lead to myocardial infarction.
1. Introduction
2. POMC Derivative Signaling
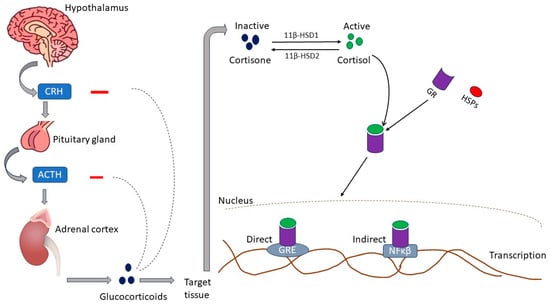
2.1. Glucocorticoids
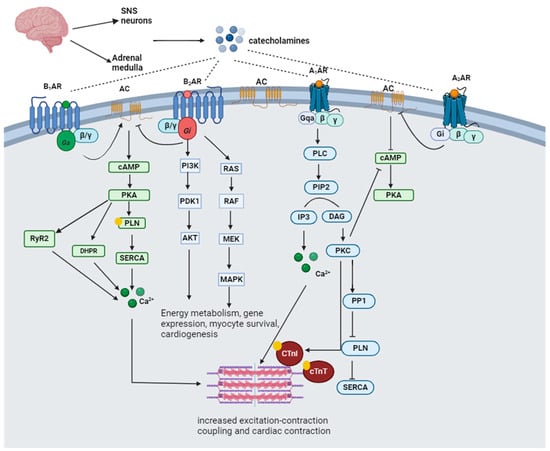
2.2. Catecholamine
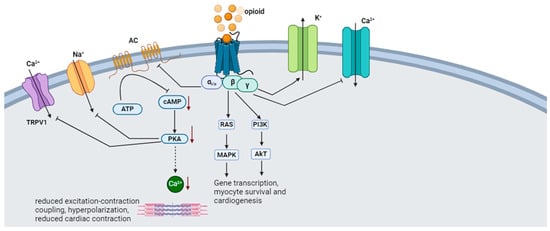
2.3. Opioids
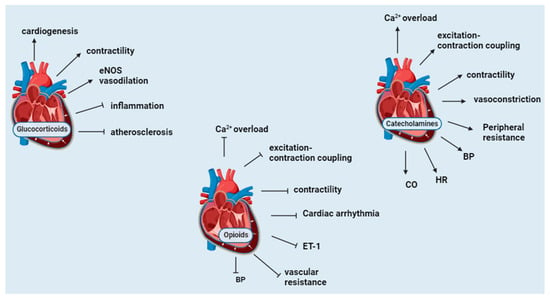
3. Role of POMC Derivatives in Cardiovascular Function
3.1. Glucocorticoids
3.2. Catecholamines
3.3. Opioids
References
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Chaitman, B.R.; Bax, J.J.; Morrow, D.A.; White, H.D. Fourth Universal Definition of Myocardial Infarction (2018). Circulation 2018, 138, e618–e651.
- Lodrini, A.M.; Goumans, M.J. Cardiomyocytes Cellular Phenotypes after Myocardial Infarction. Front. Cardiovasc. Med. 2021, 8, 750510.
- Yun, J.-S.; Ko, S.-H. Current trends in epidemiology of cardiovascular disease and cardiovascular risk management in type 2 diabetes. Metabolism 2021, 123, 154838.
- Vos, T.; Lim, S.S.; Abbafati, C.; Abbas, K.M.; Abbasi, M.; Abbasifard, M.; Abbasi-Kangevari, M.; Abbastabar, H.; Abd-Allah, F.; Abdelalim, A.; et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222.
- Safiri, S.; Karamzad, N.; Singh, K.; Carson-Chahhoud, K.; Adams, C.; Nejadghaderi, S.A.; Almasi-Hashiani, A.; Sullman, M.J.M.; Mansournia, M.A.; Bragazzi, N.L.; et al. Burden of ischemic heart disease and its attributable risk factors in 204 countries and territories, 1990–2019. Eur. J. Prev. Cardiol. 2022, 29, 420–431.
- Kelly, C.; Lan, N.S.R.; Phan, J.; Hng, C.; Matthews, A.; Rankin, J.M.; Schultz, C.J.; Hillis, G.S.; Reid, C.M.; Dwivedi, G.; et al. Characteristics and Outcomes of Young Patients with ST-Elevation Myocardial Infarction without Standard Modifiable Risk Factors. Am. J. Cardiol. 2023, 202, 81–89.
- Laakso, M.; Kuusisto, J. Insulin resistance and hyperglycaemia in cardiovascular disease development. Nat. Rev. Endocrinol. 2014, 10, 293–302.
- Steptoe, A.; Kivimäki, M. Stress and cardiovascular disease. Nat. Rev. Cardiol. 2012, 9, 360–370.
- Satyjeet, F.; Naz, S.; Kumar, V.; Aung, N.H.; Bansari, K.; Irfan, S.; Rizwan, A. Psychological Stress as a Risk Factor for Cardiovascular Disease: A Case-Control Study. Cureus 2020, 12, e10757.
- Levine, G.N. Psychological Stress and Heart Disease: Fact or Folklore? Am. J. Med. 2022, 135, 688–696.
- Gianoulakis, C. Alcohol-seeking behavior: The roles of the hypothalamic-pituitary-adrenal axis and the endogenous opioid system. Alcohol. Health Res. World 1998, 22, 202–210.
- Pilozzi, A.; Carro, C.; Huang, X. Roles of β-Endorphin in Stress, Behavior, Neuroinflammation, and Brain Energy Metabolism. Int. J. Mol. Sci. 2021, 22, 338.
- Carr, D.B.; Verrier, R.L. Opioids in pain and cardiovascular responses: Overview of common features. J. Cardiovasc. Electrophysiol. 1991, 2, s34–s42.
- Kelly, S.J.; Ismail, M. Stress and Type 2 Diabetes: A Review of How Stress Contributes to the Development of Type 2 Diabetes. Annu. Rev. Public. Health 2015, 36, 441–462.
- Rooney, M.R.; Rawlings, A.M.; Pankow, J.S.; Echouffo Tcheugui, J.B.; Coresh, J.; Sharrett, A.R.; Selvin, E. Risk of Progression to Diabetes Among Older Adults with Prediabetes. JAMA Intern. Med. 2021, 181, 511–519.
- Echouffo-Tcheugui, J.B.; Selvin, E. Prediabetes and What It Means: The Epidemiological Evidence. Annu. Rev. Public Health 2021, 42, 59–77.
- Mishra, A.; Podder, V.; Modgil, S.; Khosla, R.; Anand, A.; Nagarathna, R.; Malhotra, R.; Nagendra, H.R. Higher Perceived Stress and Poor Glycemic Changes in Prediabetics and Diabetics among Indian Population. J. Med. Life 2020, 13, 132–137.
- Sharma, K.; Akre, S.; Chakole, S.; Wanjari, M.B. Stress-Induced Diabetes: A Review. Cureus 2022, 14, e29142.
- Jyotheeswara Pillai, K.; Luo, H.; Raj, K.; Xu, S.; Thota, G. OR02-4 Prediabetes is a Risk Factor for Myocardial Infarction-A National Inpatient Sample Study. J. Endocr. Soc. 2022, 6, A248.
- Kadmiel, M.; Cidlowski, J.A. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol. Sci. 2013, 34, 518–530.
- Chrousos, G.P.; Kino, T. Glucocorticoid action networks and complex psychiatric and/or somatic disorders. Stress 2007, 10, 213–219.
- Smith, S.M.; Vale, W.W. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues Clin. Neurosci. 2006, 8, 383–395.
- Maniam, J.; Antoniadis, C.; Morris, M.J. Early-life stress, HPA axis adaptation, and mechanisms contributing to later health outcomes. Front. Endocrinol. 2014, 5, 73.
- Macfarlane, E.; Seibel, M.J.; Zhou, H. Arthritis and the role of endogenous glucocorticoids. Bone Res. 2020, 8, 33.
- Sevilla, L.M.; Jiménez-Panizo, A.; Alegre-Martí, A.; Estébanez-Perpiñá, E.; Caelles, C.; Pérez, P. Glucocorticoid Resistance: Interference between the Glucocorticoid Receptor and the MAPK Signalling Pathways. Int. J. Mol. Sci. 2021, 22, 10049.
- Qi, D.; Rodrigues, B. Glucocorticoids produce whole body insulin resistance with changes in cardiac metabolism. Am. J. Physiol. -Endocrinol. Metab. 2007, 292, E654–E667.
- Cato, A.C.; Nestl, A.; Mink, S. Rapid actions of steroid receptors in cellular signaling pathways. Sci. STKE 2002, 2002, re9.
- Nicolaides, N.C.; Kyratzi, E.; Lamprokostopoulou, A.; Chrousos, G.P.; Charmandari, E. Stress, the Stress System and the Role of Glucocorticoids. Neuroimmunomodulation 2015, 22, 6–19.
- Mihailidou, A.S.; Le, T.Y.L.; Mardini, M.; Funder, J.W. Glucocorticoids Activate Cardiac Mineralocorticoid Receptors During Experimental Myocardial Infarction. Hypertension 2009, 54, 1306–1312.
- Nicolaides, N.C.; Galata, Z.; Kino, T.; Chrousos, G.P.; Charmandari, E. The human glucocorticoid receptor: Molecular basis of biologic function. Steroids 2010, 75, 1–12.
- Girod, J.P.; Brotman, D.J. Does altered glucocorticoid homeostasis increase cardiovascular risk? Cardiovasc. Res. 2004, 64, 217–226.
- Walker, B.R. Glucocorticoids and Cardiovascular Disease. Eur. J. Endocrinol. 2007, 157, 545–559.
- Eto, K.; Mazilu-Brown, J.K.; Henderson-MacLennan, N.; Dipple, K.M.; McCabe, E.R.B. Development of catecholamine and cortisol stress responses in zebrafish. Mol. Genet. Metab. Rep. 2014, 1, 373–377.
- Kyrou, I.; Tsigos, C. Stress hormones: Physiological stress and regulation of metabolism. Curr. Opin. Pharmacol. 2009, 9, 787–793.
- Gülçin, I. Antioxidant activity of L-adrenaline: A structure-activity insight. Chem. Biol. Interact. 2009, 179, 71–80.
- Errasti, A.E.; Werneck de Avellar, M.C.; Daray, F.M.; Tramontano, J.; Luciani, L.I.; Lina Bard, M.A.J.; Maróstica, E.; Rothlin, R.P. Human umbilical vein vasoconstriction induced by epinephrine acting on α1B-adrenoceptor subtype. Am. J. Obstet. Gynecol. 2003, 189, 1472–1480.
- Foote, S.L.; Bloom, F.E.; Aston-Jones, G. Nucleus locus ceruleus: New evidence of anatomical and physiological specificity. Physiol. Rev. 1983, 63, 844–914.
- Waterhouse, B.D.; Devilbiss, D.; Fleischer, D.; Sessler, F.M.; Simpson, K.L. New perspectives on the functional organization and postsynaptic influences of the locus ceruleus efferent projection system. Adv. Pharmacol. 1998, 42, 749–754.
- Kemeny, M.E. The Psychobiology of Stress. Curr. Dir. Psychol. Sci. 2003, 12, 124–129.
- Motiejunaite, J.; Amar, L.; Vidal-Petiot, E. Adrenergic receptors and cardiovascular effects of catecholamines. Ann. D’endocrinologie 2021, 82, 193–197.
- Pavoine, C.; Defer, N. The cardiac β2-adrenergic signalling a new role for the cPLA2. Cell. Signal. 2005, 17, 141–152.
- Rozec, B.; Erfanian, M.; Laurent, K.; Trochu, J.-N.; Gauthier, C. Nebivolol, a vasodilating selective β1-blocker, is a β3-adrenoceptor agonist in the nonfailing transplanted human heart. J. Am. Coll. Cardiol. 2009, 53, 1532–1538.
- Jain, A.; Mishra, A.; Shakkarpude, J.; Lakhani, P. Beta endorphins: The natural opioids. IJCS 2019, 7, 323–332.
- Al-Hasani, R.; Bruchas, M.R. Molecular Mechanisms of Opioid Receptor-dependent Signaling and Behavior. Anesthesiology 2011, 115, 1363–1381.
- Cissom, C.; J Paris, J.; Shariat-Madar, Z. Dynorphins in development and disease: Implications for cardiovascular disease. Curr. Mol. Med. 2020, 20, 259–274.
- Mongi-Bragato, B.; Avalos, M.P.; Guzmán, A.S.; Bollati, F.A.; Cancela, L.M. Enkephalin as a pivotal player in neuroadaptations related to psychostimulant addiction. Front. Psychiatry 2018, 9, 222.
- Li, Z.; Liu, J.; Dong, F.; Chang, N.; Huang, R.; Xia, M.; Patterson, T.A.; Hong, H. Three-Dimensional Structural Insights Have Revealed the Distinct Binding Interactions of Agonists, Partial Agonists, and Antagonists with the µ Opioid Receptor. Int. J. Mol. Sci. 2023, 24, 7042.
- Bruchas, M.R.; Chavkin, C. Kinase cascades and ligand-directed signaling at the kappa opioid receptor. Psychopharmacology 2010, 210, 137–147.
- Ji, B.; Liu, H.; Zhang, R.; Jiang, Y.; Wang, C.; Li, S.; Chen, J.; Bai, B. Novel signaling of dynorphin at κ-opioid receptor/bradykinin B2 receptor heterodimers. Cell. Signal. 2017, 31, 66–78.
- Drolet, G.; Dumont, É.C.; Gosselin, I.; Kinkead, R.; Laforest, S.; Trottier, J.-F. Role of endogenous opioid system in the regulation of the stress response. Progress. Neuro-Psychopharmacol. Biol. Psychiatry 2001, 25, 729–741.
- Charmandari, E.; Kino, T.; Souvatzoglou, E.; Chrousos, G.P. Pediatric Stress: Hormonal Mediators and Human Development. Horm. Res. Paediatr. 2003, 59, 161–179.
- Xiao, R.P.; Pepe, S.; Spurgeon, H.A.; Capogrossi, M.C.; Lakatta, E.G. Opioid peptide receptor stimulation reverses beta-adrenergic effects in rat heart cells. Am. J. Physiol.-Heart Circ. Physiol. 1997, 272, H797–H805.
- Valentino, R.J.; Van Bockstaele, E. Endogenous opioids: The downside of opposing stress. Neurobiol. Stress 2015, 1, 23–32.
- Bilkei-Gorzo, A.; Racz, I.; Michel, K.; Mauer, D.; Zimmer, A.; Klingmüller, D.; Zimmer, A. Control of hormonal stress reactivity by the endogenous opioid system. Psychoneuroendocrinology 2008, 33, 425–436.
- Lefer, A.M. Influence of corticosteroids on mechanical performance of isolated rat papillary muscles. Am. J. Physiol.-Leg. Content 1968, 214, 518–524.
- Penefsky, Z.J.; Kahn, M. Inotropic effects of dexamethasone in mammalian heart muscle. Eur. J. Pharmacol. 1971, 15, 259–266.
- Whirledge, S.; DeFranco, D.B. Glucocorticoid Signaling in Health and Disease: Insights From Tissue-Specific GR Knockout Mice. Endocrinology 2017, 159, 46–64.
- Vandevyver, S.; Dejager, L.; Tuckermann, J.; Libert, C. New Insights into the Anti-inflammatory Mechanisms of Glucocorticoids: An Emerging Role for Glucocorticoid-Receptor-Mediated Transactivation. Endocrinology 2013, 154, 993–1007.
- Goodwin, J.E.; Zhang, X.; Rotllan, N.; Feng, Y.; Zhou, H.; Fernández-Hernando, C.; Yu, J.; Sessa, W.C. Endothelial glucocorticoid receptor suppresses atherogenesis—Brief report. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 779–782.
- Hafezi-Moghadam, A.; Simoncini, T.; Yang, Z.; Limbourg, F.P.; Plumier, J.-C.; Rebsamen, M.C.; Hsieh, C.-M.; Chui, D.-S.; Thomas, K.L.; Prorock, A.J.; et al. Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat. Med. 2002, 8, 473–479.
- Ziegler, M.G.; Elayan, H.; Milic, M.; Sun, P.; Gharaibeh, M. Epinephrine and the Metabolic Syndrome. Curr. Hypertens. Rep. 2012, 14, 1–7.
- Barth, E.; Albuszies, G.; Baumgart, K.; Matejovic, M.; Wachter, U.; Vogt, J.; Radermacher, P.; Calzia, E. Glucose metabolism and catecholamines. Crit. Care Med. 2007, 35, S508–S518.
- Vieira, E.; Liu, Y.-J.; Gylfe, E. Involvement of α1 and β-adrenoceptors in adrenaline stimulation of the glucagon-secreting mouse α-cell. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2004, 369, 179–183.
- Jones, B.J.; Tan, T.; Bloom, S.R. Minireview: Glucagon in Stress and Energy Homeostasis. Endocrinology 2012, 153, 1049–1054.
- Schweiger, M.; Schreiber, R.; Haemmerle, G.; Lass, A.; Fledelius, C.; Jacobsen, P.; Tornqvist, H.; Zechner, R.; Zimmermann, R. Adipose Triglyceride Lipase and Hormone-sensitive Lipase Are the Major Enzymes in Adipose Tissue Triacylglycerol Catabolism. J. Biol. Chem. 2006, 281, 40236–40241.
- Mullins, G.R.; Wang, L.; Raje, V.; Sherwood, S.G.; Grande, R.C.; Boroda, S.; Eaton, J.M.; Blancquaert, S.; Roger, P.P.; Leitinger, N.; et al. Catecholamine-induced lipolysis causes mTOR complex dissociation and inhibits glucose uptake in adipocytes. Proc. Natl. Acad. Sci. USA 2014, 111, 17450–17455.
- Jackson, L.; Williams, F.L.R.; Burchell, A.; Coughtrie, M.W.H.; Hume, R. Plasma Catecholamines and the Counterregulatory Responses to Hypoglycemia in Infants: A Critical Role for Epinephrine and Cortisol. J. Clin. Endocrinol. Metab. 2004, 89, 6251–6256.
- Gordan, R.; Gwathmey, J.K.; Xie, L.-H. Autonomic and endocrine control of cardiovascular function. World J. Cardiol. 2015, 7, 204.
- Grassi, G. Assessment of Sympathetic Cardiovascular Drive in Human Hypertension. Hypertension 2009, 54, 690–697.
- Medić, B. The role of autonomic control in cardiovascular system: Summary of basic principles. Med. Podml. 2016, 67, 14–18.
- Ventura, C.; Spurgeon, H.; Lakatta, E.G.; Guarnieri, C.; Capogrossi, M.C. Kappa and delta opioid receptor stimulation affects cardiac myocyte function and Ca2+ release from an intracellular pool in myocytes and neurons. Circ. Res. 1992, 70, 66–81.
- Forman, L.; Bagasra, O. Demonstration by in situ hybridization of the proopiomelanocortin gene in the rat heart. Brain Res. Bull. 1992, 28, 441–445.
- Weil, J.; Zolk, O.; Griepentrog, J.; Wenzel, U.; Zimmermann, W.H.; Eschenhagen, T. Alterations of the preproenkephalin system in cardiac hypertrophy and its role in atrioventricular conduction. Cardiovasc. Res. 2006, 69, 412–422.
- Millington, W.R.; Evans, V.R.; Battie, C.N.; Bagasra, O.; Forman, L.J. Proopiomelanocortin-Derived Peptides and mRNA Are Expressed in Rat Heart. Ann. N. Y. Acad. Sci. 1993, 680, 575–578.
- Forman, L.J.; Hock, C.E.; Harwell, M.; Estilow-Isabell, S. The Results of Exposure to Immobilization, Hemorrhagic Shock, and Cardiac Hypertrophy on β-Endorphin in Rat Cardiac Tissue. Proc. Soc. Exp. Biol. Med. 1994, 206, 124–129.
- He, S.F.; Jin, S.Y.; Yang, W.; Pan, Y.L.; Huang, J.; Zhang, S.J.; Zhang, L.; Zhang, Y. Cardiac μ-opioid receptor contributes to opioid-induced cardioprotection in chronic heart failure. Br. J. Anaesth. 2018, 121, 26–37.
- Cozzolino, D.; Sasso, F.C.; Cataldo, D.; Gruosso, D.; Giammarco, A.; Cavalli, A.; Di Maggio, C.; Renzo, G.; Salvatore, T.; Giugliano, D.; et al. Acute Pressor and Hormonal Effects of β-Endorphin at High Doses in Healthy and Hypertensive Subjects: Role of Opioid Receptor Agonism. J. Clin. Endocrinol. Metab. 2005, 90, 5167–5174.
- van den Brink, O.W.; Cochrane, A.D.; Rosenfeldt, F.L.; Penny, D.J.; Pepe, S. Increased myocardial methionine-enkephalin with reduced arterial oxygenation in congenital heart disease. J. Paediatr. Child. Health 2014, 50, E63–E67.
- Giersbergen, P.L.v.; Lang, H.d.; Jong, W.d. Effects of dynorphin A (1–13) and of fragments of β-endorphin on blood pressure and heart rate of anesthetized rats. Can. J. Physiol. Pharmacol. 1991, 69, 327–333.
- Rabkin, S.W. Dynorphin A (1–13) in the brain suppresses epinephrine-induced ventricular premature complexes and ventricular tachyarrhythmias. Regul. Pept. 1992, 41, 95–107.
- Lishmanov, Y.B.; Tsibul’nikov, S.Y.; Naryzhnaya, N.V.; Korobov, M.V.; Maslov, L.N. The Role of Endogenous Opioid System in the Regulation of Heart Tolerance to Stress-Induced Damage. Bull. Exp. Biol. Med. 2017, 163, 25–27.

