Myocardial infarction (MI) is a significant contributor to CVD-related mortality. T2DMType 2 diabetes mellitus (T2DM) is a risk factor for MI. Stress activates the hypothalamus–pituitary–adrenal axis (HPA axis, SNS), sympathetic nervous system (SNS), and endogenous OPS. These POMCpro-opiomelanocortin (POMC) derivatives increase the blood glucose and cardiovascular response by inhibiting the PI3K/AkT insulin signaling pathway and increasing cardiac contraction. Sustained activation of the POMC derivatives may lead to developing myocardial infarction. Suffering from T2DM and stress increases the risk of developing CVD. T2DM is preceded by prediabetes, which is a state of blood glucose level being above normal but below the level of T2DM diagnosis. Research has shown that T2DM-related complications begin during prediabetes; therefore, there is a possibility of the dysregulation of the POMC derivatives during prediabetes and pathways that could lead to myocardial infarction.
- pro-opiomelanocortin
- catecholamines
- glucocorticoids
- opioids
- type 2 diabetes
- prediabetes
- myocardial infarction
1. Introduction
2. POMC Derivative Signaling
2.1. Glucocorticoids
Glucocorticoids (GCs), such as cortisol in humans and corticosterone (CORT) in rodents, are cholesterol-derived hormones secreted by the zona fasciculata of the adrenal glands [20]. Glucocorticoids are fundamental in maintaining resting and stress-related homeostasis [21]. Animals respond to stress by activating various behavioral and physiological responses collectively called the stress response [22]. As summarized in Figure 1, the principal effectors of the stress response are localized in the paraventricular nucleus (PVN) of the hypothalamus, the anterior lobe of the pituitary gland, and the adrenal gland. This structure collection is commonly called the hypothalamic–pituitary–adrenocortical (HPA) axis. In response to stress, corticotropin-releasing hormone (CRH) is released into hypophysial portal vessels that access the anterior pituitary gland. The binding of CRH to its receptor on pituitary corticotropes induces the release of adrenocorticotropic hormone (ACTH) into the systemic circulation. The adrenal cortex is the principal target for circulating ACTH, which stimulates glucocorticoid synthesis and secretion from the zona fasciculata [22]. The cellular availability of GCs is determined by two enzymes that have opposing effects: 11β-hydroxysteroid dehydrogenase type 2 (11βHSD2) oxidizes cortisol into the inactive metabolite cortisone, whereas 11β-hydroxysteroid dehydrogenase type 1 (11βHSD1) converts cortisone to cortisol. Therefore, in normal physiology, GC levels are tightly regulated by a negative feedback loop at the level of the hypothalamus and pituitary gland, the availability of CBG in circulation, and at target tissues through the action of 11βHSD1 and 11βHSD2 [23].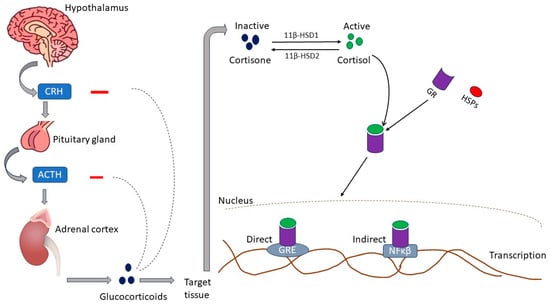
2.2. Catecholamine
Catecholamines are a group of molecules that act as neurotransmitters and hormones in the sympathetic division of the autonomic nervous system [33]. Catecholamines and glucocorticoids are the principal hormones secreted in response to extrinsic or intrinsic stressors to maintain homeostasis [34]. Catecholamines are produced from tyrosine hydroxylation to DOPA (l-3,4-dihydroxyphenylalanine) and a series of cellular reactions that ultimately produce dopamine (D), norepinephrine (NE), and epinephrine (E) from the adrenal medulla [35]. The SNS and adrenal medulla release epinephrine. It is involved in several physiological functions, including regulating blood pressure, vasoconstriction, cardiac stimulation, and blood glucose levels [36]. Norepinephrine is mainly produced by neurons within the locus coeruleus (LC) and takes part in diverse motor and mental functions, including locomotion control, motivation, attention, cognition, and memory formation [37]. Catecholamines rapidly respond to stress by binding to adrenergic receptors at the threatened site and the alerted brain, heart, and muscles [34,35,38][34][35][38]. Adrenoceptors (ARs) are categorized into alpha (α1 and α2) and beta (β1, β2 and β3) receptors. Norepinephrine activates α-AR and β1-AR, while epinephrine activates all subtypes of α- and β-AR [39]. The α1-AR receptors bind to stimulatory Gq proteins, activate phospholipase C (PLC), and induce constriction. On the contrary, α2-AR receptors are coupled to G inhibitory (Gi) proteins that inactivate adenylyl cyclase (AC), decreasing cyclic adenosine monophosphate (cAMP) production [40]. Β1-AR predominates in the heart and binds to the G stimulatory (Gs) protein–AC–cAMP–protein kinase A (PKA) signaling cascade, which results in the phosphorylation of troponin I, the L-type Ca2+ channel, phospholamban (PLN), and the cardiac ryanodine receptor (RyR), thus resulting in increased cardiac contraction and relaxation [41]. Β2-AR is distributed extensively throughout the body but is expressed predominantly in bronchial smooth muscle cells [39]. The stimulation of β2-AR activates the Gi protein, which inhibits cAMP and activates mitogen-activated protein kinase (MAPK) and cytosolic phospholipase A2 (cPLA2), thus resulting in cAMP-independent Ca2+ enhancement and reduced cardiac contraction [41]. β3-ARs are abundantly expressed in white and brown adipose tissue, increasing fat oxidation, energy expenditure, and insulin-mediated glucose uptake [39]. Β3-AR is stimulated by catecholamines only at high doses and has negative inotropy (decreased contraction) by facilitating the nitric oxide synthase (NOS) pathway. Nebivolol is reported to restore hemodynamic properties in patients with heart failure by stimulating β3-AR [42]. Figure 2 illustrates catecholamine adrenergic signaling.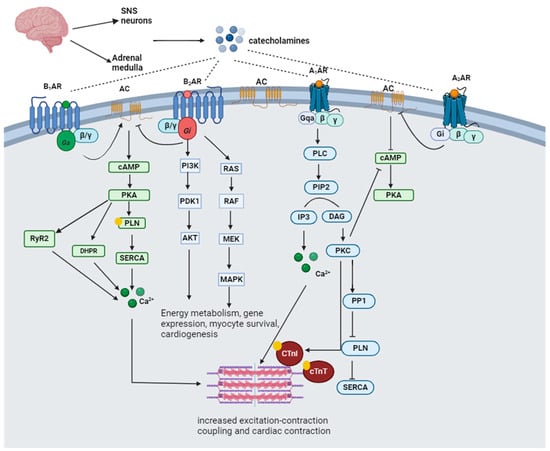
2.3. Opioids
The endogenous opioid peptides consist of endorphins, dynorphins (DYNs), and enkephalins (ENKs) [43,44][43][44]. There are four types of endorphins: alpha (α), beta (β), gamma (γ), and sigma (σ) endorphins. β-endorphins are primarily synthesized and stored in the anterior pituitary gland from their precursor protein, pro-opiomelanocortin (POMC) [43]. Dynorphin (DYN) is derived from a precursor protein, prodynorphin. Prodynorphin is cleaved to yield dynorphin-B, which has two extended forms (dynorphin-A and dynorphin-B), and leumorphin. In the peripheral circulation, dynorphin-A and dynorphin-B are further cleaved to yield dynorphin A (1–13) (DYN-A(1–13)), dynorphin A (1–8) (DYN-A (1–8)), and dynorphin B (1–13) (DYN-B (1–13)) and rimorphin, respectively [45]. Enkephalins are derived from the precursor protein proenkephalin (PENK) and interact with glutamate and dopamine in the brain reward circuit [46]. Endogenous opioids activate the mu (µ) (MOR), kappa (κ) (KOR), and delta (δ) (DOR) opioid receptors [43,44][43][44]. Opioid receptors signal through G-protein coupled receptors by stimulating inhibitory G-proteins, thus causing the Gαi subunit to dissociate from the Gβγ subunit and inhibit cAMP production. The Gαi subunit also interacts with the G-protein gated inwardly rectifying potassium channel (Kir3), thus causing hyperpolarization. The Gβγ modulates Ca2+ conduction by reducing the activation of N-type, P/Q-type, and L-type Ca2+ channels [47]. Signaling through the Gαi/o coupled proteins causes negative inotropy in rats’ ventricular myocytes by inhibiting cAMP-dependent Ca2+ [44]. The activation of KOR activates Gαi/o proteins, inhibiting the AC production of cAMP and releasing Gβγ, which modulates the conduction of Ca2+ and K+ channels [48]. Dynorphin provides cellular protection through the Gαs/cAMP/PKA signaling pathway, which causes an increase in CREB phosphorylation to enhance cell proliferation [49]. The distribution of OPR throughout the limbic system is consistent with the role of endogenous opioids in attenuating stress [50]. The stress system activates the dopaminergic reward system and the amygdala, thus forming a positive feedback loop [51]. The CRH stimulates the hypothalamus to release an α-melanocyte-stimulating hormone (α-MSH) and β-endorphin from the POMC-containing neurons in the arcuate nucleus. The α-MSH and β-endorphin inhibit CRH and the LC/NE system, thus regulating the stress response [51]. The naturally occurring δ-opioid peptide agonist, leucine enkephalin (LE), is co-released with the β-AR agonist norepinephrine (NE) from the nerve terminals in the heart during sympathetic stimulation. LE inhibits NE-induced increases in sarcolemma L-type Ca2+ current, cytosolic Ca2+ transient, and contraction [52]. The “anti-stress” activity of endogenous opioids may be mediated explicitly by the MOR [50]. During acute stress, the MOR regulation of the LC opposes the excitatory effect of CRH and protects against the detrimental NE/E hyperactivity effects. These opposing functions promote recovery after stress termination [53]. Mice with selective deletion of β-endorphin, enkephalin, or dynorphin subjected to the zero-maze test show increased anxiety-related behavioral responses [54]. Corticosterone plasma levels rapidly increased in all strains and returned to baseline after 60 minutes in b-endorphin-deficient mice [54]. In contrast, mice lacking dynorphin and enkephalin showed longer-lasting elevated corticosterone levels, which delayed the stress reaction termination [54]. Overall, upon release, opioids oppose the effect of glucocorticoids and catecholamines by inhibiting Ca2+ and Na+ channels while activating K+ channels. Figure 3 illustrates cardiac opioid signaling.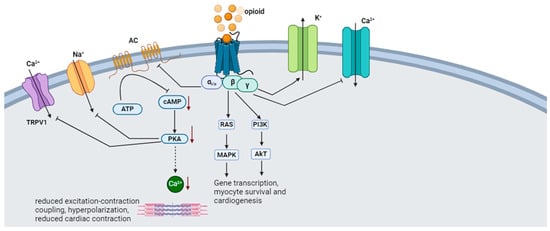
3. Role of POMC Derivatives in Cardiovascular Function
3.1. Glucocorticoids
Glucocorticoids are essential for the embryonic development of the heart and the maintenance of normal myocardial function. Adrenalectomized rats with GC insufficiency have a reduction in contractile force generation by the heart papillary muscle [55]. Dexamethasone (DEX), an exogenous GC, increases contractility tension and accelerates contraction velocity and relaxation in cardiac muscle [56]. The hearts of GR-null and smooth muscle-specific knockout (KO) mice exhibit irregularly shaped and disorganized myofibrils at the embryonic stage. Furthermore, the expression of genes that are critical for cardiac development and function is diminished in the heart [57]. GC protects from atherosclerosis and inflammation [58]. Mice lacking endothelial GR develop severe atherosclerotic lesions in the aorta and have heightened inflammation in the lesions [59]. High-dose corticosteroids are reported to exert cardiovascular protection through the non-genomic activation of eNOS. The binding of CORT to the GR stimulates PI3K and Akt, leading to eNOS activation and NO-dependent vasorelaxation. Furthermore, acute administration of pharmacological CORT concentrations in mice leads to decreased vascular inflammation and reduced myocardial infarct size following ischemia and reperfusion injury [60]. Overall, GCs are needed for the structural development of the heart and protect the heart from inflammation and atherosclerosis.3.2. Catecholamines
The activation of the SNS results in the release of catecholamines, which increase the supply of energy and oxygen delivery [61]. In fight-or-flight mode, NE/E stimulates glycogenolysis, gluconeogenesis, and aerobic glycolysis, inhibiting glycogen synthesis to supply glucose to vital organs [62]. Epinephrine increases blood glucose by stimulating glucagon secretion from mouse α-pancreatic cells through the activation of the α1 and β-AR on the α-pancreatic cell [63]. Glucagon, in turn, stimulates ACTH-induced cortisol release [64]. The stimulation of β-AR in adipocytes activates lipolysis through the activation of adenylyl cyclase and a cascade of reactions, which leads to the phosphorylation of hormone-sensitive lipase (HPL) and adipose triglyceride lipase (ATGL) [65]. It has been reported that catecholamine-induced lipolysis inhibits glucose uptake by inhibiting the PI3K–Akt–mTOR pathway [66]. Plasma concentrations of cortisol and E are significantly elevated in infants with severe hypoglycemia [67]. These mechanisms provide an acute increase in glucose levels. The spread of electrical impulses from cardiac autorhythmic cells stimulates the myocardium to contract, thus enabling the heart to pump blood to the blood vessels [68]. The electrical impulses are initiated by the sinoatrial (SA) node and result in atrial depolarization and atrial contraction; the impulse is then conducted to the internodal pathway, the AV node, the AV bundle, the left and right branches of the bundle of His, and lastly, the Purkinje fibers, which result in ventricular depolarization and contraction [68]. The activation of β1-AR increases the SA node’s firing rate, which, in turn, increases contractility because of an increased Ca2+ release from the sarcoplasmic reticulum and conduction through the AV node. The stimulation of α1-AR stimulates vasoconstriction and increases peripheral resistance [69]. The net effect is increased BP and cardiac output (CO) due to enhanced cardiac excitation, impulse conduction, and cardiac contraction [70]. Overall, catecholamines increase blood glucose by inhibiting glucose uptake while stimulating glucose release and synthesis. Catecholamines also increase blood pressure by increasing the firing rate of the SA node, Ca2+ concentration, and vasoconstriction.3.3. Opioids
The cardiovascular regulatory effects of endogenous opioids were initially considered to originate from the central nervous system. However, opioid peptides of myocardial origin have been shown to play essential roles in the local regulation of the heart [71]. A portion of POMC mRNA that contains a sequence for β-endorphin is expressed in the cardiac muscle, thus indicating that β-endorphin is produced in the heart [72]. Cell culture experiments from neonatal rat hearts revealed that myocytes and non-myocytes express ppENK mRNA [73]. Β-endorphin (1–31) is the primary endorphin form present in the cardiac muscle, although substantial amounts of N-acetylated and des-acetyl p-endorphin-(1–27) and P-endorphin-(1–26) are also detectable [74]. The cardiac tissue of rats subjected to immobilization stress has elevated β-endorphin [75]. Cardiac MOR expression is elevated in chronic heart failure and plays a cardioprotective role by reducing infarct size, the phosphorylation of ERK, and glycogen synthase-3-β (GSK3β) [76]. Infusions of high-dose β-endorphin in hypertensive subjects are reported to cause a decrease in systemic vascular resistance, blood pressure, plasma NE, and endothelin-1(ET-1) and raise atrial natriuretic factor P (ANP), thus protecting the heart [77]. The stimulation of OPR not only inhibits cardiac excitation–contraction coupling but protects the heart against hypoxic and ischemic injury [71]. Myocardial methionine-enkephalin levels increase with the severity of hypoxic stress in congenital cardiac disease. They may play an essential adaptive role in countering adrenergic over-activity and related excess demand on myocardial metabolic capacity [78]. Dynorphin provides cardiac protection during hypertension. Intracerebroventricular injections of β-endorphin and dynorphin A (1–13) in anesthetized rats result in hypotension and bradycardia [79]. Dynorphin A (1–13) also modulates epinephrine-induced cardiac arrhythmias by increasing the threshold for or suppressing the manifestation of the induced cardiac arrhythmias [80]. Receptor-dependent and independent stimulation of the adrenergic signaling pathway is reported to cause an increase in ppENK mRNA and modulate the dromotropic response to catecholamine stimulation in rat myocardial cells [73]. In stress-induced cardiac injury, the activation of central MOR with endogenous opioids is reported to aggravate stress-induced cardiomyopathy, while the stimulation of peripheral µ-opioid receptors produces a cardioprotective effect [81]. Overall, opioids counteract the effects of catecholamines on the heart. Opioid receptors are elevated during hypoxia and cardiac ischemic injury and attenuate the extent of cardiac damage. Figure 4 summarizes the cardiovascular function of POMC derivates.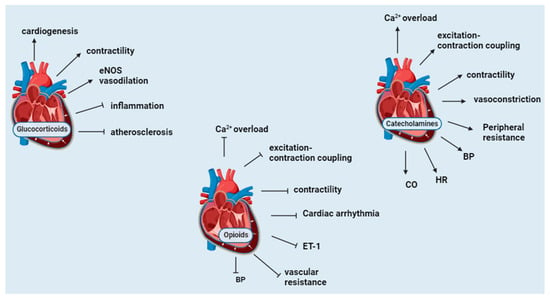
References
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Chaitman, B.R.; Bax, J.J.; Morrow, D.A.; White, H.D. Fourth Universal Definition of Myocardial Infarction (2018). Circulation 2018, 138, e618–e651.
- Lodrini, A.M.; Goumans, M.J. Cardiomyocytes Cellular Phenotypes after Myocardial Infarction. Front. Cardiovasc. Med. 2021, 8, 750510.
- Yun, J.-S.; Ko, S.-H. Current trends in epidemiology of cardiovascular disease and cardiovascular risk management in type 2 diabetes. Metabolism 2021, 123, 154838.
- Vos, T.; Lim, S.S.; Abbafati, C.; Abbas, K.M.; Abbasi, M.; Abbasifard, M.; Abbasi-Kangevari, M.; Abbastabar, H.; Abd-Allah, F.; Abdelalim, A.; et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222.
- Safiri, S.; Karamzad, N.; Singh, K.; Carson-Chahhoud, K.; Adams, C.; Nejadghaderi, S.A.; Almasi-Hashiani, A.; Sullman, M.J.M.; Mansournia, M.A.; Bragazzi, N.L.; et al. Burden of ischemic heart disease and its attributable risk factors in 204 countries and territories, 1990–2019. Eur. J. Prev. Cardiol. 2022, 29, 420–431.
- Kelly, C.; Lan, N.S.R.; Phan, J.; Hng, C.; Matthews, A.; Rankin, J.M.; Schultz, C.J.; Hillis, G.S.; Reid, C.M.; Dwivedi, G.; et al. Characteristics and Outcomes of Young Patients with ST-Elevation Myocardial Infarction without Standard Modifiable Risk Factors. Am. J. Cardiol. 2023, 202, 81–89.
- Laakso, M.; Kuusisto, J. Insulin resistance and hyperglycaemia in cardiovascular disease development. Nat. Rev. Endocrinol. 2014, 10, 293–302.
- Steptoe, A.; Kivimäki, M. Stress and cardiovascular disease. Nat. Rev. Cardiol. 2012, 9, 360–370.
- Satyjeet, F.; Naz, S.; Kumar, V.; Aung, N.H.; Bansari, K.; Irfan, S.; Rizwan, A. Psychological Stress as a Risk Factor for Cardiovascular Disease: A Case-Control Study. Cureus 2020, 12, e10757.
- Levine, G.N. Psychological Stress and Heart Disease: Fact or Folklore? Am. J. Med. 2022, 135, 688–696.
- Gianoulakis, C. Alcohol-seeking behavior: The roles of the hypothalamic-pituitary-adrenal axis and the endogenous opioid system. Alcohol. Health Res. World 1998, 22, 202–210.
- Pilozzi, A.; Carro, C.; Huang, X. Roles of β-Endorphin in Stress, Behavior, Neuroinflammation, and Brain Energy Metabolism. Int. J. Mol. Sci. 2021, 22, 338.
- Carr, D.B.; Verrier, R.L. Opioids in pain and cardiovascular responses: Overview of common features. J. Cardiovasc. Electrophysiol. 1991, 2, s34–s42.
- Kelly, S.J.; Ismail, M. Stress and Type 2 Diabetes: A Review of How Stress Contributes to the Development of Type 2 Diabetes. Annu. Rev. Public. Health 2015, 36, 441–462.
- Rooney, M.R.; Rawlings, A.M.; Pankow, J.S.; Echouffo Tcheugui, J.B.; Coresh, J.; Sharrett, A.R.; Selvin, E. Risk of Progression to Diabetes Among Older Adults with Prediabetes. JAMA Intern. Med. 2021, 181, 511–519.
- Echouffo-Tcheugui, J.B.; Selvin, E. Prediabetes and What It Means: The Epidemiological Evidence. Annu. Rev. Public Health 2021, 42, 59–77.
- Mishra, A.; Podder, V.; Modgil, S.; Khosla, R.; Anand, A.; Nagarathna, R.; Malhotra, R.; Nagendra, H.R. Higher Perceived Stress and Poor Glycemic Changes in Prediabetics and Diabetics among Indian Population. J. Med. Life 2020, 13, 132–137.
- Sharma, K.; Akre, S.; Chakole, S.; Wanjari, M.B. Stress-Induced Diabetes: A Review. Cureus 2022, 14, e29142.
- Jyotheeswara Pillai, K.; Luo, H.; Raj, K.; Xu, S.; Thota, G. OR02-4 Prediabetes is a Risk Factor for Myocardial Infarction-A National Inpatient Sample Study. J. Endocr. Soc. 2022, 6, A248.
- Kadmiel, M.; Cidlowski, J.A. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol. Sci. 2013, 34, 518–530.
- Chrousos, G.P.; Kino, T. Glucocorticoid action networks and complex psychiatric and/or somatic disorders. Stress 2007, 10, 213–219.
- Smith, S.M.; Vale, W.W. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues Clin. Neurosci. 2006, 8, 383–395.
- Maniam, J.; Antoniadis, C.; Morris, M.J. Early-life stress, HPA axis adaptation, and mechanisms contributing to later health outcomes. Front. Endocrinol. 2014, 5, 73.
- Macfarlane, E.; Seibel, M.J.; Zhou, H. Arthritis and the role of endogenous glucocorticoids. Bone Res. 2020, 8, 33.
- Sevilla, L.M.; Jiménez-Panizo, A.; Alegre-Martí, A.; Estébanez-Perpiñá, E.; Caelles, C.; Pérez, P. Glucocorticoid Resistance: Interference between the Glucocorticoid Receptor and the MAPK Signalling Pathways. Int. J. Mol. Sci. 2021, 22, 10049.
- Qi, D.; Rodrigues, B. Glucocorticoids produce whole body insulin resistance with changes in cardiac metabolism. Am. J. Physiol. -Endocrinol. Metab. 2007, 292, E654–E667.
- Cato, A.C.; Nestl, A.; Mink, S. Rapid actions of steroid receptors in cellular signaling pathways. Sci. STKE 2002, 2002, re9.
- Nicolaides, N.C.; Kyratzi, E.; Lamprokostopoulou, A.; Chrousos, G.P.; Charmandari, E. Stress, the Stress System and the Role of Glucocorticoids. Neuroimmunomodulation 2015, 22, 6–19.
- Mihailidou, A.S.; Le, T.Y.L.; Mardini, M.; Funder, J.W. Glucocorticoids Activate Cardiac Mineralocorticoid Receptors During Experimental Myocardial Infarction. Hypertension 2009, 54, 1306–1312.
- Nicolaides, N.C.; Galata, Z.; Kino, T.; Chrousos, G.P.; Charmandari, E. The human glucocorticoid receptor: Molecular basis of biologic function. Steroids 2010, 75, 1–12.
- Girod, J.P.; Brotman, D.J. Does altered glucocorticoid homeostasis increase cardiovascular risk? Cardiovasc. Res. 2004, 64, 217–226.
- Walker, B.R. Glucocorticoids and Cardiovascular Disease. Eur. J. Endocrinol. 2007, 157, 545–559.
- Eto, K.; Mazilu-Brown, J.K.; Henderson-MacLennan, N.; Dipple, K.M.; McCabe, E.R.B. Development of catecholamine and cortisol stress responses in zebrafish. Mol. Genet. Metab. Rep. 2014, 1, 373–377.
- Kyrou, I.; Tsigos, C. Stress hormones: Physiological stress and regulation of metabolism. Curr. Opin. Pharmacol. 2009, 9, 787–793.
- Gülçin, I. Antioxidant activity of L-adrenaline: A structure-activity insight. Chem. Biol. Interact. 2009, 179, 71–80.
- Errasti, A.E.; Werneck de Avellar, M.C.; Daray, F.M.; Tramontano, J.; Luciani, L.I.; Lina Bard, M.A.J.; Maróstica, E.; Rothlin, R.P. Human umbilical vein vasoconstriction induced by epinephrine acting on α1B-adrenoceptor subtype. Am. J. Obstet. Gynecol. 2003, 189, 1472–1480.
- Foote, S.L.; Bloom, F.E.; Aston-Jones, G. Nucleus locus ceruleus: New evidence of anatomical and physiological specificity. Physiol. Rev. 1983, 63, 844–914.
- Waterhouse, B.D.; Devilbiss, D.; Fleischer, D.; Sessler, F.M.; Simpson, K.L. New perspectives on the functional organization and postsynaptic influences of the locus ceruleus efferent projection system. Adv. Pharmacol. 1998, 42, 749–754.
- Kemeny, M.E. The Psychobiology of Stress. Curr. Dir. Psychol. Sci. 2003, 12, 124–129.
- Motiejunaite, J.; Amar, L.; Vidal-Petiot, E. Adrenergic receptors and cardiovascular effects of catecholamines. Ann. D’endocrinologie 2021, 82, 193–197.
- Pavoine, C.; Defer, N. The cardiac β2-adrenergic signalling a new role for the cPLA2. Cell. Signal. 2005, 17, 141–152.
- Rozec, B.; Erfanian, M.; Laurent, K.; Trochu, J.-N.; Gauthier, C. Nebivolol, a vasodilating selective β1-blocker, is a β3-adrenoceptor agonist in the nonfailing transplanted human heart. J. Am. Coll. Cardiol. 2009, 53, 1532–1538.
- Jain, A.; Mishra, A.; Shakkarpude, J.; Lakhani, P. Beta endorphins: The natural opioids. IJCS 2019, 7, 323–332.
- Al-Hasani, R.; Bruchas, M.R. Molecular Mechanisms of Opioid Receptor-dependent Signaling and Behavior. Anesthesiology 2011, 115, 1363–1381.
- Cissom, C.; J Paris, J.; Shariat-Madar, Z. Dynorphins in development and disease: Implications for cardiovascular disease. Curr. Mol. Med. 2020, 20, 259–274.
- Mongi-Bragato, B.; Avalos, M.P.; Guzmán, A.S.; Bollati, F.A.; Cancela, L.M. Enkephalin as a pivotal player in neuroadaptations related to psychostimulant addiction. Front. Psychiatry 2018, 9, 222.
- Li, Z.; Liu, J.; Dong, F.; Chang, N.; Huang, R.; Xia, M.; Patterson, T.A.; Hong, H. Three-Dimensional Structural Insights Have Revealed the Distinct Binding Interactions of Agonists, Partial Agonists, and Antagonists with the µ Opioid Receptor. Int. J. Mol. Sci. 2023, 24, 7042.
- Bruchas, M.R.; Chavkin, C. Kinase cascades and ligand-directed signaling at the kappa opioid receptor. Psychopharmacology 2010, 210, 137–147.
- Ji, B.; Liu, H.; Zhang, R.; Jiang, Y.; Wang, C.; Li, S.; Chen, J.; Bai, B. Novel signaling of dynorphin at κ-opioid receptor/bradykinin B2 receptor heterodimers. Cell. Signal. 2017, 31, 66–78.
- Drolet, G.; Dumont, É.C.; Gosselin, I.; Kinkead, R.; Laforest, S.; Trottier, J.-F. Role of endogenous opioid system in the regulation of the stress response. Progress. Neuro-Psychopharmacol. Biol. Psychiatry 2001, 25, 729–741.
- Charmandari, E.; Kino, T.; Souvatzoglou, E.; Chrousos, G.P. Pediatric Stress: Hormonal Mediators and Human Development. Horm. Res. Paediatr. 2003, 59, 161–179.
- Xiao, R.P.; Pepe, S.; Spurgeon, H.A.; Capogrossi, M.C.; Lakatta, E.G. Opioid peptide receptor stimulation reverses beta-adrenergic effects in rat heart cells. Am. J. Physiol.-Heart Circ. Physiol. 1997, 272, H797–H805.
- Valentino, R.J.; Van Bockstaele, E. Endogenous opioids: The downside of opposing stress. Neurobiol. Stress 2015, 1, 23–32.
- Bilkei-Gorzo, A.; Racz, I.; Michel, K.; Mauer, D.; Zimmer, A.; Klingmüller, D.; Zimmer, A. Control of hormonal stress reactivity by the endogenous opioid system. Psychoneuroendocrinology 2008, 33, 425–436.
- Lefer, A.M. Influence of corticosteroids on mechanical performance of isolated rat papillary muscles. Am. J. Physiol.-Leg. Content 1968, 214, 518–524.
- Penefsky, Z.J.; Kahn, M. Inotropic effects of dexamethasone in mammalian heart muscle. Eur. J. Pharmacol. 1971, 15, 259–266.
- Whirledge, S.; DeFranco, D.B. Glucocorticoid Signaling in Health and Disease: Insights From Tissue-Specific GR Knockout Mice. Endocrinology 2017, 159, 46–64.
- Vandevyver, S.; Dejager, L.; Tuckermann, J.; Libert, C. New Insights into the Anti-inflammatory Mechanisms of Glucocorticoids: An Emerging Role for Glucocorticoid-Receptor-Mediated Transactivation. Endocrinology 2013, 154, 993–1007.
- Goodwin, J.E.; Zhang, X.; Rotllan, N.; Feng, Y.; Zhou, H.; Fernández-Hernando, C.; Yu, J.; Sessa, W.C. Endothelial glucocorticoid receptor suppresses atherogenesis—Brief report. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 779–782.
- Hafezi-Moghadam, A.; Simoncini, T.; Yang, Z.; Limbourg, F.P.; Plumier, J.-C.; Rebsamen, M.C.; Hsieh, C.-M.; Chui, D.-S.; Thomas, K.L.; Prorock, A.J.; et al. Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat. Med. 2002, 8, 473–479.
- Ziegler, M.G.; Elayan, H.; Milic, M.; Sun, P.; Gharaibeh, M. Epinephrine and the Metabolic Syndrome. Curr. Hypertens. Rep. 2012, 14, 1–7.
- Barth, E.; Albuszies, G.; Baumgart, K.; Matejovic, M.; Wachter, U.; Vogt, J.; Radermacher, P.; Calzia, E. Glucose metabolism and catecholamines. Crit. Care Med. 2007, 35, S508–S518.
- Vieira, E.; Liu, Y.-J.; Gylfe, E. Involvement of α1 and β-adrenoceptors in adrenaline stimulation of the glucagon-secreting mouse α-cell. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2004, 369, 179–183.
- Jones, B.J.; Tan, T.; Bloom, S.R. Minireview: Glucagon in Stress and Energy Homeostasis. Endocrinology 2012, 153, 1049–1054.
- Schweiger, M.; Schreiber, R.; Haemmerle, G.; Lass, A.; Fledelius, C.; Jacobsen, P.; Tornqvist, H.; Zechner, R.; Zimmermann, R. Adipose Triglyceride Lipase and Hormone-sensitive Lipase Are the Major Enzymes in Adipose Tissue Triacylglycerol Catabolism. J. Biol. Chem. 2006, 281, 40236–40241.
- Mullins, G.R.; Wang, L.; Raje, V.; Sherwood, S.G.; Grande, R.C.; Boroda, S.; Eaton, J.M.; Blancquaert, S.; Roger, P.P.; Leitinger, N.; et al. Catecholamine-induced lipolysis causes mTOR complex dissociation and inhibits glucose uptake in adipocytes. Proc. Natl. Acad. Sci. USA 2014, 111, 17450–17455.
- Jackson, L.; Williams, F.L.R.; Burchell, A.; Coughtrie, M.W.H.; Hume, R. Plasma Catecholamines and the Counterregulatory Responses to Hypoglycemia in Infants: A Critical Role for Epinephrine and Cortisol. J. Clin. Endocrinol. Metab. 2004, 89, 6251–6256.
- Gordan, R.; Gwathmey, J.K.; Xie, L.-H. Autonomic and endocrine control of cardiovascular function. World J. Cardiol. 2015, 7, 204.
- Grassi, G. Assessment of Sympathetic Cardiovascular Drive in Human Hypertension. Hypertension 2009, 54, 690–697.
- Medić, B. The role of autonomic control in cardiovascular system: Summary of basic principles. Med. Podml. 2016, 67, 14–18.
- Ventura, C.; Spurgeon, H.; Lakatta, E.G.; Guarnieri, C.; Capogrossi, M.C. Kappa and delta opioid receptor stimulation affects cardiac myocyte function and Ca2+ release from an intracellular pool in myocytes and neurons. Circ. Res. 1992, 70, 66–81.
- Forman, L.; Bagasra, O. Demonstration by in situ hybridization of the proopiomelanocortin gene in the rat heart. Brain Res. Bull. 1992, 28, 441–445.
- Weil, J.; Zolk, O.; Griepentrog, J.; Wenzel, U.; Zimmermann, W.H.; Eschenhagen, T. Alterations of the preproenkephalin system in cardiac hypertrophy and its role in atrioventricular conduction. Cardiovasc. Res. 2006, 69, 412–422.
- Millington, W.R.; Evans, V.R.; Battie, C.N.; Bagasra, O.; Forman, L.J. Proopiomelanocortin-Derived Peptides and mRNA Are Expressed in Rat Heart. Ann. N. Y. Acad. Sci. 1993, 680, 575–578.
- Forman, L.J.; Hock, C.E.; Harwell, M.; Estilow-Isabell, S. The Results of Exposure to Immobilization, Hemorrhagic Shock, and Cardiac Hypertrophy on β-Endorphin in Rat Cardiac Tissue. Proc. Soc. Exp. Biol. Med. 1994, 206, 124–129.
- He, S.F.; Jin, S.Y.; Yang, W.; Pan, Y.L.; Huang, J.; Zhang, S.J.; Zhang, L.; Zhang, Y. Cardiac μ-opioid receptor contributes to opioid-induced cardioprotection in chronic heart failure. Br. J. Anaesth. 2018, 121, 26–37.
- Cozzolino, D.; Sasso, F.C.; Cataldo, D.; Gruosso, D.; Giammarco, A.; Cavalli, A.; Di Maggio, C.; Renzo, G.; Salvatore, T.; Giugliano, D.; et al. Acute Pressor and Hormonal Effects of β-Endorphin at High Doses in Healthy and Hypertensive Subjects: Role of Opioid Receptor Agonism. J. Clin. Endocrinol. Metab. 2005, 90, 5167–5174.
- van den Brink, O.W.; Cochrane, A.D.; Rosenfeldt, F.L.; Penny, D.J.; Pepe, S. Increased myocardial methionine-enkephalin with reduced arterial oxygenation in congenital heart disease. J. Paediatr. Child. Health 2014, 50, E63–E67.
- Giersbergen, P.L.v.; Lang, H.d.; Jong, W.d. Effects of dynorphin A (1–13) and of fragments of β-endorphin on blood pressure and heart rate of anesthetized rats. Can. J. Physiol. Pharmacol. 1991, 69, 327–333.
- Rabkin, S.W. Dynorphin A (1–13) in the brain suppresses epinephrine-induced ventricular premature complexes and ventricular tachyarrhythmias. Regul. Pept. 1992, 41, 95–107.
- Lishmanov, Y.B.; Tsibul’nikov, S.Y.; Naryzhnaya, N.V.; Korobov, M.V.; Maslov, L.N. The Role of Endogenous Opioid System in the Regulation of Heart Tolerance to Stress-Induced Damage. Bull. Exp. Biol. Med. 2017, 163, 25–27.