Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Zhiying Shan | -- | 2612 | 2024-02-14 16:50:51 | | | |
| 2 | Mona Zou | -3 word(s) | 2609 | 2024-02-19 10:31:15 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Chapp, A.D.; Shan, Z.; Chen, Q. Acetic Acid in Ethanol-Induced in Regulating Cardiovascular Function. Encyclopedia. Available online: https://encyclopedia.pub/entry/55047 (accessed on 24 July 2026).
Chapp AD, Shan Z, Chen Q. Acetic Acid in Ethanol-Induced in Regulating Cardiovascular Function. Encyclopedia. Available at: https://encyclopedia.pub/entry/55047. Accessed July 24, 2026.
Chapp, Andrew D., Zhiying Shan, Qing-Hui Chen. "Acetic Acid in Ethanol-Induced in Regulating Cardiovascular Function" Encyclopedia, https://encyclopedia.pub/entry/55047 (accessed July 24, 2026).
Chapp, A.D., Shan, Z., & Chen, Q. (2024, February 14). Acetic Acid in Ethanol-Induced in Regulating Cardiovascular Function. In Encyclopedia. https://encyclopedia.pub/entry/55047
Chapp, Andrew D., et al. "Acetic Acid in Ethanol-Induced in Regulating Cardiovascular Function." Encyclopedia. Web. 14 February, 2024.
Copy Citation
Acetic acid is a bioactive short-chain fatty acid produced in large quantities from ethanol metabolism. How acetic acid/acetate generates oxidative stress, alters the function of pre-sympathetic neurons, and can potentially influence cardiovascular function in both humans and rodents after ethanol consumption are described.
ethanol
acetic acid
acetate
nitric oxide
sympathetic nerve activity
cardiovascular function
1. Introduction
Ethanol metabolism produces systemic maladaptive changes through oxidative stress. Ethanol metabolism in vivo generates reactive oxygen and nitrogen species, predominantly through oxidative pathways involving NADPH/NADP+, NAD+/NADH, and/or H2O2 [1]. Ethanol is oxidized to acetic acid, which becomes acetate at physiological pH (Figure 1A) [2]. Acetate is then shuttled into the citric acid cycle, where additional oxidative stress is likely generated through electron transport chain electron leak [3]. Chronic oxidative stress from ethanol metabolism leads to liver [4][5][6][7][8], brain [9][10][11][12][13], and cardiovascular pathologies [14][15][16][17][18][19][20].
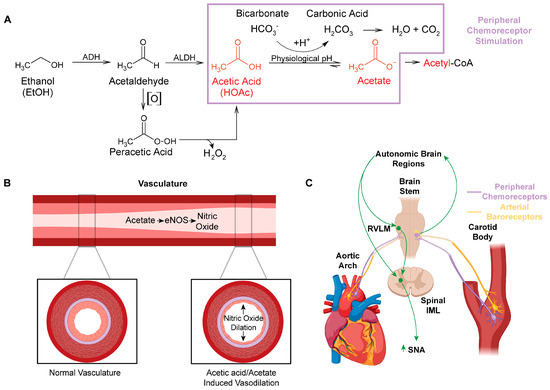
Figure 1. Impact of acetic acid/acetate on peripheral vasculature and the regulation of cardiovascular function. (A) Major metabolic pathway for ethanol metabolism (box in purple highlights potential chemoreceptor−stimulating pathways). Auto−oxidation (denoted [O]) of acetaldehyde, which can be increased by the presence of Fe3+ to form peracetic acid (peroxyacetic acid). Peracetic acid decomposes to acetic acid, generating hydrogen peroxide. (B) Acetate stimulates eNOS in the peripheral vasculature, which increases production of the powerful vasodilator, nitric oxide. (C) Schematic of the location of peripheral arterial baroreceptors and chemoreceptors, their innervation to the brainstem, and their relationship with neural control of autonomic function. Abbreviations: alcohol dehydrogenase (ADH), cytochrome P450 (CYP 450), aldehyde dehydrogenase (ALDH), endothelial nitric oxide synthase (eNOS), nitric oxide (NO), rostral ventrolateral medulla (RVLM), intermediolateral column (IML), sympathetic nerve activity (SNA).
For many years, moderate ethanol consumption was thought to provide some benefit to cardiovascular health [21][22]. This was endorsed by the American Heart Association (AHA) [23]. However, this was at odds with the literature on alcohol use disorder suggesting that chronic use of ethanol leads to the development of hypertension and cardiovascular disease [24][25][26]. By 2023, after several research articles suggested that ethanol offers no cardiovascular benefit [27][28], the AHA reversed course, warning that even one ethanol-containing drink a day can increase cardiovascular disease risk [29].
A major gap in the ethanol research field is the limited investigation of the role of acetic acid/acetate in the pathologies associated with ethanol use. Acetic acid/acetate has been assumed to be a relatively benign compound [1], as acetate is a feedstock for the generation of ATP through the citric acid cycle [30] and for the generation of acetyl-CoA and acetylation reactions [31][32]. However, acetic acid is linked to several distinct disease states, including Alzheimer’s disease [33][34], neurodegenerative disease [33][35], obesity [36], and gut–brain dysbiosis [33][35][36]. How acetic acid/acetate may influence the regulation of cardiovascular function are highlighted through several mechanisms, including the generation of reactive oxygen and nitrogen species (Figure 1 and Figure 2).
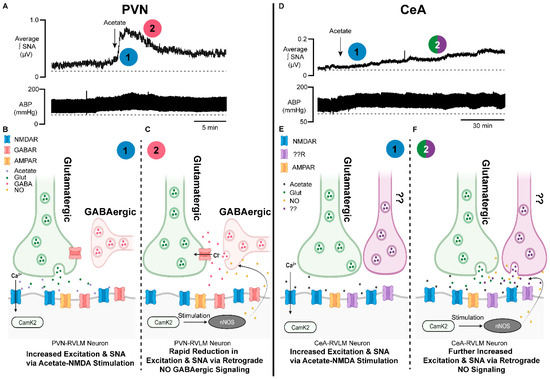
Figure 2. Brain-generated sympathoexcitatory responses to microinjections of acetate differ by autonomic region: implications for nitric oxide. (A) Representative sympathetic nerve recording and arterial blood pressure in response to PVN microinjected acetate. Note the rapid increase in sympathetic nerve activity (1, blue) and time of onset, followed by a rapid reduction in excitation (2, red). (B) Proposed schematic of the synaptic mechanisms contributing to the acetate-induced sympathoexcitatory response in PVN−RVLM neuronal circuitry. Note that (1, blue) corresponds to the response in (A). (C) Proposed schematic of NO retrograde synaptic signaling in PVN−RVLM neurons. Acetate activation of the NMDAR stimulates NO production, which increases presynaptic GABA release on glutamatergic synapses, reducing sympathoexcitation. Note that (2, red) corresponds to the response in (A). (D) Representative sympathetic nerve recording and arterial blood pressure in response to CeA−microinjected acetate. Note the steady increase in sympathetic nerve activity (1, blue) and the steady increase in excitation (2, green/purple). (E) Proposed schematic of the synaptic mechanisms contributing to the acetate-induced sympathoexcitatory response in CeA−RVLM neuronal circuitry. Note that (1, blue) corresponds to the response in (D). (F) Proposed schematic of NO retrograde synaptic signaling in CeA−RVLM neurons. Acetate activation of the NMDAR stimulates NO production, which increases the release of presynaptic glutamate and other neurotransmitters, increasing sympathoexcitation. Alternative inputs may also be propagating increased SNA. Note that (2, green/purple) corresponds to the response in (D). Abbreviations: paraventricular nucleus of the hypothalamus (PVN), rostral ventrolateral medulla (RVLM), neuronal nitric oxide synthase (nNOS), calcium calmodulin kinase 2 (CAMK2), nitric oxide (NO), calcium (Ca2+), chloride (Cl−), N-methyl-D-aspartate receptor (NMDAR), gamma aminobutyric acid receptor (GABAR), sympathetic nerve activity (SNA), glutamate (Glut), gamma aminobutyric acid (GABA), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR).
2. Acetaldehyde and Peracetic Acid: All Roads Lead to Acetic Acid/Acetate
Acetaldehyde has been one of the most highly researched ethanol metabolites and has been implicated in the generation of reactive oxygen species, protein adduct formation [37][38][39], the increased production of salsolinol [40][41][42][43][44][45][46], and neuronal toxicity [47][48][49][50]. In individuals with aldehyde dehydrogenase (ALDH) 2 deficiencies, the buildup of acetaldehyde is thought to cause flushing, nausea, and headache [51][52][53][54]. Disulfiram, an ALDH inhibitor and alcohol use deterrent, also increases the buildup of acetaldehyde, which is thought to be the mechanism of action for reducing alcohol consumption [55][56][57].
Interestingly, blood acetaldehyde concentrations are not easily measured and have been reported in the range of 10–40 nM when blood ethanol concentrations are 10–20 mM [58][59]. Part of the prevailing theory on why blood acetaldehyde concentrations are so low is due to its rapid metabolism to acetic acid [1]. And while this may be true, there are other chemical factors and conversion routes for acetaldehyde that may be contributing to its low detection.
First, acetaldehyde is a volatile liquid that is soluble in aqueous solutions [60]. Its boiling point is 20.2 °C (68 °F) [60], much lower than the physiological temperature of 37 °C (98.6 °F). Thus, it is highly likely that any acetaldehyde that is not converted to acetic acid/acetate and remains in circulation passing to the lungs is exhaled in its gaseous state. Second, in a report published in 1929 by Bowen and Tietz, they noted that acetaldehyde shaken in the presence of oxygen (auto-oxidation) produced a peroxide, identified as peracetic acid [61]. These authors also noted that this type of reaction and similar reactions produced a “long chain mechanism” [61], which would be consistent with what is now a free radical-type mechanism. Indeed, follow-up work in 1950 by Bawn and Williamson further elucidated that aqueous solutions of acetaldehyde were also capable of auto-oxidation, producing the peracetic acid reported by Bowen and Tietz [61], and that these reactions could be increased and/or induced in the presence of metals, of note, iron III (Fe3+) [62]. Interestingly, hemoglobin, the iron-containing protein that is responsible for oxygen and carbon dioxide transport in the blood, has an iron oxidation state of Fe3+ when oxygen is bound [63]. This close proximity of oxygen and Fe3+ with any circulating acetaldehyde may convert acetaldehyde into peracetic acid, and account for the low to undetectable circulating levels following ethanol consumption [59].
Peracetic acid (peroxyacetic acid) is a peroxide of acetic acid. It is utilized as a broad-spectrum disinfectant, displaying bactericidal, virucidal, fungicidal, and sporicidal properties [64]. In cell toxicity studies, peracetic acid was more cytotoxic than sodium hypochlorite (bleach) [65]. Moreover, given that peracetic acid is an organic peroxide, it undergoes similar homolysis reactions as other peroxides do [66]. As such, the spontaneous or metal-catalyzed homolysis reactions of peroxides generate free radicals [66][67] and these free radicals are thought to induce cellular and tissue damage [68][69]. Hydrogen peroxide is a classic example of a peroxide used as a source of oxygen-derived free radicals in research studies [70]. Additionally, hydrogen peroxide also interacts with iron in the well-known Fenton reaction to generate additional sources of free radicals that can lead to tissue damage [71].
3. Peripheral Actions of Acetic Acid/Acetate: Impact on Nitric Oxide Synthase (NOS) and Nitric Oxide (NO)
Ethanol in humans has been found to influence heart rate, cardiac output, stroke volume, and sympathetic nerve activity [72][73][74][75][76][77][78]. It causes both vasoconstriction and vasodilation [14][79][80]. In rodent models, ethanol has been shown to increase the bioavailability of nitric oxide [80][81][82]. However, these studies did not account for ethanol metabolism or the production of acetic acid/acetate.
In studies investigating the effect of buffering agents in hemodialysis solutions, it is observed that acetate-based solutions create greater cardiovascular and hemodynamic instability than bicarbonate-based solutions do [83][84]. These instabilities include vasodilation, hypoxemia, increased cardiac output, and angina pectoris [85][86][87]. Methodical investigation identified that these acetate-based hemodialysis solutions increased levels of nitric oxide synthase (NOS) and nitric oxide (NO) [88][89], a powerful radical which stimulates vasodilation [90][91][92]. Several studies have also identified acetate-induced elevations in tumor necrosis factor alpha (TNFa) and cyclic adenosine monophosphate (cAMP), both mediators of NOS [89]. This was seen in dialysis buffers containing as little as 4 mM acetate [89]. A likely mechanism through which ethanol produces vasodilation is through ethanol’s metabolism to acetic acid/acetate, which stimulates production of NOS and NO (Figure 1B). This finding was also substantiated by Sakakibara S et al., who found that consumption of vinegar increased eNOS production with as little as 200 uM acetate in both early-phase detection (20 min) as well as late-phase detection (4 h) [93]. Furthermore, they found increased forearm vasodilation in adults who had consumed acetic acid compared to a control. This time course of action for acetic acid/acetate suggests a profound impact on peripheral eNOS even with minimal concentrations of and exposure to acetate [93].
4. Direct Effects of Ethanol and Acetic Acid/Acetate on Cardiac Function
The potential of ethanol consumption to influence cardiac function has been well documented. In the acute setting, ethanol can increase cardiac output [94] and reduce plasma potassium [95], which may contribute to ethanol-induced arrhythmia or “holiday heart” [96]. The acute effects of ethanol on contractility appear to be mixed, with some studies suggesting either no change [94][97][98] or a decrease [99][100] in cardiac contractility. Chronic ethanol use also contributes to cardiomyopathy [101][102] and arrhythmias [20].
Similar to the effects of acute ethanol, acute acetate is also capable of altering cardiac function. Acute acetate administration has been found to increase cardiac output [103][104], precipitate hemodynamic instability [84][88][103], and reduce myocardial contractility [105][106]. Interestingly, some of the direct cardiac effects of acetate were speculated to be tied to acetate-induced NO release [88]. Indeed, the direct cardiac effects of NO are bimodal, with lower concentrations of NO increasing the ionotropic effects [107] and high concentrations of NO reducing cardiac myocyte contractility [108]. Whether the mixed effects of ethanol on cardiac function are a result of acetate and/or acetate-induced NO release remains to be determined. Furthermore, the effect of chronically elevated acetate from chronic ethanol use and metabolism on cardiac function has yet to be investigated. It is provocative to postulate that the major compound driving alcohol-induced cardiomyopathy [109][110] may be acetate rather than ethanol, and as such, future studies of chronic acetate seem warranted.
5. Ethanol Metabolism to Acetic Acid Alters Acid/Base Homeostasis and Likely Engages Peripheral Chemoreceptors
Although ethanol itself is not acidic (pKa value ~16) [111], ethanol acts as an exogenous source of acidic hydrogens. Once ethanol is fully oxidized to acetic acid (pKa value ~4.78) [112], the labile acidic hydrogen becomes >95% dissociated at physiological pH [113]. pKa is a measure of hydrogen acidity, as a review of pKa (see General Chemistry, sixth edition, Julia Burdge). The higher the pKa value, the less acidic the hydrogen. At physiological pH, acetic acid likely reacts with the bicarbonate buffering system (i.e., the vinegar and baking soda reaction), forming acetate and carbonic acid (Figure 1A). Carbonic acid is then broken down into water and carbon dioxide. The acidic hydrogen on acetic acid and the excess carbon dioxide cause activation of (1) peripheral chemoreceptors located in the carotid sinus and aortic arches, and (2) neural control centers in the brain, which control sympathetic outflow and increase sympathetic nerve activity (SNA) (Figure 1C).
In the United States, humans are considered legally intoxicated at a blood alcohol concentration (BAC) of 0.08% or a blood serum concentration of ~17 mM ethanol [114]. The conversion of ethanol to acetic acid is nearly 1:1, with ~95% of ethanol converted to acetic acid [115]. Thus, for 17 mM of ethanol consumed, the predicted amount of acetic acid produced would be ~16–17 mM. To buffer or neutralize this concentration of protons, roughly 17 mM of bicarbonate would be consumed. The normal concentration of bicarbonate in the serum is ~23–28 mM [116][117]. This bicarbonate consumption creates a high acid load, generating compensatory mechanisms within the kidneys and lungs to maintain pH homeostasis [117][118][119].
6. Integration of Arterial Baroreceptors, Chemoreceptors, and NO Signaling in Response to Acetic Acid Generated by Ethanol Metabolism
The integration of the peripheral cardiovascular regulatory systems engaged by acetic acid likely explains the complex phenomenon of mixed vasodilation and vasoconstriction induced by ethanol. First, acetic acid increases NOS activity and NO production, which leads to vasodilation [85][88][89]. Arterial baroreceptors located in the carotid sinus and aortic arch [120] would relay the resultant drop in blood pressure to neural control centers, which regulate cardiovascular function [121] (Figure 1C). These neural control centers then increase SNA to constrict the vasculature in an attempt to maintain homeostasis [120]. Third, the acid load generated from the acetic acid and/or excess carbon dioxide would similarly be sensed by the peripheral chemoreceptors, also located in the carotid sinus and aortic arch. And, in the same type of neural feedback loop as the arterial baroreceptor activation, chemoreceptor activation would also increase SNA and constrict the peripheral vasculature [122][123][124][125]. Thus, the complex vasoconstriction and vasodilation effects of ethanol consumption [80] can be explained when looking at the effects being driven by acetic acid and not ethanol per se.
7. Acetic Acid-Induced Changes in Neural Control of Cardiovascular Regulation
Acute oral consumption of ethanol consistently increases arterial blood pressure (ABP) and SNA in humans [72][76][77][78] and rodents [126][127][128]. Likewise, chronic ethanol use has been shown to increase norepinephrine, SNA, and ABP [98][129][130][131][132][133]. In one acute human study, dexamethasone was found to blunt ethanol’s sympathoexcitatory effect [77] through a reduction in corticotropin-releasing hormones (CRHs), suggesting that this response is at least partially mediated by central effects. In rodent studies, oral ethanol has been less consistent [79], with some studies showing an increase in SNA and ABP [130][134] and other studies showing decreases in ABP [95][97]. As some other reviews have noted, the timing of measurements in rodents may be important [24]. In one rodent study, Crandall et al. observed that ABP was normal at peak blood ethanol concentrations but was significantly elevated 24 h post ethanol dosing [135]. Interestingly, we now know from pharmacokinetic studies that this time point corresponds to when serum acetate is still elevated, typically remaining elevated for 12–24 h post ethanol metabolism [136][137]. As such, reasonable evidence exists that suggests acetate may be a lead compound in driving the ethanol-induced effects on cardiovascular function from a centrally mediated standpoint.
When attempting to elucidate the neuronal mechanisms of ethanol-induced changes in cardiovascular regulation, several brain regions that regulate autonomic function have been studied. The rostral ventrolateral medulla (RVLM) is a significant brain region involved in the integration of upstream brain regional sympathoexcitatory outputs [126][138][139][140][141][142][143][144][145]. The RVLM projects to the spinal intermediolateral column (IML), which is the first synapse in the central to peripheral sympathetic output pathway [145][146]. In direct application studies of the RVLM conducted by El-Mas and colleagues, ethanol microinjection dose-dependently increased RVLM norepinephrine and ABP in spontaneously hypertensive rats (SHRs) [128][147]. Additional follow-up studies by the same group suggested that this ethanol response was due to enhanced catabolism of ethanol to acetaldehyde [128]. They however indirectly speculated that acetate had little effect on increasing ABP levels [128]. Interestingly, when investigating catalase and ALDH enzyme activity in the RVLM between SHRs and non-hypertensive control (Wistar–Kyoto, WKY) rats, the SHRs had higher levels of catalase activity compared to the WKY rats, with no differences in ALDH activity, suggesting a potential genetic component in relation to differences in brain ethanol metabolism [128]. While this study described above did not detect differences in ALDH activity, a recent and exciting report has highlighted brain region differences in ALDH2 expression in mice [148]. This previous finding suggests that investigators must be cognizant of how brain regions and/or genetic differences in ethanol metabolism may influence behavior and/or cardiovascular function.
References
- Zakhari, S. Overview: How is alcohol metabolized by the body? Alcohol. Res. Health 2006, 29, 245–254.
- Chapp, A.D.; Nwakama, C.A.; Collins, A.R.; Mermelstein, P.G.; Thomas, M.J. Physiological acetic acid concentrations from ethanol metabolism stimulate accumbens shell medium spiny neurons via NMDAR activation in a sex-dependent manner. Neuropsychopharmacology 2023.
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial proton and electron leaks. Essays Biochem. 2010, 47, 53–67.
- Fernández-Checa, J.C.; Kaplowitz, N.; Colell, A.; García-Ruiz, C. Oxidative stress and alcoholic liver disease. Alcohol. Health Res. World 1997, 21, 321–324.
- Tan, H.K.; Yates, E.; Lilly, K.; Dhanda, A.D. Oxidative stress in alcohol-related liver disease. World J. Hepatol. 2020, 12, 332–349.
- Ambade, A.; Mandrekar, P. Oxidative Stress and Inflammation: Essential Partners in Alcoholic Liver Disease. Int. J. Hepatol. 2012, 2012, 853175.
- Cederbaum, A.I.; Lu, Y.; Wu, D. Role of oxidative stress in alcohol-induced liver injury. Arch. Toxicol. 2009, 83, 519–548.
- Kendrick, S.F.; O’Boyle, G.; Mann, J.; Zeybel, M.; Palmer, J.; Jones, D.E.; Day, C.P. Acetate, the key modulator of inflammatory responses in acute alcoholic hepatitis. Hepatology 2010, 51, 1988–1997.
- Comporti, M.; Signorini, C.; Leoncini, S.; Gardi, C.; Ciccoli, L.; Giardini, A.; Vecchio, D.; Arezzini, B. Ethanol-induced oxidative stress: Basic knowledge. Genes. Nutr. 2010, 5, 101–109.
- Sun, A.Y.; Sun, G.Y. Ethanol and oxidative mechanisms in the brain. J. Biomed. Sci. 2001, 8, 37–43.
- Crews, F.T. Alcohol-related neurodegeneration and recovery: Mechanisms from animal models. Alcohol. Res. Health 2008, 31, 377–388.
- Kamal, H.; Tan, G.C.; Ibrahim, S.F.; Shaikh, M.F.; Mohamed, I.N.; Mohamed, R.M.P.; Hamid, A.A.; Ugusman, A.; Kumar, J. Alcohol Use Disorder, Neurodegeneration, Alzheimer’s and Parkinson’s Disease: Interplay Between Oxidative Stress, Neuroimmune Response and Excitotoxicity. Front. Cell Neurosci. 2020, 14, 282.
- Crews, F.T.; Nixon, K. Mechanisms of Neurodegeneration and Regeneration in Alcoholism. Alcohol. Alcohol. 2008, 44, 115–127.
- Piano, M.R. Alcohol’s Effects on the Cardiovascular System. Alcohol. Res. 2017, 38, 219–241.
- Biddinger, K.J.; Emdin, C.A.; Haas, M.E.; Wang, M.; Hindy, G.; Ellinor, P.T.; Kathiresan, S.; Khera, A.V.; Aragam, K.G. Association of Habitual Alcohol Intake With Risk of Cardiovascular Disease. JAMA Netw. Open 2022, 5, e223849.
- Larsson, S.C.; Burgess, S.; Mason, A.M.; Michaëlsson, K. Alcohol Consumption and Cardiovascular Disease. Circ. Genom. Precis. Med. 2020, 13, e002814.
- Toma, A.; Paré, G.; Leong, D.P. Alcohol and Cardiovascular Disease: How Much is Too Much? Curr. Atheroscler. Rep. 2017, 19, 13.
- Sutanto, H.; Cluitmans, M.J.M.; Dobrev, D.; Volders, P.G.A.; Bébarová, M.; Heijman, J. Acute effects of alcohol on cardiac electrophysiology and arrhythmogenesis: Insights from multiscale in silico analyses. J. Mol. Cell Cardiol. 2020, 146, 69–83.
- Kupari, M.; Koskinen, P. Alcohol, cardiac arrhythmias and sudden death. Novartis Found. Symp. 1998, 216, 68–79; discussion 79–85.
- Mustroph, J.; Baier, M.J.; Unsin, D.; Provaznik, Z.; Kozakov, K.; Lebek, S.; Tarnowski, D.; Schildt, S.; Voigt, N.; Wagner, S.; et al. Ethanol-Induced Atrial Fibrillation Results From Late INa and Can Be Prevented by Ranolazine. Circulation 2023, 148, 698–700.
- Agarwal, D.P. Cardioprotective effects of light–moderate consumption of alcohol: A review of putative mechanisms. Alcohol. Alcohol. 2002, 37, 409–415.
- Chen, C.H.; Gray, M.O.; Mochly-Rosen, D. Cardioprotection from ischemia by a brief exposure to physiological levels of ethanol: Role of epsilon protein kinase C. Proc. Natl. Acad. Sci. USA 1999, 96, 12784–12789.
- Pearson, T.A. AHA Science Advisory. Alcohol and heart disease. Nutrition Committee of the American Heart Association. Am. J. Clin. Nutr. 1997, 65, 1567–1569.
- Husain, K.; Ansari, R.A.; Ferder, L. Alcohol-induced hypertension: Mechanism and prevention. World J. Cardiol. 2014, 6, 245–252.
- Beilin, L.J.; Puddey, I.B. Alcohol and Hypertension. Hypertension 2006, 47, 1035–1038.
- Tsuruta, M.; Adachi, H.; Hirai, Y.; Fujiura, Y.; Imaizumi, T. Association Between Alcohol Intake and Development of Hypertension in Japanese Normotensive Men: 12-Year Follow-Up Study. Am. J. Hypertens. 2000, 13, 482–487.
- Roerecke, M.; Kaczorowski, J.; Tobe, S.W.; Gmel, G.; Hasan, O.S.M.; Rehm, J. The effect of a reduction in alcohol consumption on blood pressure: A systematic review and meta-analysis. Lancet Public. Health 2017, 2, e108–e120.
- Federico, S.D.; Filippini, T.; Whelton, P.K.; Cecchini, M.; Iamandii, I.; Boriani, G.; Vinceti, M. Alcohol Intake and Blood Pressure Levels: A Dose-Response Meta-Analysis of Nonexperimental Cohort Studies. Hypertension 2023, 80, 1961–1969.
- Association, A.H. Even Just 1 Alcoholic Drink a Day May Increase Blood Pressure. Available online: https://www.heart.org/en/news/2023/07/31/even-just-1-alcoholic-drink-a-day-may-increase-blood-pressure (accessed on 19 December 2023).
- Zakhari, S. Alcohol metabolism and epigenetics changes. Alcohol. Res. 2013, 35, 6–16.
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 2015, 21, 805–821.
- Mews, P.; Egervari, G.; Nativio, R.; Sidoli, S.; Donahue, G.; Lombroso, S.I.; Alexander, D.C.; Riesche, S.L.; Heller, E.A.; Nestler, E.J.; et al. Alcohol metabolism contributes to brain histone acetylation. Nature 2019, 574, 717–721.
- Erny, D.; Dokalis, N.; Mezö, C.; Castoldi, A.; Mossad, O.; Staszewski, O.; Frosch, M.; Villa, M.; Fuchs, V.; Mayer, A.; et al. Microbiota-derived acetate enables the metabolic fitness of the brain innate immune system during health and disease. Cell Metab. 2021, 33, 2260–2276.e7.
- Colombo, A.V.; Sadler, R.K.; Llovera, G.; Singh, V.; Roth, S.; Heindl, S.; Sebastian Monasor, L.; Verhoeven, A.; Peters, F.; Parhizkar, S.; et al. Microbiota-derived short chain fatty acids modulate microglia and promote Aβ plaque deposition. eLife 2021, 10, e59826.
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480.e12.
- Perry, R.J.; Peng, L.; Barry, N.A.; Cline, G.W.; Zhang, D.; Cardone, R.L.; Petersen, K.F.; Kibbey, R.G.; Goodman, A.L.; Shulman, G.I. Acetate mediates a microbiome-brain-β-cell axis to promote metabolic syndrome. Nature 2016, 534, 213–217.
- Setshedi, M.; Wands, J.R.; Monte, S.M. Acetaldehyde adducts in alcoholic liver disease. Oxid. Med. Cell Longev. 2010, 3, 178–185.
- Tuma, D.J.; Hoffman, T.; Sorrell, M.F. The chemistry of acetaldehyde-protein adducts. Alcohol. Alcohol. Suppl. 1991, 1, 271–276.
- Rintala, J.; Jaatinen, P.; Parkkila, S.; Sarviharju, M.; Kiianmaa, K.; Hervonen, A.; Niemelä, O. Evidence of acetaldehyde–protein adduct formation in rat brain after lifelong consumption of ethanol. Alcohol. Alcohol. 2000, 35, 458–463.
- Ito, A.; Jamal, M.; Ameno, K.; Tanaka, N.; Takakura, A.; Kawamoto, T.; Kitagawa, K.; Nakayama, K.; Matsumoto, A.; Miki, T.; et al. Acetaldehyde administration induces salsolinol formation in vivo in the dorsal striatum of Aldh2-knockout and C57BL/6N mice. Neurosci. Lett. 2018, 685, 50–54.
- Peana, A.T.; Rosas, M.; Porru, S.; Acquas, E. From Ethanol to Salsolinol: Role of Ethanol Metabolites in the Effects of Ethanol. J. Exp. Neurosci. 2016, 10, 137–146.
- Bassareo, V.; Frau, R.; Maccioni, R.; Caboni, P.; Manis, C.; Peana, A.T.; Migheli, R.; Porru, S.; Acquas, E. Ethanol-Dependent Synthesis of Salsolinol in the Posterior Ventral Tegmental Area as Key Mechanism of Ethanol’s Action on Mesolimbic Dopamine. Front. Neurosci. 2021, 15, 675061.
- Correa, M.; Acquas, E.; Salamone, J.D. The renaissance of acetaldehyde as a psychoactive compound: Decades in the making. Front. Behav. Neurosci. 2014, 8, 249.
- Kuriyama, K.; Ohkuma, S.; Taguchi, J.-I.; Hashimoto, T. Alcohol, acetaldehyde and salsolinol-induced alterations in functions of cerebral GABA/benzodiazepine receptor complex. Physiol. Behav. 1987, 40, 393–399.
- Deehan Jr, G.A.; Engleman, E.A.; Ding, Z.M.; McBride, W.J.; Rodd, Z.A. Microinjections of acetaldehyde or salsolinol into the posterior ventral tegmental area increase dopamine release in the nucleus accumbens shell. Alcohol. Clin. Exp. Res. 2013, 37, 722–729.
- Jamal, M.; Ameno, K.; Ameno, S.; Okada, N.; Ijiri, I. In vivo study of salsolinol produced by a high concentration of acetaldehyde in the striatum and nucleus accumbens of free-moving rats. Alcohol. Clin. Exp. Res. 2003, 27, 79S–84S.
- Zuddas, A.; Corsini, G.U.; Schinelli, S.; Barker, J.L.; Kopin, I.J.; di Porzio, U. Acetaldehyde directly enhances MPP+ neurotoxicity and delays its elimination from the striatum. Brain Res. 1989, 501, 11–22.
- Cui, J.; Liu, Y.; Chang, X.; Gou, W.; Zhou, X.; Liu, Z.; Li, Z.; Wu, Y.; Zuo, D. Acetaldehyde Induces Neurotoxicity In Vitro via Oxidative Stress- and Ca(2+) Imbalance-Mediated Endoplasmic Reticulum Stress. Oxid. Med. Cell Longev. 2019, 2019, 2593742.
- Yan, T.; Zhao, Y. Acetaldehyde induces phosphorylation of dynamin-related protein 1 and mitochondrial dysfunction via elevating intracellular ROS and Ca2+ levels. Redox Biol. 2020, 28, 101381.
- Peana, A.T.; Sánchez-Catalán, M.J.; Hipólito, L.; Rosas, M.; Porru, S.; Bennardini, F.; Romualdi, P.; Caputi, F.F.; Candeletti, S.; Polache, A.; et al. Mystic Acetaldehyde: The Never-Ending Story on Alcoholism. Front. Behav. Neurosci. 2017, 11, 81.
- Matsumura, Y.; Stiles, K.M.; Reid, J.; Frenk, E.Z.; Cronin, S.; Pagovich, O.E.; Crystal, R.G. Gene Therapy Correction of Aldehyde Dehydrogenase 2 Deficiency. Mol. Ther. Methods Clin. Dev. 2019, 15, 72–82.
- Goldman, D. Aldehyde Dehydrogenase Deficiency as Cause of Facial Flushing Reaction to Alcohol in Japanese. Alcohol. Health Res. World 1995, 19, 48–49.
- Chen, C.H.; Kraemer, B.R.; Mochly-Rosen, D. ALDH2 variance in disease and populations. Dis. Model. Mech. 2022, 15, dmm049601.
- Ohsawa, I.; Kamino, K.; Nagasaka, K.; Ando, F.; Niino, N.; Shimokata, H.; Ohta, S. Genetic deficiency of a mitochondrial aldehyde dehydrogenase increases serum lipid peroxides in community-dwelling females. J. Hum. Genet. 2003, 48, 404–409.
- Stokes, M.; Abdijadid, S. Disulfiram. In StatPearls; StatPearls Publishing: St. Petersburg, FL, USA, 2023.
- Rosman, A.S.; Waraich, A.; Baraona, E.; Lieber, C.S. Disulfiram treatment increases plasma and red blood cell acetaldehyde in abstinent alcoholics. Alcohol. Clin. Exp. Res. 2000, 24, 958–964.
- Kleczkowska, P.; Sulejczak, D.; Zaremba, M. Advantages and disadvantages of disulfiram coadministered with popular addictive substances. Eur. J. Pharmacol. 2021, 904, 174143.
- Eriksson, C.J. Measurement of acetaldehyde: What levels occur naturally and in response to alcohol? Novartis Found. Symp. 2007, 285, 247–255; discussion 256–260.
- Lindros, K.O. Human blood acetaldehyde levels: With improved methods, a clearer picture emerges. Alcohol. Clin. Exp. Res. 1983, 7, 70–75.
- Lide, D.R. (Ed.) CRC Handbook of Chemistry and Physics, 78th ed.; CRC Press: Boca Raton, FL, USA, 1997; p. 3.
- Bowen, E.J.; Tietz, E.L. The Oxidation of Acetaldehyde by Oxygen. Nature 1929, 124, 914.
- Bawn, C.E.H.; Williamson, J.B. The oxidation of acetaldehyde in solution. Part I.—The chemistry of the intermediate stages. Trans. Faraday Soc. 1951, 47, 721–734.
- Sánchez, M.; Sabio, L.; Gálvez, N.; Capdevila, M.; Dominguez-Vera, J.M. Iron chemistry at the service of life. IUBMB Life 2017, 69, 382–388.
- Koivunen, J.; Heinonen-Tanski, H. Peracetic acid (PAA) disinfection of primary, secondary and tertiary treated municipal wastewaters. Water Res. 2005, 39, 4445–4453.
- Viola, K.S.; Rodrigues, E.M.; Tanomaru-Filho, M.; Carlos, I.Z.; Ramos, S.G.; Guerreiro-Tanomaru, J.M.; Faria, G. Cytotoxicity of peracetic acid: Evaluation of effects on metabolism, structure and cell death. Int. Endod. J. 2018, 51, e264–e277.
- Turrà, N.; Neuenschwander, U.; Hermans, I. Molecule-induced peroxide homolysis. Chemphyschem 2013, 14, 1666–1669.
- Hiatt, R.R.; Irwin, K.C.; Gould, C.W. Homolytic decompositions of hydroperoxides. IV. Metal-catalyzed decompositions. J. Org. Chem. 1968, 33, 1430–1435.
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74.
- Machlin, L.J.; Bendich, A. Free radical tissue damage: Protective role of antioxidant nutrients. Faseb J. 1987, 1, 441–445.
- Wu, M.-L.; Tsai, K.-L.; Wang, S.-M.; Wu, J.-C.; Wang, B.-S.; Lee, Y.-T. Mechanism of Hydrogen Peroxide and Hydroxyl Free Radical–Induced Intracellular Acidification in Cultured Rat Cardiac Myoblasts. Circ. Res. 1996, 78, 564–572.
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82–83, 969–974.
- van de Borne, P.; Mark, A.L.; Montano, N.; Mion, D.; Somers, V.K. Effects of alcohol on sympathetic activity, hemodynamics, and chemoreflex sensitivity. Hypertension 1997, 29, 1278–1283.
- Morvai, V.; Nádházi, Z.; Molnár, G.Y.; Ungváry, G.Y.; Folly, G. Acute effects of low doses of alcohol on the cardiovascular system in young men. Acta Med. Hung. 1988, 45, 339–348.
- Sagawa, Y.; Kondo, H.; Matsubuchi, N.; Takemura, T.; Kanayama, H.; Kaneko, Y.; Kanbayashi, T.; Hishikawa, Y.; Shimizu, T. Alcohol has a dose-related effect on parasympathetic nerve activity during sleep. Alcohol. Clin. Exp. Res. 2011, 35, 2093–2100.
- Julian, T.H.; Syeed, R.; Glascow, N.; Zis, P. Alcohol-induced autonomic dysfunction: A systematic review. Clin. Auton. Res. 2020, 30, 29–41.
- Greenlund, I.M.; Cunningham, H.A.; Tikkanen, A.L.; Bigalke, J.A.; Smoot, C.A.; Durocher, J.J.; Carter, J.R. Morning sympathetic activity after evening binge alcohol consumption. Am. J. Physiol.-Heart Circ. Physiol. 2021, 320, H305–H315.
- Randin, D.; Vollenweider, P.; Tappy, L.; Jéquier, E.; Nicod, P.; Scherrer, U. Suppression of Alcohol-Induced Hypertension by Dexamethasone. N. Engl. J. Med. 1995, 332, 1733–1738.
- Carter, J.R.; Stream, S.F.; Durocher, J.J.; Larson, R.A. Influence of acute alcohol ingestion on sympathetic neural responses to orthostatic stress in humans. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E771–E778.
- Kawano, Y. Physio-pathological effects of alcohol on the cardiovascular system: Its role in hypertension and cardiovascular disease. Hypertens. Res. 2010, 33, 181–191.
- Tawakol, A.; Omland, T.; Creager, M.A. Direct effect of ethanol on human vascular function. Am. J. Physiol.-Heart Circ. Physiol. 2004, 286, H2468–H2473.
- Rezvani, A.H.; Grady, D.R.; Peek, A.E.; Pucilowski, O. Inhibition of nitric oxide synthesis attenuates alcohol consumption in two strains of alcohol-preferring rats. Pharmacol. Biochem. Behav. 1995, 50, 265–270.
- Baraona, E.; Zeballos, G.A.; Shoichet, L.; Mak, K.M.; Lieber, C.S. Ethanol Consumption Increases Nitric Oxide Production in Rats, and Its Peroxynitrite-Mediated Toxicity Is Attenuated by Polyenylphosphatidylcholine. Alcohol. Clin. Exp. Res. 2002, 26, 883–889.
- Hakim, R.M.; Pontzer, M.A.; Tilton, D.; Lazarus, J.M.; Gottlieb, M.N. Effects of acetate and bicarbonate dialysate in stable chronic dialysis patients. Kidney Int. 1985, 28, 535–540.
- Leunissen, K.M.L.; Hoorntje, S.J.; Fiers, H.A.; Dekkers, W.T.; Mulder, A.W. Acetate versus Bicarbonate Hemodialysis in Critically Ill Patients. Nephron 2008, 42, 146–151.
- Keshaviah, P.R. The role of acetate in the etiology of symptomatic hypotension. Artif. Organs 1982, 6, 378–387.
- Vinay, P.; Cardoso, M.; Tejedor, A.; Prud’homme, M.; Levelillee, M.; Vinet, B.; Courteau, M.; Gougoux, A.; Rengel, M.; Lapierre, L.; et al. Acetate metabolism during hemodialysis: Metabolic considerations. Am. J. Nephrol. 1987, 7, 337–354.
- Keshaviah, P.; Shapiro, F.L. A critical examination of dialysis-induced hypotension. Am. J. Kidney Dis. 1982, 2, 290–301.
- Noris, M.; Todeschini, M.; Casiraghi, F.; Roccatello, D.; Martina, G.; Minetti, L.; Imberti, B.; Gaspari, F.; Atti, M.; Remuzzi, G. Effect of acetate, bicarbonate dialysis, and acetate-free biofiltration on nitric oxide synthesis: Implications for dialysis hypotension. Am. J. Kidney Dis. 1998, 32, 115–124.
- Amore, A.; Cirina, P.; Mitola, S.; Peruzzi, L.; Bonaudo, R.; Gianoglio, B.; Coppo, R. Acetate intolerance is mediated by enhanced synthesis of nitric oxide by endothelial cells. J. Am. Soc. Nephrol. 1997, 8, 1431–1436.
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376.
- Moncada, S.; Higgs, E.A. The discovery of nitric oxide and its role in vascular biology. Br. J. Pharmacol. 2006, 147 (Suppl. S1), S193–S201.
- Moncada, S.; Palmer, R.M.; Higgs, E.A. Biosynthesis of nitric oxide from L-arginine. A pathway for the regulation of cell function and communication. Biochem. Pharmacol. 1989, 38, 1709–1715.
- Sakakibara, S.; Murakami, R.; Takahashi, M.; Fushimi, T.; Murohara, T.; Kishi, M.; Kajimoto, Y.; Kitakaze, M.; Kaga, T. Vinegar intake enhances flow-mediated vasodilatation via upregulation of endothelial nitric oxide synthase activity. Biosci. Biotechnol. Biochem. 2010, 74, 1055–1061.
- Riff, D.P.; Jain, A.C.; Doyle, J.T. Acute hemodynamic effects of ethanol on normal human volunteers. Am. Heart J. 1969, 78, 592–597.
- Puddey, I.B.; Vandongen, R.; Beilin, L.J.; Rouse, I.L. Alcohol stimulation of renin release in man: Its relation to the hemodynamic, electrolyte, and sympatho-adrenal responses to drinking. J. Clin. Endocrinol. Metab. 1985, 61, 37–42.
- Jain, A.; Yelamanchili, V.S.; Brown, K.N.; Goel, A. Holiday Heart Syndrome. In StatPearls; StatPearls Publishing: St. Petersburg, FL, USA, 2023.
- Kupari, M. Acute cardiovascular effects of ethanol A controlled non-invasive study. Br. Heart J. 1983, 49, 174–182.
- Iwase, S.; Matsukawa, T.; Ishihara, S.; Tanaka, A.; Tanabe, K.; Danbara, A.; Matsuo, M.; Sugiyama, Y.; Mano, T. Effect of oral ethanol intake on muscle sympathetic nerve activity and cardiovascular functions in humans. J. Auton. Nerv. Syst. 1995, 54, 206–214.
- Delbridge, L.M.; Connell, P.J.; Harris, P.J.; Morgan, T.O. Ethanol effects on cardiomyocyte contractility. Clin. Sci. 2000, 98, 401–407.
- Ahmed, S.S.; Levinson, G.E.; Regan, T.J. Depression of myocardial contractility with low doses of ethanol in normal man. Circulation 1973, 48, 378–385.
- Fernández-Solà, J. The Effects of Ethanol on the Heart: Alcoholic Cardiomyopathy. Nutrients 2020, 12, 572.
- Figueredo, V.M.; Chang, K.C.; Baker, A.J.; Camacho, S.A. Chronic alcohol-induced changes in cardiac contractility are not due to changes in the cytosolic Ca2+ transient. Am. J. Physiol. 1998, 275, H122–H130.
- Kiviluoma, K.T.; Karhunen, M.; Lapinlampi, T.; Peuhkurinen, K.J.; Hassinen, I.E. Acetate-induced changes in cardiac energy metabolism and hemodynamics in the rat. Basic. Res. Cardiol. 1988, 83, 431–444.
- Suokas, A.; Kupari, M.; Heikkilä, J.; Lindros, K.; Ylikahri, R. Acute Cardiovascular and Metabolic Effects of Acetate in Men. Alcohol. Clin. Exp. Res. 1988, 12, 52–58.
- Jiang, X.; Zhang, Y.; Zhang, H.; Zhang, X.; Yin, X.; Yuan, F.; Wang, S.; Tian, Y. Acetate suppresses myocardial contraction via the short-chain fatty acid receptor GPR43. Front. Physiol. 2022, 13, 1111156.
- Poll, B.G.; Xu, J.; Jun, S.; Sanchez, J.; Zaidman, N.A.; He, X.; Lester, L.; Berkowitz, D.E.; Paolocci, N.; Gao, W.D.; et al. Acetate, a Short-Chain Fatty Acid, Acutely Lowers Heart Rate and Cardiac Contractility Along with Blood Pressure. J. Pharmacol. Exp. Ther. 2021, 377, 39–50.
- Massion, P.B.; Feron, O.; Dessy, C.; Balligand, J.-L. Nitric Oxide and Cardiac Function. Circ. Res. 2003, 93, 388–398.
- Finkel, M.S.; Oddis, C.V.; Jacob, T.D.; Watkins, S.C.; Hattler, B.G.; Simmons, R.L. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science 1992, 257, 387–389.
- Shaaban, A.; Gangwani, M.K.; Pendela, V.S.; Vindhyal, M.R. Alcoholic Cardiomyopathy. In StatPearls; StatPearls Publishing: St. Petersburg, FL, USA, 2023.
- Andersson, C.; Schou, M.; Gustafsson, F.; Torp-Pedersen, C. Alcohol Intake in Patients With Cardiomyopathy and Heart Failure: Consensus and Controversy. Circ. Heart Fail. 2022, 15, e009459.
- Riddick, J.A.; Bunger, W.B.; Sakano, T.K. Organic Solvents: Physical Properties and Methods of Purification, 4th ed.; Wiley: Hoboken, NJ, USA, 1986.
- Serjeant, E.P.; Dempsey, B. Ionisation constants of organic acids in aqueous solution. IUPAC Chem. Data Ser. 1979, 23, 160–190.
- Bjarnsholt, T.; Alhede, M.; Jensen, P.; Nielsen, A.K.; Johansen, H.K.; Homøe, P.; Høiby, N.; Givskov, M.; Kirketerp-Møller, K. Antibiofilm Properties of Acetic Acid. Adv. Wound Care 2015, 4, 363–372.
- Koob, G.F.; Arends, M.A.; Le Moal, M. Chapter 6—Alcohol. In Drugs, Addiction, and the Brain; Koob, G.F., Arends, M.A., Le Moal, M., Eds.; Academic Press: San Diego, CA, USA, 2014; pp. 173–219.
- Gemma, S.; Vichi, S.; Testai, E. Individual susceptibility and alcohol effects:biochemical and genetic aspects. Ann. Ist. Super. Sanita 2006, 42, 8–16.
- Raphael, K.L.; Murphy, R.A.; Shlipak, M.G.; Satterfield, S.; Huston, H.K.; Sebastian, A.; Sellmeyer, D.E.; Patel, K.V.; Newman, A.B.; Sarnak, M.J.; et al. Bicarbonate Concentration, Acid-Base Status, and Mortality in the Health, Aging, and Body Composition Study. Clin. J. Am. Soc. Nephrol. 2016, 11, 308–316.
- Burger, M.; Schaller, D.J. Metabolic Acidosis. In StatPearls; StatPearls Publishing: St. Petersburg, FL, USA, 2023.
- Hopkins, E.; Sanvictores, T.; Sharma, S. Physiology, Acid Base Balance. In StatPearls; StatPearls Publishing: St. Petersburg, FL, USA, 2023.
- Raphael, K.L. Metabolic Acidosis in CKD: Core Curriculum 2019. Am. J. Kidney Dis. 2019, 74, 263–275.
- Armstrong, M.; Kerndt, C.C.; Moore, R.A. Physiology, Baroreceptors. In StatPearls; StatPearls Publishing: St. Petersburg, FL, USA, 2023.
- Suarez-Roca, H.; Mamoun, N.; Sigurdson, M.I.; Maixner, W. Baroreceptor Modulation of the Cardiovascular System, Pain, Consciousness, and Cognition. Compr. Physiol. 2021, 11, 1373–1423.
- Caverson, M.M.; Ciriello, J.; Calaresu, F.R. Chemoreceptor and baroreceptor inputs to ventrolateral medullary neurons. Am. J. Physiol. 1984, 247, R872–R879.
- Cooper, V.L.; Pearson, S.B.; Bowker, C.M.; Elliott, M.W.; Hainsworth, R. Interaction of chemoreceptor and baroreceptor reflexes by hypoxia and hypercapnia—A mechanism for promoting hypertension in obstructive sleep apnoea. J. Physiol. 2005, 568, 677–687.
- Guyenet, P.G.; Bayliss, D.A. Neural Control of Breathing and CO2 Homeostasis. Neuron 2015, 87, 946–961.
- Toledo, C.; Andrade, D.C.; Lucero, C.; Schultz, H.D.; Marcus, N.; Retamal, M.; Madrid, C.; Del Rio, R. Contribution of peripheral and central chemoreceptors to sympatho-excitation in heart failure. J. Physiol. 2017, 595, 43–51.
- Chapp, A.D.; Gui, L.; Huber, M.J.; Liu, J.; Larson, R.A.; Zhu, J.; Carter, J.R.; Chen, Q.H. Sympathoexcitation and pressor responses induced by ethanol in the central nucleus of amygdala involves activation of NMDA receptors in rats. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H701–H709.
- Brizzolara, A.L.; Morris, D.G.; Burnstock, G. Ethanol affects sympathetic cotransmission and endothelium-dependent relaxation in the rat. Eur. J. Pharmacol. 1994, 254, 175–181.
- El-Mas, M.M.; Abdel-Rahman, A.A. Enhanced catabolism to acetaldehyde in rostral ventrolateral medullary neurons accounts for the pressor effect of ethanol in spontaneously hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H837–H844.
- Chan, T.C.; Wall, R.A.; Sutter, M.C. Chronic ethanol consumption, stress, and hypertension. Hypertension 1985, 7, 519–524.
- Grassi, G.M.; Somers, V.K.; Renk, W.S.; Abboud, F.M.; Mark, A.L. Effects of alcohol intake on blood pressure and sympathetic nerve activity in normotensive humans: A preliminary report. J. Hypertens. Suppl. 1989, 7, S20–S21.
- Hering, D.; Kucharska, W.; Kara, T.; Somers, V.K.; Narkiewicz, K. Potentiated sympathetic and hemodynamic responses to alcohol in hypertensive vs. normotensive individuals. J. Hypertens. 2011, 29, 537–541.
- Resstel, L.B.; Scopinho, A.A.; Lopes da Silva, A.; Antunes-Rodrigues, J.; Corrêa, F.M. Increased circulating vasopressin may account for ethanol-induced hypertension in rats. Am. J. Hypertens. 2008, 21, 930–935.
- Tirapelli, C.R.; Leone, A.F.; Coelho, E.B.; Resstel, L.B.; Corrêa, F.M.; Lanchote, V.L.; Uyemura, S.A.; Padovan, C.M.; de Oliveira, A.M. Effect of ethanol consumption on blood pressure and rat mesenteric arterial bed, aorta and carotid responsiveness. J. Pharm. Pharmacol. 2007, 59, 985–993.
- Ireland, M.A.; Vandongen, R.; Davidson, L.; Beilin, L.J.; Rouse, I.L. Acute effects of moderate alcohol consumption on blood pressure and plasma catecholamines. Clin. Sci. 1984, 66, 643–648.
- Crandall, D.L.; Ferraro, G.D.; Lozito, R.J.; Cervoni, P.; Clark, L.T. Cardiovascular effects of intermittent drinking: Assessment of a novel animal model of human alcoholism. J. Hypertens. 1989, 7, 683–687.
- Pronko, P.S.; Velichko, M.G.; Moroz, A.R.; Rubanovich, N.N. Low-molecular-weight metabolites relevant to ethanol metabolism: Correlation with alcohol withdrawal severity and utility for identification of alcoholics. Alcohol. Alcohol. 1997, 32, 761–768.
- Jiang, L.; Gulanski, B.I.; De Feyter, H.M.; Weinzimer, S.A.; Pittman, B.; Guidone, E.; Koretski, J.; Harman, S.; Petrakis, I.L.; Krystal, J.H.; et al. Increased brain uptake and oxidation of acetate in heavy drinkers. J. Clin. Investig. 2013, 123, 1605–1614.
- Chapp, A.D.; Collins, A.R.; Driscoll, K.M.; Behnke, J.E.; Shan, Z.; Zhang, L.; Chen, Q.-H. Ethanol Metabolite, Acetate, Increases Excitability of the Central Nucleus of Amygdala Neurons through Activation of NMDA Receptors. ACS Chem. Neurosci. 2023, 14, 1278–1290.
- Chapp, A.D.; Wang, R.; Cheng, Z.J.; Shan, Z.; Chen, Q.H. Long-Term High Salt Intake Involves Reduced SK Currents and Increased Excitability of PVN Neurons with Projections to the Rostral Ventrolateral Medulla in Rats. Neural Plast. 2017, 2017, 7282834.
- Larson, R.A.; Chapp, A.D.; Gui, L.; Huber, M.J.; Cheng, Z.J.; Shan, Z.; Chen, Q.-H. High Salt Intake Augments Excitability of PVN Neurons in Rats: Role of the Endoplasmic Reticulum Ca2+ Store. Front. Neurosci. 2017, 11, 182.
- Larson, R.A.; Gui, L.; Huber, M.J.; Chapp, A.D.; Zhu, J.; LaGrange, L.P.; Shan, Z.; Chen, Q.H. Sympathoexcitation in ANG II-salt hypertension involves reduced SK channel function in the hypothalamic paraventricular nucleus. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H1547–H1555.
- Chen, Q.H.; Haywood, J.R.; Toney, G.M. Sympathoexcitation by PVN-injected bicuculline requires activation of excitatory amino acid receptors. Hypertension 2003, 42, 725–731.
- Chen, Q.H.; Toney, G.M. Excitability of paraventricular nucleus neurones that project to the rostral ventrolateral medulla is regulated by small-conductance Ca2+-activated K+ channels. J. Physiol. 2009, 587, 4235–4247.
- Chen, Q.H.; Toney, G.M. In vivo discharge properties of hypothalamic paraventricular nucleus neurons with axonal projections to the rostral ventrolateral medulla. J. Neurophysiol. 2010, 103, 4–15.
- Adams, J.M.; McCarthy, J.J.; Stocker, S.D. Excess dietary salt alters angiotensinergic regulation of neurons in the rostral ventrolateral medulla. Hypertension 2008, 52, 932–937.
- Kumagai, H.; Oshima, N.; Matsuura, T.; Iigaya, K.; Imai, M.; Onimaru, H.; Sakata, K.; Osaka, M.; Onami, T.; Takimoto, C.; et al. Importance of rostral ventrolateral medulla neurons in determining efferent sympathetic nerve activity and blood pressure. Hypertens. Res. 2012, 35, 132–141.
- El-Mas, M.M.; Fan, M.; Abdel-Rahman, A.A. Role of rostral ventrolateral medullary ERK/JNK/p38 MAPK signaling in the pressor effects of ethanol and its oxidative product acetaldehyde. Alcohol. Clin. Exp. Res. 2013, 37, 1827–1837.
- Jin, S.; Cao, Q.; Yang, F.; Zhu, H.; Xu, S.; Chen, Q.; Wang, Z.; Lin, Y.; Cinar, R.; Pawlosky, R.J.; et al. Brain ethanol metabolism by astrocytic ALDH2 drives the behavioural effects of ethanol intoxication. Nat. Metab. 2021, 3, 337–351.
More
Information
Subjects:
Physiology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
684
Revisions:
2 times
(View History)
Update Date:
19 Feb 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No