 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Claudio Graziano | -- | 2205 | 2024-02-07 14:59:18 | | | |
| 2 | Jason Zhu | Meta information modification | 2205 | 2024-02-08 03:01:38 | | |
Video Upload Options
SOX proteins are transcription factors which play a role in regulating the development of progenitor cells and tissue differentiation. Twenty members are known, clustered in eight groups named A through H and sharing a common DNA-binding domain called the HMG (high-mobility-group) box. Eleven of the SOX genes have been associated with genetic disorders so far, covering a broad spectrum of developmental diseases. SOX4 is a single-exon gene and belongs to the SOXC group, together with SOX11 and SOX12. SOX4 variants have been recently described to cause a highly penetrant but heterogeneous disorder, with a phenotypic spectrum ranging from mild developmental delays and learning difficulties to intellectual disabilities with congenital anomalies. Nineteen pathogenic variants have been reported to date, generally de novo, heterozygous, and inactivating, either stop–gain or missense, the latter ones primarily targeting the HMG domain.
1. Introduction
2. SOX4 Structure and Function
3. SOX4 Single Nucleotide Variants
3.1. Heterozygous Pathogenic Variants
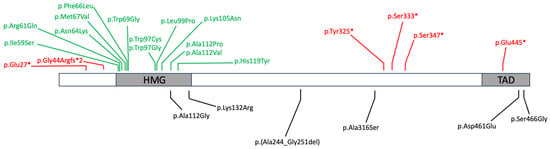
3.2. Variants of Uncertain Significance and Possible Bi-Allelic Inheritance
3.3. Co-Occurrence of Variants in Other Genes
References
- Schepers, G.E.; Teasdale, R.D.; Koopman, P. Twenty Pairs of Sox: Extent, Homology, and Nomenclature of the Mouse and Human Sox Transcription Factor Gene Families. Dev. Cell 2002, 3, 167–170.
- Harley, V.R.; Clarkson, M.J.; Argentaro, A. The Molecular Action and Regulation of the Testis-Determining Factors, SRY (Sex-Determining Region on the Y Chromosome) and SOX9 . Endocr. Rev. 2003, 24, 466–487.
- Sutton, E.; Hughes, J.; White, S.; Sekido, R.; Tan, J.; Arboleda, V.; Rogers, N.; Knower, K.; Rowley, L.; Eyre, H.; et al. Identification of SOX3 as an XX Male Sex Reversal Gene in Mice and Humans. J. Clin. Investig. 2011, 121, 328–341.
- Fantes, J.; Ragge, N.K.; Lynch, S.-A.; McGill, N.I.; Collin, J.R.O.; Howard-Peebles, P.N.; Hayward, C.; Vivian, A.J.; Williamson, K.; van Heyningen, V.; et al. Mutations in SOX2 Cause Anophthalmia. Nat. Genet. 2003, 33, 462–463.
- Wagner, T.; Wirth, J.; Meyer, J.; Zabel, B.; Held, M.; Zimmer, J.; Pasantes, J.; Bricarelli, F.D.; Keutel, J.; Hustert, E.; et al. Autosomal Sex Reversal and Campomelic Dysplasia Are Caused by Mutations in and around the SRY-Related Gene SOX9. Cell 1994, 79, 1111–1120.
- Lamb, A.N.; Rosenfeld, J.A.; Neill, N.J.; Talkowski, M.E.; Blumenthal, I.; Girirajan, S.; Keelean-Fuller, D.; Fan, Z.; Pouncey, J.; Stevens, C.; et al. Haploinsufficiency of SOX5 at 12p12.1 Is Associated with Developmental Delays with Prominent Language Delay, Behavior Problems, and Mild Dysmorphic Features. Hum. Mutat. 2012, 33, 728–740.
- Pingault, V.; Zerad, L.; Bertani-Torres, W.; Bondurand, N. SOX10: 20 Years of Phenotypic Plurality and Current Understanding of Its Developmental Function. J. Med. Genet. 2022, 59, 105–114.
- Angelozzi, M.; Lefebvre, V. SOXopathies: Growing Family of Developmental Disorders Due to SOX Mutations. Trends Genet. 2019, 35, 658–671.
- Kavyanifar, A.; Turan, S.; Lie, D.C. SoxC Transcription Factors: Multifunctional Regulators of Neurodevelopment. Cell Tissue Res. 2018, 371, 91–103.
- Schilham, M.W.; Oosterwegel, M.A.; Moerer, P.; Ya, J.; de Boer, P.A.J.; van de Wetering, M.; Verbeek, S.; Lamers, W.H.; Kruisbeek, A.M.; Cumano, A.; et al. Defects in Cardiac Outflow Tract Formation and Pro-B-Lymphocyte Expansion in Mice Lacking Sox-4. Nature 1996, 380, 711–714.
- Sock, E.; Rettig, S.D.; Enderich, J.; Bösl, M.R.; Tamm, E.R.; Wegner, M. Gene Targeting Reveals a Widespread Role for the High-Mobility-Group Transcription Factor Sox11 in Tissue Remodeling. Mol. Cell. Biol. 2004, 24, 6635–6644.
- Hoser, M.; Potzner, M.R.; Koch, J.M.C.; Bösl, M.R.; Wegner, M.; Sock, E. Sox12 Deletion in the Mouse Reveals Nonreciprocal Redundancy with the Related Sox4 and Sox11 Transcription Factors. Mol. Cell. Biol. 2008, 28, 4675–4687.
- Tsurusaki, Y.; Koshimizu, E.; Ohashi, H.; Phadke, S.; Kou, I.; Shiina, M.; Suzuki, T.; Okamoto, N.; Imamura, S.; Yamashita, M.; et al. De Novo SOX11 Mutations Cause Coffin–Siris Syndrome. Nat. Commun. 2014, 5, 4011.
- Hempel, A.; Pagnamenta, A.T.; Blyth, M.; Mansour, S.; McConnell, V.; Kou, I.; Ikegawa, S.; Tsurusaki, Y.; Matsumoto, N.; Lo-Castro, A.; et al. Deletions and de Novo Mutations of SOX11 Are Associated with a Neurodevelopmental Disorder with Features of Coffin–Siris Syndrome. J. Med. Genet. 2016, 53, 152–162.
- Al-Jawahiri, R.; Foroutan, A.; Kerkhof, J.; McConkey, H.; Levy, M.; Haghshenas, S.; Rooney, K.; Turner, J.; Shears, D.; Holder, M.; et al. SOX11 Variants Cause a Neurodevelopmental Disorder with Infrequent Ocular Malformations and Hypogonadotropic Hypogonadism and with Distinct DNA Methylation Profile. Genet. Med. 2022, 24, 1261–1273.
- Van de Wetering, M.; Oosterwegel, M.; van Norren, K.; Clevers, H. Sox-4, an Sry-like HMG Box Protein, Is a Transcriptional Activator in Lymphocytes. EMBO J. 1993, 12, 3847–3854.
- Bhattaram, P.; Penzo-Méndez, A.; Sock, E.; Colmenares, C.; Kaneko, K.J.; Vassilev, A.; DePamphilis, M.L.; Wegner, M.; Lefebvre, V. Organogenesis Relies on SoxC Transcription Factors for the Survival of Neural and Mesenchymal Progenitors. Nat. Commun. 2010, 1, 9.
- Shim, S.; Kwan, K.Y.; Li, M.; Lefebvre, V.; Šestan, N. Cis-Regulatory Control of Corticospinal System Development and Evolution. Nature 2012, 486, 74–79.
- Bhattaram, P.; Penzo-Méndez, A.; Kato, K.; Bandyopadhyay, K.; Gadi, A.; Taketo, M.M.; Lefebvre, V. SOXC Proteins Amplify Canonical WNT Signaling to Secure Nonchondrocytic Fates in Skeletogenesis. J. Cell Biol. 2014, 207, 657–671.
- Sun, B.; Mallampati, S.; Gong, Y.; Wang, D.; Lefebvre, V.; Sun, X. Sox4 Is Required for the Survival of Pro-B Cells. J. Immunol. 2013, 190, 2080–2089.
- Wang, X.; Llamas, J.; Trecek, T.; Shi, T.; Tao, L.; Makmura, W.; Crump, J.G.; Segil, N.; Gnedeva, K. SoxC Transcription Factors Shape the Epigenetic Landscape to Establish Competence for Sensory Differentiation in the Mammalian Organ of Corti. Proc. Natl. Acad. Sci. USA 2023, 120, e2301301120.
- Scharer, C.D.; McCabe, C.D.; Ali-Seyed, M.; Berger, M.F.; Bulyk, M.L.; Moreno, C.S. Genome-Wide Promoter Analysis of the SOX4 Transcriptional Network in Prostate Cancer Cells. Cancer Res. 2009, 69, 709–717.
- Zawerton, A.; Yao, B.; Yeager, J.P.; Pippucci, T.; Haseeb, A.; Smith, J.D.; Wischmann, L.; Kühl, S.J.; Dean, J.C.S.; Pilz, D.T.; et al. De Novo SOX4 Variants Cause a Neurodevelopmental Disease Associated with Mild Dysmorphism. Am. J. Hum. Genet. 2019, 104, 246–259.
- Angelozzi, M.; Karvande, A.; Molin, A.N.; Ritter, A.L.; Leonard, J.M.M.; Savatt, J.M.; Douglass, K.; Myers, S.M.; Grippa, M.; Tolchin, D.; et al. Consolidation of the Clinical and Genetic Definition of a SOX4-Related Neurodevelopmental Syndrome. J. Med. Genet. 2022, 59, 1058–1068.
- Grosse, M.; Kuechler, A.; Dabir, T.; Spranger, S.; Beck-Wödl, S.; Bertrand, M.; Haack, T.B.; Grasemann, C.; Manka, E.; Depienne, C.; et al. Novel Variants of SOX4 in Patients with Intellectual Disability. Int. J. Mol. Sci. 2023, 24, 3519.
- Ghaffar, A.; Rasheed, F.; Rashid, M.; van Bokhoven, H.; Ahmed, Z.M.; Riazuddin, S.; Riazuddin, S. Biallelic In-Frame Deletion of SOX4 Is Associated with Developmental Delay, Hypotonia and Intellectual Disability. Eur. J. Hum. Genet. 2022, 30, 243–247.
- Stevenson, R.E.; Vincent, V.; Spellicy, C.J.; Friez, M.J.; Chaubey, A. Biallelic Deletions of the Waardenburg II Syndrome Gene, SOX10, Cause a Recognizable Arthrogryposis Syndrome. Am. J. Med. Genet. Part A 2018, 176, 1968–1971.
- Irrthum, A.; Devriendt, K.; Chitayat, D.; Matthijs, G.; Glade, C.; Steijlen, P.M.; Fryns, J.-P.; Van Steensel, M.A.M.; Vikkula, M. Mutations in the Transcription Factor Gene SOX18 Underlie Recessive and Dominant Forms of Hypotrichosis-Lymphedema-Telangiectasia. Am. J. Hum. Genet. 2003, 72, 1470–1478.
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Coban Akdemir, Z.H.; Walkiewicz, M.; Bi, W.; Xiao, R.; Ding, Y.; et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N. Engl. J. Med. 2017, 376, 21–31.
- Smith, E.D.; Blanco, K.; Sajan, S.A.; Hunter, J.M.; Shinde, D.N.; Wayburn, B.; Rossi, M.; Huang, J.; Stevens, C.A.; Muss, C.; et al. A Retrospective Review of Multiple Findings in Diagnostic Exome Sequencing: Half Are Distinct and Half Are Overlapping Diagnoses. Genet. Med. 2019, 21, 2199–2207.
- Sobering, A.K.; Bryant, L.M.; Li, D.; McGaughran, J.; Maystadt, I.; Moortgat, S.; Graham, J.M.; van Haeringen, A.; Ruivenkamp, C.; Cuperus, R.; et al. Variants in PHF8 Cause a Spectrum of X-Linked Neurodevelopmental Disorders and Facial Dysmorphology. HGG Adv. 2022, 3, 100102.
- Weiss, K.; Terhal, P.A.; Cohen, L.; Bruccoleri, M.; Irving, M.; Martinez, A.F.; Rosenfeld, J.A.; Machol, K.; Yang, Y.; Liu, P.; et al. De Novo Mutations in CHD4, an ATP-Dependent Chromatin Remodeler Gene, Cause an Intellectual Disability Syndrome with Distinctive Dysmorphisms. Am. J. Hum. Genet. 2016, 99, 934–941.


