Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | William Stone | -- | 2801 | 2024-02-06 14:36:01 | | | |
| 2 | Lindsay Dong | + 4 word(s) | 2805 | 2024-02-07 02:14:12 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Tuell, D.; Ford, G.; Los, E.; Stone, W. Glutathione and Its Precursors in Type 2 Diabetes. Encyclopedia. Available online: https://encyclopedia.pub/entry/54803 (accessed on 23 July 2026).
Tuell D, Ford G, Los E, Stone W. Glutathione and Its Precursors in Type 2 Diabetes. Encyclopedia. Available at: https://encyclopedia.pub/entry/54803. Accessed July 23, 2026.
Tuell, Dawn, George Ford, Evan Los, William Stone. "Glutathione and Its Precursors in Type 2 Diabetes" Encyclopedia, https://encyclopedia.pub/entry/54803 (accessed July 23, 2026).
Tuell, D., Ford, G., Los, E., & Stone, W. (2024, February 06). Glutathione and Its Precursors in Type 2 Diabetes. In Encyclopedia. https://encyclopedia.pub/entry/54803
Tuell, Dawn, et al. "Glutathione and Its Precursors in Type 2 Diabetes." Encyclopedia. Web. 06 February, 2024.
Copy Citation
Type 2 diabetes (T2D) is a major worldwide health crisis affecting about 6.2% of the world’s population. Alarmingly, about one in five children in the USA have prediabetes. Glutathione (GSH) and its precursors play a promising role in the prevention and management of type T2D. Oxidative stress (OxS) is a probable factor in both T2D initiation and progression. GSH is the major cytosolic water-soluble chemical antioxidant and emerging evidence supports its role in improving T2D outcomes. Dietary supplementation with N-acetyl-cysteine (NAC) and/or glycine (GLY), which are GSH precursors, has also been studied for possible beneficial effects on T2D.
glutathione
oxidative stress
type 2 diabetes
1. Introduction
Type 2 diabetes (T2D) is a chronic disease in which blood glucose levels are high enough and of sufficient duration to cause damage to susceptible organs/tissues. Insulin resistance, pancreatic beta-cell dysfunction, and abnormally high glucagon levels are the major etiological factors that cause high blood glucose levels [1][2]. The progression of T2D starts with insulin resistance followed by prediabetes, overt T2D with fasting blood glucose level above 126 mg/dL, and finally vascular damage [2][3]. T2D is, however, a multifactorial disease with alterations in lipid and protein metabolism as well as complex interactions between organs, tissues, cell types, and subcellular compartments/organelles. In this regard, it is significant that alterations in lipid metabolism (e.g., high triglyceride levels) have been found to occur years before overt T2D diagnosis in adults and can improve the prediction of T2D progression [4]. Gummesson et al. [5] have observed distinct alterations in plasma protein profiles in newly diagnosed adult T2D patients in comparison to healthy adult controls. These data suggest that early-stage T2D is accompanied by plasma protein alterations that may also prove useful in helping to predict T2D progression.
The complexity of T2D is reflected by the fact that it is a polygenic disorder with an association with over 120 genetic loci [6]. In addition to insulin resistance, obesity (and intraorgan adipose tissue), poor-quality high-calorie diets, and lack of physical exercise are major risk factors for T2D [1]. The consumption of high-calorie/high-fat ultra-processed foods by children and adolescents is strongly associated with obesity [7][8] and obesity, in turn, is associated with chronic oxidative stress (OxS) [9]. OxS has emerged as an important mechanism for the initiation of prediabetes and for promoting T2D progression [3][10][11].
2. The Biochemistry and Roles of GSH (and Its Precursors) in OxS and T2D
GSH is a key water-soluble thiol (R-SH) antioxidant that may play an important role in preventing, slowing, and perhaps reversing T2D progression [12]. In 1969, Kosower and Kosower wrote a review of GSH with the title “Least I Forget Thee, Glutathione” [13]. This admonition has been well heeded as evidenced by an eight-fold increase in the number of GSH publications in 2022 compared to 1969 [14]. GSH (gamma-glutamylcysteinylglycine) is most often described as a “tripeptide” composed of CYS, GLY, and glutamic acid (GLU) as shown in Figure 1. Both GLY and CYS are considered conditionally essential amino acids in the context of T2D [15][16].
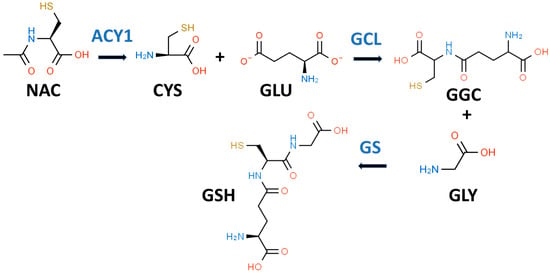
Figure 1. Glutathione biosynthetic scheme. N-acetyl-L-cysteine (NAC) can supply cysteine (CYS) for the biosynthesis of reduced glutathione (GSH). NAC must first be hydrolyzed by aminoacylase 1 (ACY1) to release CYS. In the first step of GSH synthesis, L-glutamate-L-cysteine ligase (GCL) catalyzes the formation of gamma-L-glutamyl-L-cysteine (GGC) by linking CYS and L-glutamate (GLU). In the second step, glutathione synthetase (GS) catalyzes the formation of GSH by linking GGC to GLY.
While it is true that GSH is a tripeptide, it is not a “eu-tripeptide”. Eupeptides have amide bonds that are formed between the C-l of one amino acid and the N-2 of another amino acid as occurs in typical proteins. In GSH, there is an iso-peptide bond between the gamma-carboxyl group of the GLU side chain and CYS (see Figure 1). This iso-peptide bond is important because it renders GSH relatively resistant to intracellular proteases that cleave eu-peptide bonds [17]. This intracellular protease resistance enables GSH to reach unusually high intracellular concentrations (5 mM) for an organic compound and thereby contributes to its effectiveness as an intracellular antioxidant [18]. In contrast, the plasma level of GSH in healthy individuals is about 0.0034 mM [19].
2.1. Diminished GSH Synthesis in T2D Results in OxS
Patients with uncontrolled T2D (i.e., persistent hyperglycemia) have been found by Sekhar et al. to have severely diminished in vivo GSH synthesis, which can be restored by dietary supplementation with “GSH plus GLY” [20]. It should be noted that the dietary “GSH” used in this research was provided as NAC, which is a xenobiotic precursor to GSH; it would be more accurate, therefore, to state that “NAC plus GLY” was used as the dietary supplement. Surprisingly, the mechanism by which NAC enters cells is not completely understood but NAC cannot simply be considered the equivalent of GSH [14]. Significantly, Sekhar et al. found that supplementation with “NAC plus GLY” reduced measures of systemic OxS, e.g., serum lipid peroxides (see Section 2.2.1 below) [20].
GSH is present and synthesized in the cytosol of all mammalian cells by a two-step regulated process (see Figure 1) [21]. In the first step, L-glutamate-L-cysteine ligase (GCL) catalyzes the formation of gamma-L-glutamyl-L-cysteine (GGC) by linking CYS (a thiol-containing amino acid) and GLU. In the second step, glutathione synthetase (GS) catalyzes the formation of GSH by linking GGC to GLY [17][22]. The first step is rate-limiting for GSH synthesis, and the availability of CYS and the activity of GCL are key determinants of GSH synthesis [21]. CYS, and its thiol group, are essential for the antioxidant properties of GSH [23].
2.2. GSH and the Glutathione Peroxidase (GPX) System
As outlined in Figure 2, GPX enzymes reduce hydroperoxides utilizing hydrogen ions donated by the thiol group of GSH with the formation of oxidized GSH (GSSG). There are eight known GPXs (GPX1-GPX8) with GPX1 being the most abundant and this isoform is expressed in the cytoplasm and mitochondria of most cells [24][25]. GPX1-4 and GPX6 are selenoenzymes with selenocysteine (selenium replacing sulfur in CYS) at the active site. Selenium (Se) is an essential trace element and in the absence of dietary Se, total GPX activity in most mammalian tissues is markedly decreased [26]. GPX1-4 and GPX6 can reduce either organic hydroperoxides (ROOH) or hydrogen peroxide (H2O2) to the corresponding organic alcohol or H2O in the case of H2O2.
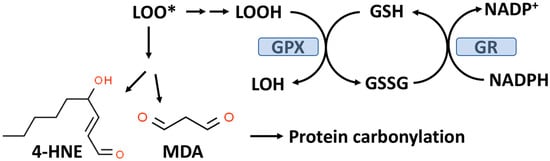
Figure 2. The glutathione peroxidase (GPX) system and lipid peroxidation. GPX catalyzes the conversion of lipid hydroperoxides (LOOH), formed from lipid peroxidation to a lipid alcohol (LOH) utilizing GSH as a reducing agent. The oxidized GSH (GSSG) formed by this reaction is reduced back to GSH by glutathione reductase (GR) with the consumption of NADPH. The lipid peroxyl radical (LOO*) formed from lipid peroxidation can undergo chemical decomposition to 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA), which are reactive aldehydes that can react with proteins to form carbonylation products.
GPX1, due to its affinity for H2O2 and its abundance in most cells, is the key enzyme responsible for minimizing cytosolic H2O2 levels as well as mitochondrial H2O2 levels where no catalase is present in most mammalian cells [25][27][28]. Mitochondria, in addition to GPX1, also contain GPX4, which is unique in its ability to reduce phospholipid hydroperoxides (PLOOH), which are not water-soluble [25].
While Se is required for most forms of GSPX, this trace element is present in some 25 other selenoproteins [29]. The role of Se in T2D has been studied for many decades yet remains controversial [30]. A study by Laralis using a small population of patients (N = 94) with T2D (no diabetic complications and consuming a Mediterranean diet) suggests that 200 micrograms/day (chemical form not specified) can improve glycemic control (after three or six months) [31].
Blood levels of GPX are lower in patients with T2D compared to healthy controls and the decrement in GPX activity was more severe in T2D patients with obesity compared to T2D patients without obesity [32]. Compared to healthy controls, patients with T2D have lower levels of red blood cell GSH levels as well as lower GSH synthesis rates, particularly in T2D patients with microvascular complications [33].
2.2.1. Lipid Peroxidation, Protein Carbonylation, and T2D
Lipid hydroperoxides (LOOH) formed by the process of lipid peroxidation are also a biomarker of OxS. If not reduced by the GPX system, lipid hydroperoxides can accumulate and decompose, yielding lipid peroxyl radicals (LOO*) and/or lipid alkoxyl radicals (LO*), which can amplify lipid peroxidation with the formation of additional lipid hydroperoxides [34]. The positive associations between lipid peroxidation and T2D progression have been comprehensively reviewed by Shabalala et al. [35]. Both malondialdehyde (MDA) and 4-hydroxy-2-nonenal (4-HNE) are reactive aldehyde–lipid peroxidation by-products (see Figure 2) that have been widely used as biomarkers of OxS [34][36].
As indicated in Figure 2, MDA and 4-HNE can covalently modify proteins by carbonylation of CYS, histidine (HIS), and lysine (LYS) residues, thereby potentially altering their structure and function(s), e.g., signal transduction pathways [37][38]. CYS, HIS, and LYS are often present at the active sites of many enzymes.
2.3. NAC Metabolism and Its Role as an “Antioxidant” in T2D Management
NAC has been described as the “most frequently used” antioxidant supplement but recent research suggests that its precise mechanism of action is less certain [14][39][40]. NAC supplements are traditionally thought to provide the CYS residues needed to support GSH synthesis [14]. It is likely that NAC is not an effective direct antioxidant and must be converted into GSH and/or hydrogen sulfide and sulfane sulfur species [14][41]. NAC is hydrolyzed to CYS by aminoacylase1 (ACY1) (Figure 1), which is found in many tissues including the liver and intestines [42]. CYS, in turn, promotes GSH synthesis primarily under circumstances in which tissue GSH is depleted, e.g., T2D [14]. The newly synthesized GSH can act as an antioxidant via the GPX system (Figure 2) [43].
2.4. GLY Alone Has Been Found to Play a Role in Promoting Insulin Resistance
It has long been noted that GLY deficiency (hypoglycinemia) is associated with obesity or T2D and that improvement in insulin resistance is associated with increased plasma GLY [44]. In a comprehensive literature review, McCarty et al. [45] concluded that dietary GLY is rate-limiting for GSH synthesis and that supplemental GLY might promote GSH synthesis and be clinically effective (and safe) in health disorders in which OxS is relevant, e.g., T2D. For over a decade, it has been known that dietary collagen supplementation (rich in GLY) strongly potentiates glucose-stimulated insulin secretion in patients with T2D [46].
A second mechanism by which GLY could affect glucose homeostasis lies in its potential ability to modulate insulin secretion from pancreatic beta-cells by activating ligand-gated chloride channels [45][47]. A progressive decline in insulin secretion from beta-cells is a hallmark of T2D progression [2]. A small clinical study (N = 9) in 2002 found that GLY supplementation increased plasma insulin levels in healthy subjects but did not establish a mechanism [46].
3. Interconnections between OxS, T2D Risk Factors, and GSH Metabolism
Insulin resistance and impaired beta-cell secretion of insulin both contribute to hyperglycemia-induced OxS and diabetic complications [48]. A paper by Boyaci et al. [49] in 2021 raised the question of whether OxS is a consequence of hyperglycemia or if hyperglycemia is a result of OxS. It is likely, however, that multiple “positive feedback loops” between T2D risk factors and OxS are at play and drive T2D progression in susceptible individuals.
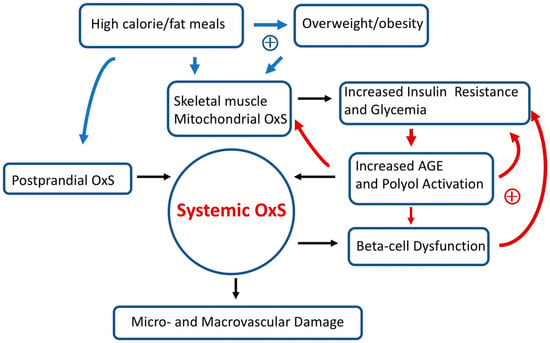
Over long time intervals, T2D positive feedback loops can eventually result in irreversible OxS micro- and/or macro-vascular damage such as diabetic retinopathy (a microvascular disease) [50]. Figure 3 provides a simplified scheme showing the interconnections between OxS and T2D risk factors as well as some likely positive feedback loops. High-calorie meals, overweight/obesity, gastrointestinal postprandial oxidative stress (POS), insulin resistance, hyperglycemia, and pancreatic beta-cell dysfunction are examples of risk factors that can modulate both local and systemic OxS in T2D. Encouragingly, accumulating evidence suggests that T2D progression is not “inevitable” and possibly reversible at an early stage [1]. As will be detailed below, GSH metabolism plays a central role in modulating T2D OxS and may play a role in reversing/slowing early T2D progression.
Figure 3. Simplified scheme connecting oxidative stress (OxS) and T2D risk factors. Two sets of potential positive feedback loops (circles with +) are indicated (blue and red arrows). The text describes these positive feedback loops in more detail. Over prolonged time intervals, systemic OxS can cause beta-cell dysfunction as well as irreversible micro- and macro-vascular damage.
In addition to high-calorie meals, it should be mentioned that there are other common exogenous sources of OxS. As comprehensively reviewed by Bhattacharyya et al. [51], some pollutants, radiation, cigarette smoking, and some drugs/xenobiotics can contribute to OxS. As recently noted by the World Health Organization, quitting cigarette smoking can decrease the risk of developing T2D by 30–40% [52]. In addition to mitochondrial OxS (as detailed below), there are numerous sources of endogenous OxS (e.g., inflammatory responses) that can be relevant to T2D progression [51].
3.1. High-Fat/High-Calorie Diets Promote Postprandial Oxidative Stress (POS), Mitochondrial OxS, and Insulin Resistance
In healthy young adults, the consumption of a lipid (saturated fat)-rich meal results in robust postprandial oxidative stress (POS) compared to an isocaloric carbohydrate-rich meal (dextrose) [53]. When fed a standard meal, patients with T2D show a greater POS compared to matched healthy subjects [54]. POS has been proposed as a probable mechanism contributing to systemic OxS, T2D progression, and vascular damage (see Figure 3) [55][56]. Moreover, supplementation (for 15 days) with dietary antioxidants (including NAC) has been found to reduce POS and improve markers of endothelial dysfunction in subjects with T2D or insulin resistance [57].
3.2. Skeletal Muscle Mitochondrial OxS and Insulin Resistance
It has long been recognized that skeletal muscle insulin resistance is the primary defect in T2D [58]. The insulin-sensitive glucose transporter 4 (GLUT4) is the primary means by which skeletal muscle takes up glucose. Insulin stimulates the translocation of GLUT4 from intracellular GLUT4-containing vesicles to the cell surface where this transporter can actively support facilitated glucose transport [59]. The insulin signaling pathway is complex but requires the activation of Akt (a serine–threonine kinase) by phosphorylation [60]. The reduced ability of insulin to activate the GLUT4 glucose transport system in skeletal muscle is a primary cause of insulin resistance [59][61].
In pioneering work, Anderson et al. [62] found that a high-calorie diet (in healthy adults) promoted skeletal muscle mitochondrial OxS which, in turn, resulted in transient insulin resistance. While both a high-calorie fat or carbohydrate meal could induce this transient insulin resistance, dietary fat was more effective than dietary carbohydrates [62].
3.3. Hyperglycemia Promotes Protein Glycation, Formation of AGEs, Activation of the Polyol Pathway, OxS, and T2D Progression
Chronic bouts of transient insulin resistance will result in chronic increases in postprandial glucose (PPG) as indicated in Figure 3. Glucose can covalently react with lysine, arginine, and the N-terminal residues on proteins to form glycation products, which can further react to produce advanced glycation end products (AGEs) [63]. Glycation can modify the structure and functions of proteins. It has been found, for example, that glycation of the GPX results in a loss of enzymatic activity [64]. This is a hypothetical example of a positive feedback loop, i.e., increased mitochondrial OxS promotes increased glycemia, promoting increased glycation–inactivation of GPX enzymes with a further increase in skeletal muscle mitochondrial OxS and glycemia.
High plasma glucose also promotes OxS by activation of the polyol pathway in which aldose reductase (AR) reduces glucose to sorbitol followed by the conversion of sorbitol to fructose by sorbitol dehydrogenase (SDH). In the first reaction, NADPH is consumed by AR, which can result in OxS since NADPH is required for reducing GSSG to GSH (by GR) and GSH is required for the reducing peroxides by the GPX system (see Figure 2). It has been estimated that in diabetes, as much as 30% of body glucose can be consumed by the polyol pathway [63].
3.4. Chronic Inflammation, OxS, and T2D
OxS arising from chronic inflammation has long been recognized as a potential driver of T2D progression [65][66]. Chronic inflammation is associated with increased levels of ROS, reactive nitrogen oxide species (RNOS), and C-reactive protein. As reviewed by Son et al. [67], the increased production of superoxide radicals (O2•−) resulting from T2D hyperglycemia can rapidly react with nitric oxide (NO) to produce peroxynitrite (ONOO−), which can subsequently react with protein (and apolipoprotein) tyrosine residues to form 3-nitro-tyrosine (3-NT). 3-NT levels are biomarkers for inflammation and patients with T2D have increased serum 3-NT levels compared to controls [68]. ROS and RNOS are both thought to contribute to T2D macro- and micro-vascular damage [67]. GSH is thought to play a key role in NO biochemistry [69]. GSH, for example, can react with NO to form S-nitrosoglutathione, which may be relevant to T2D by promoting insulin sensitivity [70].
References
- Taylor, R. Type 2 diabetes: Etiology and reversibility. Diabetes Care 2013, 36, 1047–1055.
- Fonseca, V.A. Defining and characterizing the progression of type 2 diabetes. Diabetes Care 2009, 32 (Suppl. S2), S151–S156.
- Tuell, D.S.; Los, E.A.; Ford, G.A.; Stone, W.L. The Role of Natural Antioxidant Products That Optimize Redox Status in the Prevention and Management of Type 2 Diabetes. Antioxidants 2023, 12, 1139.
- Suvitaival, T.; Bondia-Pons, I.; Yetukuri, L.; Pöhö, P.; Nolan, J.J.; Hyötyläinen, T.; Kuusisto, J.; Orešič, M. Lipidome as a predictive tool in progression to type 2 diabetes in Finnish men. Metabolism 2018, 78, 1–12.
- Gummesson, A.; Björnson, E.; Fagerberg, L.; Zhong, W.; Tebani, A.; Edfors, F.; Schmidt, C.; Lundqvist, A.; Adiels, M.; Bäckhed, F.; et al. Longitudinal plasma protein profiling of newly diagnosed type 2 diabetes. EBioMedicine 2021, 63, 103147.
- Lyssenko, V.; Groop, L.; Prasad, R.B. Genetics of Type 2 Diabetes: It Matters from Which Parent We Inherit the Risk. Rev. Diabet. Stud. 2015, 12, 233–242.
- Neri, D.; Steele, E.M.; Khandpur, N.; Cediel, G.; Zapata, M.E.; Rauber, F.; Marrón-Ponce, J.A.; Machado, P.; da Costa Louzada, M.L.; Andrade, G.C.; et al. Ultraprocessed food consumption and dietary nutrient profiles associated with obesity: A multicountry study of children and adolescents. Obes. Rev. 2022, 23 (Suppl. S1), e13387.
- Poti, J.M.; Braga, B.; Qin, B. Ultra-processed Food Intake and Obesity: What Really Matters for Health-Processing or Nutrient Content? Curr. Obes. Rep. 2017, 6, 420–431.
- Savini, I.; Catani, M.V.; Evangelista, D.; Gasperi, V.; Avigliano, L. Obesity-associated oxidative stress: Strategies finalized to improve redox state. Int. J. Mol. Sci. 2013, 14, 10497–10538.
- Fatima, M.T.; Bhat, A.A.; Nisar, S.; Fakhro, K.A.; Ammira, S. The role of dietary antioxidants in type 2 diabetes and neurodegenerative disorders: An assessment of the benefit profile. Heliyon 2023, 9, e12698.
- Alu, S.N.; Los, E.A.; Ford, G.A.; Stone, W.L. Oxidative Stress in Type 2 Diabetes: The Case for Future Pediatric Redoxomics Studies. Antioxidants 2022, 11, 1336.
- Sekhar, R.V. GlyNAC (Glycine and N-Acetylcysteine) Supplementation Improves Impaired Mitochondrial Fuel Oxidation and Lowers Insulin Resistance in Patients with Type 2 Diabetes: Results of a Pilot Study. Antioxidants 2022, 11, 154.
- Ong, T.P.; Cardozo, M.T.; de Conti, A.; Moreno, F.S. Chemoprevention of Hepatocarcinogenesis with Dietary Isoprenic Derivatives: Cellular and Molecular Aspects. Curr. Cancer Drug Targets 2012, 12, 1173–1190.
- Pedre, B.; Barayeu, U.; Ezeriņa, D.; Dick, T.P. The mechanism of action of N-acetylcysteine (NAC): The emerging role of H. Pharmacol. Ther. 2021, 228, 107916.
- Achari, A.E.; Jain, S.K. l-Cysteine supplementation increases insulin sensitivity mediated by upregulation of GSH and adiponectin in high glucose treated 3T3-L1 adipocytes. Arch. Biochem. Biophys. 2017, 630, 54–65.
- Razak, M.A.; Begum, P.S.; Viswanath, B.; Rajagopal, S. Multifarious Beneficial Effect of Nonessential Amino Acid, Glycine: A Review. Oxid. Med. Cell. Longev. 2017, 2017, 1716701.
- Lushchak, V.I. Glutathione homeostasis and functions: Potential targets for medical interventions. J. Amino Acids 2012, 2012, 736837.
- Pizzorno, J. Glutathione! Integr. Med. (Encinitas) 2014, 13, 8–12.
- Michelet, F.; Gueguen, R.; Leroy, P.; Wellman, M.; Nicolas, A.; Siest, G. Blood and plasma glutathione measured in healthy subjects by HPLC: Relation to sex, aging, biological variables, and life habits. Clin. Chem. 1995, 41, 1509–1517.
- Sekhar, R.V.; McKay, S.V.; Patel, S.G.; Guthikonda, A.P.; Reddy, V.T.; Balasubramanyam, A.; Jahoor, F. Glutathione synthesis is diminished in patients with uncontrolled diabetes and restored by dietary supplementation with cysteine and glycine. Diabetes Care 2011, 34, 162–167.
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153.
- Lu, S.C. Regulation of hepatic glutathione synthesis. Semin. Liver Dis. 1998, 18, 331–343.
- Asantewaa, G.; Harris, I.S. Glutathione and its precursors in cancer. Curr. Opin. Biotechnol. 2021, 68, 292–299.
- Brigelius-Flohé, R.; Flohé, L. Regulatory Phenomena in the Glutathione Peroxidase Superfamily. Antioxid. Redox Signal. 2020, 33, 498–516.
- Handy, D.E.; Lubos, E.; Yang, Y.; Galbraith, J.D.; Kelly, N.; Zhang, Y.Y.; Leopold, J.A.; Loscalzo, J. Glutathione peroxidase-1 regulates mitochondrial function to modulate redox-dependent cellular responses. J. Biol. Chem. 2009, 284, 11913–11921.
- Stone, W.L.; Dratz, E.A. Selenium and non-selenium glutathione peroxidase activities in selected ocular and non-ocular rat tissues. Exp. Eye Res. 1982, 35, 405–412.
- Powers, S.K.; Ji, L.L.; Kavazis, A.N.; Jackson, M.J. Reactive oxygen species: Impact on skeletal muscle. Compr. Physiol. 2011, 1, 941–969.
- Bai, J.; Cederbaum, A.I. Mitochondrial catalase and oxidative injury. Biol. Signals Recept. 2001, 10, 189–199.
- Shimada, B.K.; Swanson, S.; Toh, P.; Seale, L.A. Metabolism of Selenium, Selenocysteine, and Selenoproteins in Ferroptosis in Solid Tumor Cancers. Biomolecules 2022, 12, 1581.
- Casanova, P.; Monleon, D. Role of selenium in type 2 diabetes, insulin resistance and insulin secretion. World J. Diabetes 2023, 14, 147–158.
- Karalis, D.T. The Beneficiary Role of Selenium in Type II Diabetes: A Longitudinal Study. Cureus 2019, 11, e6443.
- Goyal, R.; Singhai, M.; Faizy, A.F. Glutathione peroxidase activity in obese and nonobese diabetic patients and role of hyperglycemia in oxidative stress. J. Midlife Health 2011, 2, 72–76.
- Lutchmansingh, F.K.; Hsu, J.W.; Bennett, F.I.; Badaloo, A.V.; McFarlane-Anderson, N.; Gordon-Strachan, G.M.; Wright-Pascoe, R.A.; Jahoor, F.; Boyne, M.S. Glutathione metabolism in type 2 diabetes and its relationship with microvascular complications and glycemia. PLoS ONE 2018, 13, e0198626.
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438.
- Shabalala, S.C.; Johnson, R.; Basson, A.K.; Ziqubu, K.; Hlengwa, N.; Mthembu, S.X.H.; Mabhida, S.E.; Mazibuko-Mbeje, S.E.; Hanser, S.; Cirilli, I.; et al. Detrimental Effects of Lipid Peroxidation in Type 2 Diabetes: Exploring the Neutralizing Influence of Antioxidants. Antioxidants 2022, 11, 2071.
- Dham, D.; Roy, B.; Gowda, A.; Pan, G.; Sridhar, A.; Zeng, X.; Thandavarayan, R.A.; Palaniyandi, S.S. 4-Hydroxy-2-nonenal, a lipid peroxidation product, as a biomarker in diabetes and its complications: Challenges and opportunities. Free Radic. Res. 2021, 55, 547–561.
- Sun, L.; Wu, Q.; Mao, X. Effects of Oxidation Modification by Malondialdehyde on the Structure and Functional Properties of Walnut Protein. Foods 2022, 11, 2432.
- Milkovic, L.; Zarkovic, N.; Marusic, Z.; Zarkovic, K.; Jaganjac, M. The 4-Hydroxynonenal-Protein Adducts and Their Biological Relevance: Are Some Proteins Preferred Targets? Antioxidants 2023, 12, 856.
- Zhitkovich, A. Acetylcysteine: Antioxidant, Aldehyde Scavenger, and More. Chem. Res. Toxicol. 2019, 32, 1318–1319.
- Mlejnek, P. Direct Interaction between N-Acetylcysteine and Cytotoxic Electrophile—An Overlooked In Vitro Mechanism of Protection. Antioxidants 2022, 11, 1485.
- Rushworth, G.F.; Megson, I.L. Existing and potential therapeutic uses for N-acetylcysteine: The need for conversion to intracellular glutathione for antioxidant benefits. Pharmacol. Ther. 2014, 141, 150–159.
- Uttamsingh, V.; Baggs, R.B.; Krenitsky, D.M.; Anders, M.W. Immunohistochemical localization of the acylases that catalyze the deacetylation of N-acetyl-L-cysteine and haloalkene-derived mercapturates. Drug Metab. Dispos. 2000, 28, 625–632.
- Lasram, M.M.; Dhouib, I.B.; Annabi, A.; El Fazaa, S.; Gharbi, N. A review on the possible molecular mechanism of action of N-acetylcysteine against insulin resistance and type-2 diabetes development. Clin. Biochem. 2015, 48, 1200–1208.
- Adeva-Andany, M.; Souto-Adeva, G.; Ameneiros-Rodríguez, E.; Fernández-Fernández, C.; Donapetry-García, C.; Domínguez-Montero, A. Insulin resistance and glycine metabolism in humans. Amino Acids 2018, 50, 11–27.
- McCarty, M.F.; O’Keefe, J.H.; DiNicolantonio, J.J. Dietary Glycine Is Rate-Limiting for Glutathione Synthesis and May Have Broad Potential for Health Protection. Ochsner J. 2018, 18, 81–87.
- Gannon, M.C.; Nuttall, J.A.; Nuttall, F.Q. The metabolic response to ingested glycine. Am. J. Clin. Nutr. 2002, 76, 1302–1307.
- Yan-Do, R.; MacDonald, P.E. Impaired “Glycine”-mia in Type 2 Diabetes and Potential Mechanisms Contributing to Glucose Homeostasis. Endocrinology 2017, 158, 1064–1073.
- King, J.C.; Fabro, S. Alcohol consumption and cigarette smoking: Effect on pregnancy. Clin. Obs. Gynecol. 1983, 26, 437–448.
- Boyacı1, İ.; Yiğitbaşı, T.; Ankaralı, H. Is Oxidative Stress a Consequence of Hyperglycemia? Or Is Hyperglycemia the Consequence of Oxidative Stress? Or Are Both Caused by Insulin Resistance? Int. Arch. Endocrinol. Clin. Res. 2021, 7, 023.
- Safi, S.Z.; Qvist, R.; Kumar, S.; Batumalaie, K.; Ismail, I.S. Molecular mechanisms of diabetic retinopathy, general preventive strategies, and novel therapeutic targets. BioMed Res. Int. 2014, 2014, 801269.
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative stress: An essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol. Rev. 2014, 94, 329–354.
- World Health Organization. Quitting Smoking Cuts Your Risk of Developing Type 2 Diabetes by 30–40%. Available online: www.who.int/news/item/14-11-2023-quitting-smoking-cuts-your-risk-of-developing-type-2-diabetes-by-30-40 (accessed on 9 January 2023).
- Bloomer, R.J.; Kabir, M.M.; Marshall, K.E.; Canale, R.E.; Farney, T.M. Postprandial oxidative stress in response to dextrose and lipid meals of differing size. Lipids Health Dis. 2010, 9, 79.
- Ceriello, A.; Bortolotti, N.; Motz, E.; Crescentini, A.; Lizzio, S.; Russo, A.; Tonutti, L.; Taboga, C. Meal-generated oxidative stress in type 2 diabetic patients. Diabetes Care 1998, 21, 1529–1533.
- Sies, H.; Stahl, W.; Sevanian, A. Nutritional, dietary and postprandial oxidative stress. J. Nutr. 2005, 135, 969–972.
- Sottero, B.; Gargiulo, S.; Russo, I.; Barale, C.; Poli, G.; Cavalot, F. Postprandial Dysmetabolism and Oxidative Stress in Type 2 Diabetes: Pathogenetic Mechanisms and Therapeutic Strategies. Med. Res. Rev. 2015, 35, 968–1031.
- Neri, S.; Calvagno, S.; Mauceri, B.; Misseri, M.; Tsami, A.; Vecchio, C.; Mastrosimone, G.; Di Pino, A.; Maiorca, D.; Judica, A.; et al. Effects of antioxidants on postprandial oxidative stress and endothelial dysfunction in subjects with impaired glucose tolerance and type 2 diabetes. Eur. J. Nutr. 2010, 49, 409–416.
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32 (Suppl. S2), S157–S163.
- Henriksen, E.J.; Diamond-Stanic, M.K.; Marchionne, E.M. Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic. Biol. Med. 2011, 51, 993–999.
- Carmichael, R.E.; Wilkinson, K.A.; Craig, T.J. Insulin-dependent GLUT4 trafficking is not regulated by protein SUMOylation in L6 myocytes. Sci. Rep. 2019, 9, 6477.
- Mueckler, M. Insulin resistance and the disruption of Glut4 trafficking in skeletal muscle. J. Clin. Investig. 2001, 107, 1211–1213.
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.T.; Price, J.W.; Kang, L.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investig. 2009, 119, 573–581.
- Yan, L.J. Redox imbalance stress in diabetes mellitus: Role of the polyol pathway. Anim. Model. Exp. Med. 2018, 1, 7–13.
- Suravajjala, S.; Cohenford, M.; Frost, L.R.; Pampati, P.K.; Dain, J.A. Glycation of human erythrocyte glutathione peroxidase: Effect on the physical and kinetic properties. Clin. Chim. Acta 2013, 421, 170–176.
- Oguntibeju, O.O. Type 2 diabetes mellitus, oxidative stress and inflammation: Examining the links. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 45–63.
- Stone, W.L.; Basit, H.; Burns, B. Pathology, Inflammation. In StatPearls ; StatPearls Publishing LLC: Treasure Island, FL, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK534820/ (accessed on 1 January 2024).
- Son, S.M. Reactive oxygen and nitrogen species in pathogenesis of vascular complications of diabetes. Diabetes Metab. J. 2012, 36, 190–198.
- Jakubiak, G.K.; Cieślar, G.; Stanek, A. Nitrotyrosine, Nitrated Lipoproteins, and Cardiovascular Dysfunction in Patients with Type 2 Diabetes: What Do We Know and What Remains to Be Explained? Antioxidants 2022, 11, 856.
- Baldelli, S.; Ciccarone, F.; Limongi, D.; Checconi, P.; Palamara, A.T.; Ciriolo, M.R. Glutathione and Nitric Oxide: Key Team Players in Use and Disuse of Skeletal Muscle. Nutrients 2019, 11, 2318.
- Sousa-Lima, I.; Fernandes, A.B.; Patarrão, R.S.; Kim, Y.B.; Macedo, M.P. S-Nitrosoglutathione Reverts Dietary Sucrose-Induced Insulin Resistance. Antioxidants 2020, 9, 870.
- Stanimirovic, J.; Radovanovic, J.; Banjac, K.; Obradovic, M.; Essack, M.; Zafirovic, S.; Gluvic, Z.; Gojobori, T.; Isenovic, E.R. Role of C-Reactive Protein in Diabetic Inflammation. Mediat. Inflamm. 2022, 2022, 3706508.
- Lin, C.-C.; Li, C.-I.; Liu, C.-S.; Liao, L.-N.; Yang, C.-W.; Lin, C.-H.; Yang, S.-Y.; Li, T.-C. Association of high-sensitivity C-reactive protein and diabetic nephropathy in patients with type 2 diabetes: A Mendelian randomization study. BMJ Open Diabetes Res. Care 2023, 11, e003197.
- Askari, M.; Faryabi, R.; Mozaffari, H.; Darooghegi Mofrad, M. The effects of N-Acetylcysteine on serum level of inflammatory biomarkers in adults. Findings from a systematic review and meta-analysis of randomized clinical trials. Cytokine 2020, 135, 155239.
More
Information
Subjects:
Integrative & Complementary Medicine
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
930
Revisions:
2 times
(View History)
Update Date:
07 Feb 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No