Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | TOHRU KITADA | -- | 2467 | 2024-02-06 10:30:36 | | | |
| 2 | Rita Xu | -36 word(s) | 2431 | 2024-02-06 10:40:02 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kitada, T.; Ardah, M.T.; Haque, M.E. Parkinson’s Disease-Associated Gene, Parkin. Encyclopedia. Available online: https://encyclopedia.pub/entry/54796 (accessed on 28 June 2026).
Kitada T, Ardah MT, Haque ME. Parkinson’s Disease-Associated Gene, Parkin. Encyclopedia. Available at: https://encyclopedia.pub/entry/54796. Accessed June 28, 2026.
Kitada, Tohru, Mustafa T. Ardah, M. Emdadul Haque. "Parkinson’s Disease-Associated Gene, Parkin" Encyclopedia, https://encyclopedia.pub/entry/54796 (accessed June 28, 2026).
Kitada, T., Ardah, M.T., & Haque, M.E. (2024, February 06). Parkinson’s Disease-Associated Gene, Parkin. In Encyclopedia. https://encyclopedia.pub/entry/54796
Kitada, Tohru, et al. "Parkinson’s Disease-Associated Gene, Parkin." Encyclopedia. Web. 06 February, 2024.
Copy Citation
Parkin, the gene responsible for hereditary Parkinson’s disease (PD) called “Autosomal Recessive Juvenile Parkinsonism (AR-JP)” was discovered a quarter of a century ago.
Lewy bodies
Parkinson’s disease
Parkin
1. Cloning of Parkin Gene in a Short Period: Three Lucky Breaks
In the late 1990s, the global sequencing teams in the Human Genome Project were still in the process of deciphering the entire genome sequence, while the genomic information itself remained unclear.
First, a polymorphism analysis was performed for the manganese superoxide dismutase (Mn-SOD), a mitochondrial enzyme that has an antioxidant effect, as a candidate gene for hereditary Parkinson’s disease (PD). In one patient’s family, all patient-specific genetic polymorphisms were observed [1], and, ultimately, it became clear that the Mn-SOD was not the causative gene. Nevertheless, this linkage analysis result convinced that the causative gene was located near the Mn-SOD gene, which was the first lucky break. Among the 46 chromosomes, it was possible to concentrate on linkage analysis of the long arm of chromosome 6, where the Mn-SOD gene resides.
In the second lucky break, following a series of linkage analyses, the microsatellite marker D6S305, located on the long arm of chromosome 6, was linked to the patient’s pedigree, and the patient was found to lack this marker. Thus, it was found that the causative gene exists in the gene region in chromosome 6 containing D6S305. Nevertheless, identifying the causative genes from hundreds of genes ranging from the 3 Mb is important. Exons were searched from the BAC DNA library using the exon trap method with the D6S305 as a probe; however, only a few exons were identified.
Ultimately, parkin was the only gene in the vicinity and was found to be an unknown jumbo gene of >1 Mb, consisting of only 12 exons. Due to this, the reason why exon trapping can identify only a few exons became clear. This was the third time lucky, as it was enough to end up with only one genetic analysis.
Thus, a gene that is second in size to the muscle disease gene dystrophin (approximately 2.5 Mb), which was previously known as a giant gene, has been discovered. Kitada proposed the name “parkin” to represent the primary disease, and, above all, he hoped that the discovery of this gene could promote pioneering research on the etiology of PD [2]. Kitada et al. (1998) also reported that mutations of the parkin gene are associated with autosomal recessive juvenile parkinsonism (AR-JP).
2. Why Abnormal Mitochondrial Accumulation Theory Has Been Supported for So Long: Three Turning Points
2.1. The First Turning Point—Year 2000
The first turning point was the proposal of the assumption that the etiology of the AR-JP is a high intracellular accumulation of substrate proteins due to parkin deficiency. This assumption is in direct conflict with the pathological reports of pathologists and neurologists [3][4][5].
Since the Parkin protein was recognized as an E3 ligase in 2000 [6], a reasonable but simple idea was raised that Parkin must have substrates and its deficiency would cause their accumulation due to proteasomal failure (Figure 1A). Subsequently, similar to bamboo shoots in rain, countless candidates for Parkin’s substrates have been reported over approximately 10 years. However, neither the characteristic structure nor function common to these candidates has been reported, nor has the high accumulation of specific proteins been pathologically explained or identified.
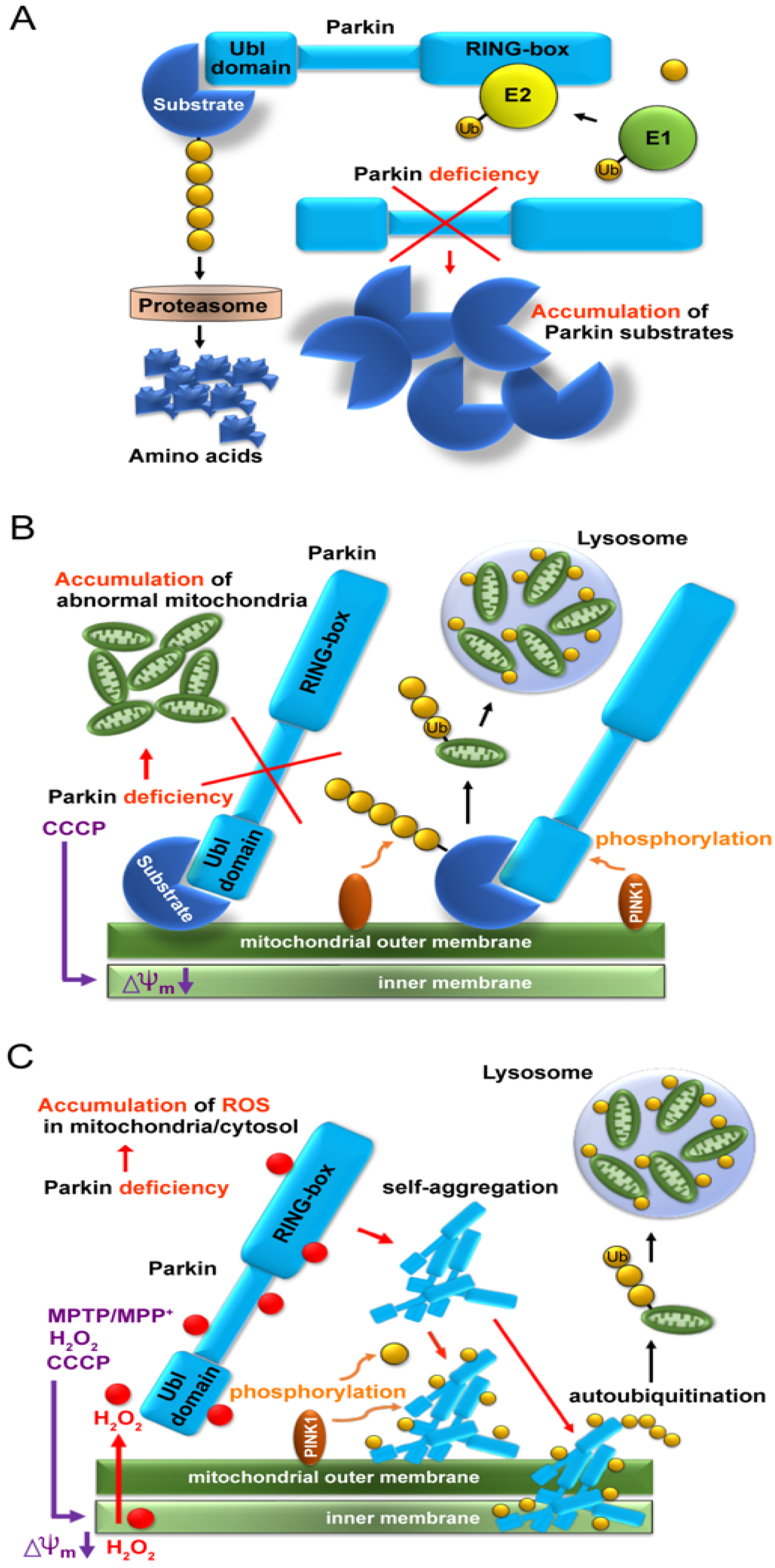
Figure 1. Schema of three leading hypotheses. (A) Accumulation of substrate proteins due to lack of proteasomal degradation caused by parkin deficiency. (B) Accumulation of aberrant mitochondria due to failure of mitochondrial ubiquitination caused by parkin deficiency. (C) Accumulation of aggregated and insoluble Parkin proteins in mitochondria due to reaction with hydrogen peroxide leaked from aberrant mitochondria.
2.2. Genetic Interaction between Parkin and PINK1 Genes: Knowledge Gathered from Fruit Fly Models
In 2004, the PTEN-induced kinase 1 (PINK1) gene was identified as the third causative gene of autosomal recessive Parkinson’s disease (ARPD) [7]. The PINK1 protein is thought to be a serine/threonine protein kinase localized in the mitochondrial inner membrane. Recent in vitro and cultured cell studies have reported that PINK1 phosphorylates and activates Parkin and ubiquitin [8].
In 2006, genetic studies on the Drosophila melanogaster reported that the PINK1 functions upstream of Parkin. The parkin- and PINK1-deficient Drosophila melanogaster has been observed to exhibit flight muscle degeneration, short lifespan, male sterility, and decreased energy [9][10]. Overexpression of the PINK1 in parkin knockout flies did not improve the above phenotype; however, the parkin overexpression did improve the phenotype in the PINK1 knockout flies.
2.3. The Second Turning Point—Year 2008
Biological cell research on the Parkin flourished in the years leading up to 2008, when a surprising phenomenon was reported in which the Parkin localized defective mitochondria with reduced membrane potential, leading to mitochondrial degradation via autophagy called “mitophagy” [11].
In 2010, two groups demonstrated a cooperative relationship between Parkin and PINK1 in abnormal mitochondrial processing (Figure 1B) [12][13]. When the mitochondria are artificially depolarized using a mitochondrial uncoupler, such as carbonyl cyanide m-chlorophenyl hydrazone (CCCP), after the overexpression of the tagged Parkin in cultured cells, PINK1 recognizes depolarization and moves from the inner membrane to the outer membrane, where it recruits Parkin, which polyubiquitinates various mitochondrial substrates and induces mitophagy. Thus, defects in Parkin or PINK1 are thought to prevent the degradation of these abnormal mitochondria, which accumulate intracellularly and cause neuronal cell death. The substrates on the mitochondria themselves do not appear to be defective; however, Parkin ubiquitinates them non-specifically in depolarized mitochondria.
One of the two groups that presented the results argued for approximately 10 years that Parkin deficiency failed to degrade the Parkin substrates and that the accumulation of those substrates caused neuronal cell death [6]. In this case, specimens from patients with AR-JP should have been thoroughly examined for abnormal mitochondrial deposition using a variety of orthodox staining methods, such as hematoxylin-eosin (HE) staining, protein staining, or electron microscopy.
More than a decade later, the intracellular number of abnormal mitochondria, their morphology, and tissue distribution remained unknown. In contrast to the beta-amyloid deposits found in Alzheimer’s disease and alpha-synuclein deposits seen in sporadic PD, the inability to pathologically diagnose accumulated abnormal mitochondria is paradoxical.
In conclusion, the primary problem with this hypothesis is that it cannot be confirmed pathologically or histologically. Unfortunately, we used the same route as before. No abnormal mitochondrial accumulation has been reported in autopsy or biopsy cases of the parkin or PINK1 mutant patients. Details are described in the clinical and pathological aspects of AR-JP.
2.4. The Third Turning Point: “Parkin Dilemma”
An important functional and structural feature of the Parkin protein has been neglected, which is the third turning point (unfortunate) of Parkin research. The Parkin protein contains 35 cysteine residues (7.5%) out of 465 amino acids. Such cysteine-rich proteins are rare among the E3 ligases. The cysteine residue contains a thiol group (-SH) that scavenges superoxide (O2−) and hydrogen peroxide (H2O2) and forms a disulfide bond (S-S). It was observed that Parkin reacts directly with and reduces H2O2 and aggregates into dimers and multimers, resulting in poor solubility [14][15][16].
When CCCP is used to collapse the inner membrane potential of mitochondria, the membrane potential of all mitochondria decreases, and ions, proteins, and reactive oxygen species (ROS) leak into the cytoplasm through the collapse. It is extremely disappointing that many researchers claiming mitochondrial ubiquitination and mitophagy deficiency due to the lack of Parkin or PINK1 have not taken into account Parkin’s function as a redox molecule and its secondary consequences. The E3 ligase function of Parkin and its function as a redox molecule are essential, but not simultaneous. Thus, when Parkin scavenges H2O2 as a redox molecule, it aggregates and loses E3 activity, which is called the “Parkin dilemma”.
3. Clinical and Pathological Aspects of Parkin (AR-JP, PARK2) and PINK1 (PARK6) Mutations
The focus here was on studying the PINK1/Parkin pathway using molecular and cell biological techniques, and the clinical and pathological aspects of the two autosomal recessive Parkinson’s Disease (ARPDs) were referred to. First, the two inherited diseases are genetically independent.
Compared to the sporadic form, its prominent clinical feature is Parkinsonism with marked diurnal fluctuations. In addition, AR-JP has a generally benign clinical course, with marked effects of levodopa, high frequency of dyskinesia and dystonia, and hyperreflexia. If present, autonomic symptoms are mild [3][4][5][17][18]. AR-JP autopsy studies typically show the loss of neurons without Lewy bodies, gliosis, relatively localized lesions in the substantia nigra and locus coeruleus, and the presence of low or immature melanin in residual neurons. However, several cases of Lewy bodies have also been reported [19][20].
In contrast to the AR-JP phenotype, patients with PINK1 mutations rarely present with symptoms specific to patients with AR-JP described above. Families with PINK1 (PARK-6) mutations have a wide age range of onset (up to 68 years) and slow progression [21]. Features typical of AR-JP, such as dystonia at onset and sleep effects, are not observed in PARK6-related families; thus, the clinical manifestations of late-onset cases are indistinguishable from those of sporadic PD [21]. However, sporadic PD with heterozygous variants of PINK1 has been reported to overlap with some of the characteristic clinical features of AR-JP and should be noted [22].
Pathologically, as of 2020, three autopsy brain reports of PINK1 gene mutations have been reported, including three patients with Lewy-related pathology and one patient without Lewy-related pathology [23]. Thus, it cannot be concluded that Parkin and PINK1 are involved in the same pathway in both clinical and pathological aspects.
To date, there have been many reports of brain autopsies or biopsies of tissues of parkin mutants and several reports of brain autopsies of the PINK1 mutants. However, none of them exhibited abnormal mitochondrial accumulation. There are hundreds to thousands of mitochondria in a single cell, and they have a very short lifespan of seven to 10 days. If these mitochondria are not processed, thousands of unnecessary mitochondria can accumulate in just one week, and many abnormal mitochondria can be expected to accumulate in the cell every day.
In the Parkin/PINK1 pathway, PINK1 is upstream of Parkin, and the current theory is that the phosphorylation of Parkin by PINK1 is essential for Parkin recruitment. However, this contradicts the results of the two fruit fly studies. The phenotype of PINK1-deficient fruit flies should be recovered by overexpression of parkin [9][10].
However, the two studies described below have reported muscle biopsies of patients with AR-JP [24][25][26]. Although two studies on fruit flies, the starting point of the Parkin/PINK1 pathway theory, acknowledged significant findings, only minor changes in muscle fibers were observed histologically. These reports are valuable because mitochondria undergo progressive degeneration in post-mortem specimens. Tissue biopsies can provide a tissue environment similar to that of the living environment.
In an electron microscopy study by van der Merwe et al., muscle biopsies were obtained from two patients [24]. Muscle fibers showed subtle abnormalities such as slightly swollen mitochondria in the focal areas of the fibers and some folding of the sarcolemma.
The laboratory of Murat Emre at Istanbul University performed immunohistochemistry on biceps brachii muscles from three patients with exon 9/intron 9 junction “G” deletion or exon 3 deletion of the parkin gene and observed only mild histopathological changes in the muscles [25]. Researchers have also reported a patient with a homozygous IVS-9-1 deletion in the parkin gene [26]. The patient had bilateral thigh muscle hypertrophy and a muscle biopsy of the biceps revealed abundant cytochrome oxidase (COX) (-) fibers.
Nevertheless, only minor pathological changes have been reported in muscle biopsies of parkin mutants at both electron and light microscopic levels, and abnormal mitochondrial accumulation has not been reported.
4. Studies Focusing on Parkin as a Redox Molecule Alongside E3 Ligases
In the early 2000s, Parkin was found to be oxidized to form aggregates [27]. Meng et al. showed that oxidative stress causes sulfation/sulfonation of cysteine-rich regions of Parkin, resulting in decreased E3 activity [28]. In addition, since the discovery of the parkin gene, numerous chemical and histological reports of increased oxidative stress in the parkin-knockout cultured cells and animal models have been reported [29][30]. Furthermore, it has been reported that iron staining in the substantia nigra of AR-JP is more intense than in controls or sporadic PD, suggesting that oxidative stress may play an important role in the neurodegeneration that occurs in AR-JP [31]. Recently, Tokarew et al. have reported a series of Parkin protein analyses using the following human autopsy brains [16]. First, Parkin solubility is decreased in aged human brains, including the substantia nigra. Decreased Parkin solubility is correlated with increased levels of hydrogen peroxide in human brains. They also demonstrated elevated hydrogen peroxide in the AR-JP brain. While it has been reported that Parkin reacts with dopamine and its metabolites and reduces their toxicity [27]. Tokarew et al. have also experimentally shown a protective role for Parkin as a redox molecule, including conjugation of reactive dopamine metabolites, containment of radicals within insoluble aggregates, and increased melanin formation [16]. Hypoplasia of neuromelanin in the substantia nigra was mentioned as a characteristic pathological finding of AR-JP. From these findings, it is understood that neuromelanin formation is not successful without Parkin.
The precise mechanism by which the Parkin protects itself from oxidative stress has long been unclear. Kitada et al. proved that Parkin reacts directly with hydrogen peroxide and reduces it significantly in a very simple experiment in which Parkin protein and hydrogen peroxide solution were mixed in a test tube [14]. Until then, Parkin was perceived negatively because its E3 function was impaired by oxidative stress, but another important function, the antioxidant effect, of Parkin is now being recognized. However, these two aspects cannot exist simultaneously, and this fact should be called the “Parkin dilemma”, as mentioned above.
After overexpression of various combinations of Parkin, PINK1, and Ubiquitin in HEK293 cultured cells for 24 h and CCCP treatment, mitochondrial fractions were extracted and Western blot (WB) analysis was performed using a reducing agent-free loading dye [32]. Surprisingly, the E3 Parkin monomer was not recruited into the mitochondria by PINK1 but was deposited as aggregates on the outer and inner mitochondrial membranes and was self-ubiquitinated (auto-ubiquitination). Rather than phosphorylating Parkin monomers to activate them, PINK1 seemed to phosphorylate Parkin aggregates of various sizes and promote Parkin aggregation, making them insoluble (Figure 1C). Interestingly, when WB was performed using a loading dye containing the reducing agent (dithiothreitol [DTT]), the poorly soluble Parkin aggregates were broken down to the monomer level. When cultured cells were treated with glutathione before adding CCCP, some bands at the monomer level were observed even when WB was performed using DTT-free loading dye. These results suggest that at the cellular level, glutathione, which is the most abundant reducing substance in the body, partially converts insoluble Parkin into an E3 monomer. Intracellular regulation between Parkin aggregates and Parkin monomers needs to be investigated in the future.
References
- Matsumine, H.; Yamamurab, Y.; Hattoria, N.; Kobayashia, T.; Kitadaa, T.; Yoritakaa, A.; Mizunoa, Y. A Microdeletion of D6S305 in a Family of Autosomal Recessive Juvenile Parkinsonism (PARK2). Genomics 1998, 49, 143–146.
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608.
- Takahashi, H.; Ohama, E.; Suzuki, S.; Horikawa, Y.; Ishikawa, A.; Morita, T.; Tsuji, S.; Ikuta, F. Familial juvenile parkinsonism: Clinical and pathologic study in a family. Neurology 1994, 44, 437.
- Yamamura, Y.; Kohriyama, T.; Kawakami, H.; Kaseda, Y.; Kuzuhara, S.; Nakamura, S. Autosomal recessive early-onset parkinsonism with diurnal fluctuation (AR-EPDF)—Clinical characteristics. Rinsho Shinkeigaku 1996, 36, 944–950. (In Japanese)
- Yamamura, Y.; Kuzuhara, S.; Kondo, K.; Yanagi, T.; Uchida, M.; Matsumine, H.; Mizuno, Y. Clinical, pathologic and genetic studies on autosomal recessive early-onset parkinsonism with diurnal fluctuation. Park. Relat. Disord. 1998, 4, 65–72.
- Shimura, H.; Hattori, N.; Kubo, S.-I.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000, 25, 302–305.
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary Early-Onset Parkinson’s Disease Caused by Mutations in PINK1. Science 2004, 304, 1158–1160.
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166.
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166.
- Park, J.; Lee, S.B.; Lee, S.; Kim, Y.; Song, S.; Kim, S.; Bae, E.; Kim, J.; Shong, M.; Kim, J.-M.; et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006, 441, 1157–1161.
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803.
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, e1000298.
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.-S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221.
- Kitada, T.; Hikita, R.; Hirose, H. Parkin, Parkinson disease gene product, directly reduces hydrogen peroxide (mitochondrial oxidant), and forms dimerization reversibly. Int. J. Latest Res. Sci. Technol. 2016, 5, 1–3.
- Kitada, T.; Hikita, R.; Hirose, H. Parkinson’s disease gene product, Parkin, has alternative and reversible functions, both as an E3 ligase and a redox molecule. Int. J. Latest Res. Sci. Technol. 2016, 5, 20–22.
- Tokarew, J.M.; El-Kodsi, D.N.; Lengacher, N.A.; Fehr, T.K.; Nguyen, A.P.; Shutinoski, B.; O’nuallain, B.; Jin, M.; Khan, J.M.; Ng, A.C.H.; et al. Age-associated insolubility of parkin in human midbrain is linked to redox balance and sequestration of reactive dopamine metabolites. Acta Neuropathol. 2021, 141, 725–754.
- Yamamura, Y.; Sobue, I.; Ando, K.; Iida, M.; Yanagi, T.; Kono, C. Paralysis agitans of early onset with marked diurnal fluctuation of symptoms. Neurology 1973, 23, 239.
- Yamamura, Y.; Arihiro, K.; Kohriyama, T.; Nakamura, S. Early-onset parkinsonism with diurnal fluctuation—Clinical and pathological studies. Rinsho Shinkeigaku 1993, 33, 491–496. (In Japanese)
- Schneider, S.A.; Alcalay, R.N. Neuropathology of genetic synucleinopathies with parkinsonism: Review of the literature. Mov. Disord. 2017, 32, 1504–1523.
- Johansen, K.K.; Torp, S.H.; Farrer, M.J.; Gustavsson, E.K.; Aasly, J.O. A Case of Parkinson’s Disease with No Lewy Body Pathology due to a Homozygous Exon Deletion in Parkin. Case Rep. Neurol. Med. 2018, 2018, 6838965.
- Valente, E.M.; Brancati, F.; Ferraris, A.; Graham, E.A.; Davis, M.B.; Breteler, M.M.; Gasser, T.; Bonifati, V.; Bentivoglio, A.R.; De Michele, G.; et al. PARK6-linked parkinsonism occurs in several European families. Ann. Neurol. 2002, 51, 14–18.
- Hayashida, A.; Li, Y.; Yoshino, H.; Daida, K.; Ikeda, A.; Ogaki, K.; Fuse, A.; Mori, A.; Takanashi, M.; Nakahara, T.; et al. The identified clinical features of Parkinson’s disease in homo-, heterozygous and digenic variants of PINK1. Neurobiol. Aging 2021, 97, 146.e1–146.e13.
- Nybø, C.J.; Gustavsson, E.K.; Farrer, M.J.; Aasly, J.O. Neuropathological findings in PINK1-associated Parkinson’s disease. Park. Relat. Disord. 2020, 78, 105–108.
- Van der Merwe, C.; Loos, B.; Swart, C.; Kinnear, C.; Henning, F.; van der Merwe, L.; Pillay, K.; Muller, N.; Zaharie, D.; Engelbrecht, L.; et al. Mitochondrial impairment observed in fibroblasts from South African Parkinson’s disease patients with parkin mutations. Biochem. Biophys. Res. Commun. 2014, 447, 334–340.
- Serdaroglu, P.; Hanagasi, H.; Tasli, H.; Emre, M. Parkin expression in muscle from three patients with autosomal recessive Parkinson’s disease carrying parkin mutation. Acta Myol. 2005, 24, 2–5.
- Hanagasi, H.A.; Serdaroglu, P.; Ozansoy, M.; Basak, N.; Tasli, H.; Emre, M. Mitochondrial pathology in muscle of a patient with a novel parkin mutation. Int. J. Neurosci. 2009, 119, 1572–1583.
- Lavoie, M.J.; Ostaszewski, B.L.; Weihofen, A.; Schlossmacher, M.G.; Selkoe, D.J. Dopamine covalently modifies and functionally inactivates parkin. Nat. Med. 2005, 11, 1214–1221.
- Meng, F.; Yao, D.; Shi, Y.; Kabakoff, J.; Wu, W.; Reicher, J.; Ma, Y.; Moosmann, B.; Masliah, E.; Lipton, S.A.; et al. Oxidation of the cysteine-rich regions of parkin perturbs its E3 ligase activity and contributes to protein aggregation. Mol. Neurodegener. 2011, 6, 34.
- Palacino, J.J.; Sagi, D.; Goldberg, M.S.; Krauss, S.; Motz, C.; Wacker, M.; Klose, J.; Shen, J. Mitochondrial Dysfunction and Oxidative Damage in parkin-deficient Mice. J. Biol. Chem. 2004, 279, 18614–18622.
- Rosen, K.M.; Veereshwarayya, V.; Moussa, C.E.-H.; Fu, Q.; Goldberg, M.S.; Schlossmacher, M.G.; Shen, J.; Querfurth, H.W. Parkin Protects against Mitochondrial Toxins and β-Amyloid Accumulation in Skeletal Muscle Cells. J. Biol. Chem. 2006, 281, 12809–12816.
- Takanashi, M.; Mochizuki, H.; Yokomizo, K.; Hattori, N.; Mori, H.; Yamamura, Y.; Mizuno, Y. Iron accumulation in the substantia nigra of autosomal recessive juvenile parkinsonism (ARJP). Park. Relat. Disord. 2001, 7, 311–314.
- Ardah, M.T.; Radwan, N.; Khan, E.; Kitada, T.; Haque, M.E. Parkin Precipitates on Mitochondria via Aggregation and Autoubiquitination. Int. J. Mol. Sci. 2023, 24, 9027.
More
Information
Subjects:
Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
654
Revisions:
2 times
(View History)
Update Date:
06 Feb 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No