 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yunxiao Wei | -- | 2614 | 2024-02-02 16:22:55 | | | |
| 2 | Rita Xu | -85 word(s) | 2529 | 2024-02-04 02:55:38 | | |
Video Upload Options
The Single-cell Assay for Transposase-Accessible Chromatin with high throughput sequencing (scATAC-seq) has gained increasing popularity in recent years, allowing for chromatin accessibility to be deciphered and gene regulatory networks (GRNs) to be inferred at single-cell resolution. This cutting-edge technology now enables the genome-wide profiling of chromatin accessibility at the cellular level and the capturing of cell-type-specific cis-regulatory elements (CREs) that are masked by cellular heterogeneity in bulk assays. Additionally, it can also facilitate the identification of rare and new cell types based on differences in chromatin accessibility and charting of cellular developmental trajectories within lineage-related cell clusters. Due to technical challenges and limitations, the data generated from scATAC-seq exhibits unique features, often characterized by high sparsity and noise, even within the same cell type. To address these challenges, various bioinformatic tools have been developed. Furthermore, the application of scATAC-seq in plant science is still in its infancy, with most research focusing on root tissues and model plant species.
1. Introduction
2. Challenges for Application of scATAC-seq in Plants
2.1. Preparation of Nuclei Suspensions Compatible with scATAC-seq
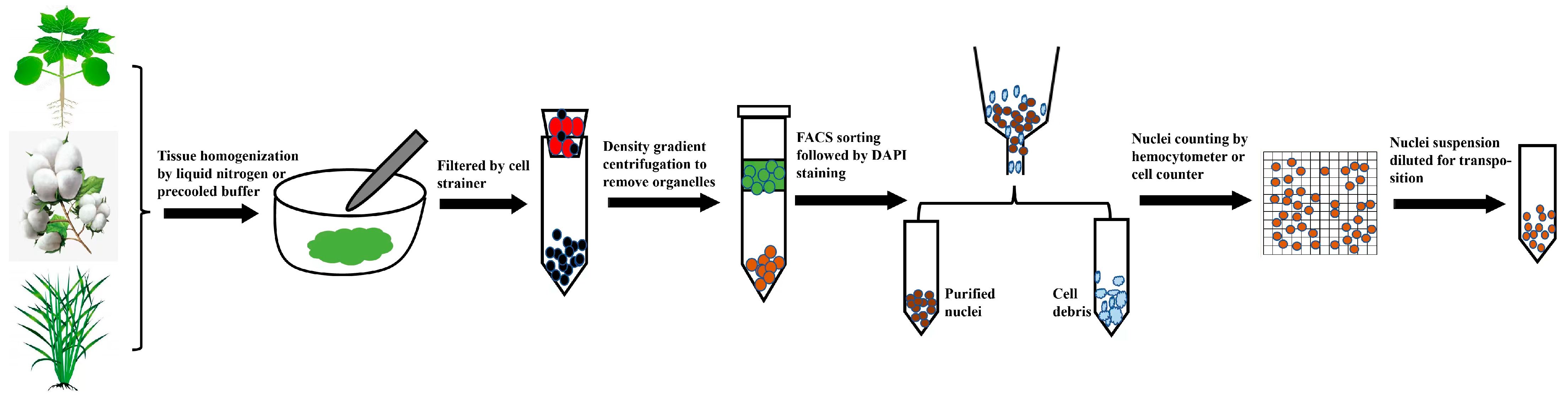
2.2. Analytical Tools Compatible with Plants
2.3. Challenges for Cell Type Annotation in scATAC-seq
2.4. The Annotation Quality of Reference Genomes
References
- Preissl, S.; Gaulton, K.J.; Ren, B. Characterizing cis-regulatory elements using single-cell epigenomics. Nat. Rev. Genet. 2023, 24, 21–43.
- Swinnen, G.; Goossens, A.; Pauwels, L. Lessons from Domestication: Targeting Cis-Regulatory Elements for Crop Improvement. Trends Plant Sci. 2019, 24, 1065.
- Meyer, R.S.; Purugganan, M.D. Evolution of crop species: Genetics of domestication and diversification. Nat. Rev. Genet. 2013, 14, 840–852.
- Springer, N.; de Leon, N.; Grotewold, E. Challenges of Translating Gene Regulatory Information into Agronomic Improvements. Trends Plant Sci. 2019, 24, 1075–1082.
- Huang, C.; Sun, H.; Xu, D.; Chen, Q.; Liang, Y.; Wang, X.; Xu, G.; Tian, J.; Wang, C.; Li, D.; et al. ZmCCT9 enhances maize adaptation to higher latitudes. Proc. Natl. Acad. Sci. USA 2018, 115, E334–E341.
- Studer, A.; Zhao, Q.; Ross-Ibarra, J.; Doebley, J. Identification of a functional transposon insertion in the maize domestication gene tb1. Nat. Genet. 2011, 43, U1160–U1164.
- Jiang, S.; Huang, Z.; Li, Y.; Yu, C.; Yu, H.; Ke, Y.; Jiang, L.; Liu, J. Single-cell chromatin accessibility and transcriptome atlas of mouse embryos. Cell Rep. 2023, 42, 112210.
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220.
- Grandi, F.C.; Modi, H.; Kampman, L.; Corces, M.R. Chromatin accessibility profiling by ATAC-seq. Nat. Protoc. 2022, 17, 1518–1552.
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218.
- Buenrostro, J.D.; Wu, B.; Litzenburger, U.M.; Ruff, D.; Gonzales, M.L.; Snyder, M.P.; Chang, H.Y.; Greenleaf, W.J. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 2015, 523, 486–490.
- You, M.; Chen, L.; Zhang, D.; Zhao, P.; Chen, Z.; Qin, E.Q.; Gao, Y.; Davis, M.M.; Yang, P. Single-cell epigenomic landscape of peripheral immune cells reveals establishment of trained immunity in individuals convalescing from COVID-19. Nat. Cell Biol. 2021, 23, 620–630.
- Wimmers, F.; Donato, M.; Kuo, A.; Ashuach, T.; Gupta, S.; Li, C.; Dvorak, M.; Foecke, M.H.; Chang, S.E.; Hagan, T.; et al. The single-cell epigenomic and transcriptional landscape of immunity to influenza vaccination. Cell 2021, 184, 3915–3935.e21.
- Ranzoni, A.M.; Tangherloni, A.; Berest, I.; Riva, S.G.; Myers, B.; Strzelecka, P.M.; Xu, J.; Panada, E.; Mohorianu, I.; Zaugg, J.B.; et al. Integrative Single-Cell RNA-Seq and ATAC-Seq Analysis of Human Developmental Hematopoiesis. Cell Stem Cell 2021, 28, 472–487.e7.
- Takayama, N.; Murison, A.; Takayanagi, S.I.; Arlidge, C.; Zhou, S.; Garcia-Prat, L.; Chan-Seng-Yue, M.; Zandi, S.; Gan, O.I.; Boutzen, H.; et al. The Transition from Quiescent to Activated States in Human Hematopoietic Stem Cells Is Governed by Dynamic 3D Genome Reorganization. Cell Stem Cell 2021, 28, 488–501.e10.
- Alonso-Curbelo, D.; Ho, Y.J.; Burdziak, C.; Maag, J.L.V.; Morris, J.P.t.; Chandwani, R.; Chen, H.A.; Tsanov, K.M.; Barriga, F.M.; Luan, W.; et al. A gene-environment-induced epigenetic program initiates tumorigenesis. Nature 2021, 590, 642–648.
- Regner, M.J.; Wisniewska, K.; Garcia-Recio, S.; Thennavan, A.; Mendez-Giraldez, R.; Malladi, V.S.; Hawkins, G.; Parker, J.S.; Perou, C.M.; Bae-Jump, V.L.; et al. A multi-omic single-cell landscape of human gynecologic malignancies. Mol. Cell 2021, 81, 4924–4941.e10.
- Feng, D.; Liang, Z.; Wang, Y.; Yao, J.; Yuan, Z.; Hu, G.; Qu, R.; Xie, S.; Li, D.; Yang, L.; et al. Chromatin accessibility illuminates single-cell regulatory dynamics of rice root tips. BMC Biol. 2022, 20, 274.
- Dorrity, M.W.; Alexandre, C.M.; Hamm, M.O.; Vigil, A.L.; Fields, S.; Queitsch, C.; Cuperus, J.T. The regulatory landscape of Arabidopsis thaliana roots at single-cell resolution. Nat. Commun. 2021, 12, 3334.
- Marand, A.P.; Chen, Z.L.; Gallavotti, A.; Schmitz, R.J. A cis-regulatory atlas in maize at single-cell resolution. Cell 2021, 184, 3041–3055.e21.
- Thibivilliers, S.B.; Anderson, D.K.; Libault, M.Y. Isolation of Plant Nuclei Compatible with Microfluidic Single-nucleus ATAC-sequencing. Bio-Protocol 2021, 11, e4240.
- Farmer, A.; Thibivilliers, S.; Ryu, K.H.; Schiefelbein, J.; Libault, M. Single-nucleus RNA and ATAC sequencing reveals the impact of chromatin accessibility on gene expression in Arabidopsis roots at the single-cell level. Mol. Plant 2021, 14, 372–383.
- Neumann, M.; Xu, X.C.; Smaczniak, C.; Schumacher, J.; Yan, W.H.; Bluthgen, N.; Greb, T.; Jonsson, H.; Traas, J.; Kaufmann, K.; et al. A 3D gene expression atlas of the floral meristem based on spatial reconstruction of single nucleus RNA sequencing data. Nat. Commun. 2022, 13, 2838.
- Conde, D.; Triozzi, P.M.; Pereira, W.J.; Schmidt, H.W.; Balmant, K.M.; Knaack, S.A.; Redondo-Lopez, A.; Roy, S.; Dervinis, C.; Kirst, M. Single-nuclei transcriptome analysis of the shoot apex vascular system differentiation in Populus. Development 2022, 149, dev200632.
- Pliner, H.A.; Packer, J.S.; McFaline-Figueroa, J.L.; Cusanovich, D.A.; Daza, R.M.; Aghamirzaie, D.; Srivatsan, S.; Qiu, X.; Jackson, D.; Minkina, A.; et al. Cicero Predicts cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data. Mol. Cell 2018, 71, 858–871.e8.
- Stuart, T.; Srivastava, A.; Madad, S.; Lareau, C.A.; Satija, R. Single-cell chromatin state analysis with Signac. Nat. Methods 2021, 18, 1333–1341.
- Yan, F.; Powell, D.R.; Curtis, D.J.; Wong, N.C. From reads to insight: A hitchhiker’s guide to ATAC-seq data analysis. Genome Biol. 2020, 21, 22.
- Nair, V.D.; Vasoya, M.; Nair, V.; Smith, G.R.; Pincas, H.; Ge, Y.C.; Douglas, C.M.; Esser, K.A.; Sealfon, S.C. Optimization of the Omni-ATAC protocol to chromatin accessibility profiling in snap-frozen rat adipose and muscle tissues. Methodsx 2022, 9, 101681.
- Nadelmann, E.R.; Gorham, J.M.; Reichart, D.; Delaughter, D.M.; Wakimoto, H.; Lindberg, E.L.; Litvinukova, M.; Maatz, H.; Curran, J.J.; Ischiu Gutierrez, D.; et al. Isolation of Nuclei from Mammalian Cells and Tissues for Single-Nucleus Molecular Profiling. Curr. Protoc. 2021, 1, e132.
- Wiegleb, G.; Reinhardt, S.; Dahl, A.; Posnien, N. Tissue dissociation for single-cell and single-nuclei RNA sequencing for low amounts of input material. Front. Zool. 2022, 19, 27.
- Narayanan, A.; Blanco-Carmona, E.; Demirdizen, E.; Sun, X.Y.; Herold-Mende, C.; Schlesner, M.; Turcan, S. Nuclei Isolation from Fresh Frozen Brain Tumors for Single-Nucleus RNA-seq and ATAC-seq. Jove-J. Vis. Exp. 2020, 162, e61542.
- Corces, M.R.; Trevino, A.E.; Hamilton, E.G.; Greenside, P.G.; Sinnott-Armstrong, N.A.; Vesuna, S.; Satpathy, A.T.; Rubin, A.J.; Montine, K.S.; Wu, B.; et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 2017, 14, 959–962.
- Sikorskaite, S.; Rajamaki, M.L.; Baniulis, D.; Stanys, V.; Valkonen, J.P.T. Protocol: Optimised methodology for isolation of nuclei from leaves of species in the Solanaceae and Rosaceae families. Plant Methods 2013, 9, 31.
- Loureiro, J.; Kron, P.; Temsch, E.M.; Koutecky, P.; Lopes, S.; Castro, M.; Castro, S. Isolation of plant nuclei for estimation of nuclear DNA content: Overview and best practices. Cytom. Part A 2021, 99, 318–327.
- Thibivilliers, S.; Anderson, D.; Libault, M. Isolation of Plant Root Nuclei for Single Cell RNA Sequencing. Curr. Protoc. Plant Biol. 2020, 5, e20120.
- Wang, K.; Zhao, C.; Xiang, S.; Duan, K.; Chen, X.; Guo, X.; Sahu, S.K. An optimized FACS-free single-nucleus RNA sequencing (snRNA-seq) method for plant science research. Plant Sci. 2023, 326, 111535.
- Conde, D.; Triozzi, P.M.; Balmant, K.M.; Doty, A.L.; Miranda, M.; Boullosa, A.; Schmidt, H.W.; Pereira, W.J.; Dervinis, C.; Kirst, M. A robust method of nuclei isolation for single-cell RNA sequencing of solid tissues from the plant genus Populus. PLoS ONE 2021, 16, e0251149.
- Tu, X.Y.; Marand, A.P.; Schmitz, R.J.; Zhong, S.L. A combinatorial indexing strategy for low-cost epigenomic profiling of plant single cells. Plant Commun. 2022, 3, 100308.
- Wang, C.; Sun, D.; Huang, X.; Wan, C.; Li, Z.; Han, Y.; Qin, Q.; Fan, J.; Qiu, X.; Xie, Y.; et al. Integrative analyses of single-cell transcriptome and regulome using MAESTRO. Genome Biol. 2020, 21, 198.
- Granja, J.M.; Corces, M.R.; Pierce, S.E.; Bagdatli, S.T.; Choudhry, H.; Chang, H.Y.; Greenleaf, W.J. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 2021, 53, 403–411.
- Fang, R.; Preissl, S.; Li, Y.; Hou, X.; Lucero, J.; Wang, X.; Motamedi, A.; Shiau, A.K.; Zhou, X.; Xie, F.; et al. Comprehensive analysis of single cell ATAC-seq data with SnapATAC. Nat. Commun. 2021, 12, 1337.
- Vlot, A.H.C.; Maghsudi, S.; Ohler, U. Cluster-independent marker feature identification from single-cell omics data using SEMITONES. Nucleic Acids Res. 2022, 50, e107.
- Shahan, R.; Hsu, C.W.; Nolan, T.M.; Cole, B.J.; Taylor, I.W.; Greenstreet, L.; Zhang, S.; Afanassiev, A.; Vlot, A.H.C.; Schiebinger, G.; et al. A single-cell Arabidopsis root atlas reveals developmental trajectories in wild-type and cell identity mutants. Dev. Cell 2022, 57, 543–560.e9.
- Dai, M.; Pei, X.; Wang, X.J. Accurate and fast cell marker gene identification with COSG. Brief. Bioinform. 2022, 23, bbab579.
- Sun, S.; Shen, X.; Li, Y.; Li, Y.; Wang, S.; Li, R.; Zhang, H.; Shen, G.; Guo, B.; Wei, J.; et al. Single-cell RNA sequencing provides a high-resolution roadmap for understanding the multicellular compartmentation of specialized metabolism. Nat. Plants 2023, 9, 179–190.

