Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Margarita L. Martinez-Fierro | -- | 1918 | 2024-02-02 11:17:19 | | | |
| 2 | Jason Zhu | Meta information modification | 1918 | 2024-02-04 02:39:14 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Perez-Favila, A.; Garza-Veloz, I.; Hernandez-Marquez, L.D.S.; Gutierrez-Vela, E.F.; Flores-Morales, V.; Martinez-Fierro, M.L. Mechanisms in Development of Idiopathic Pulmonary Fibrosis. Encyclopedia. Available online: https://encyclopedia.pub/entry/54704 (accessed on 18 June 2026).
Perez-Favila A, Garza-Veloz I, Hernandez-Marquez LDS, Gutierrez-Vela EF, Flores-Morales V, Martinez-Fierro ML. Mechanisms in Development of Idiopathic Pulmonary Fibrosis. Encyclopedia. Available at: https://encyclopedia.pub/entry/54704. Accessed June 18, 2026.
Perez-Favila, Aurelio, Idalia Garza-Veloz, Lucia Del Socorro Hernandez-Marquez, Edgar Fernando Gutierrez-Vela, Virginia Flores-Morales, Margarita L. Martinez-Fierro. "Mechanisms in Development of Idiopathic Pulmonary Fibrosis" Encyclopedia, https://encyclopedia.pub/entry/54704 (accessed June 18, 2026).
Perez-Favila, A., Garza-Veloz, I., Hernandez-Marquez, L.D.S., Gutierrez-Vela, E.F., Flores-Morales, V., & Martinez-Fierro, M.L. (2024, February 02). Mechanisms in Development of Idiopathic Pulmonary Fibrosis. In Encyclopedia. https://encyclopedia.pub/entry/54704
Perez-Favila, Aurelio, et al. "Mechanisms in Development of Idiopathic Pulmonary Fibrosis." Encyclopedia. Web. 02 February, 2024.
Copy Citation
Fibroblasts synthesize collagen, fibronectin, and extracellular matrix (ECM). Myofibroblasts—other cells involved in fibrosis—secrete factors such as VEGF and TGF-β, produce denser but more disorganized ECM than fibroblasts, and persist longer at the injury site. One cytokine involved in tissue repair is TGF-β. Sources of TGF-β include platelet granules and macrophages. TGF-β is predominantly expressed in PF and helps stimulate the formation of ECM, collagen, fibronectin, elastic fibers, and matrix substances.
antifibrotic drugs
pulmonary fibrosis
COVID-19
1. Cells Involved in the Development of IPF
Several cells contribute to the development of PF by producing growth factors and cytokines, which contribute to the fibrotic process and pathogenesis of the disease.
1.1. Macrophage Activation
Macrophages are a type of cell that plays an essential role in innate immunity and inflammation [1]. There are two main macrophage phenotypes: M1, which is associated with proinflammatory responses, and M2 macrophages (M2a, M2b, M2c, and M2d), which play crucial roles in anti-inflammatory processes [2]. The transition from one phenotype to another is highly variable, and activation can be a rapid and reversible process [3]. Macrophage activation occurs through several pathways. The canonical IRF/STAT signaling pathways are activated by IFN and TLR to activate the M1 macrophage phenotype (STAT1) or to activate the M2 phenotype. IL-4 and IL-3 (STAT6) are required [4]. M1 macrophages increase the regulation of IRF5, thereby stimulating cytokines (IL-12, IL-23, TNF) involved in Th1 and Th17 responses [5]. On the other hand, it has been seen that interleukin IL-10 can activate the expression of genes mediated by STAT3, which can be associated with the M2 phenotype [6].
Two populations of macrophages have been identified in the regulation of pulmonary homeostasis: alveolar macrophages (AMs), which are located in the lumen of the airways, whose markers are CD11blow, CD11c++, and CD169+, and interstitial macrophages (IMs), which are located in the lung parenchyma, with markers CD11b+, CD11clow, CD169 [7]. Macrophage signaling pathways in PF are TGF-β/Smad, Wnt/β-catenin, and interleukin signaling [8][9][10]. In a study conducted with patients with severe COVID-19, the accumulation of CD163+ macrophages were reported, some of which co-expressed the chemokine receptor CXCR3 and complement factor C1Q. In addition, collagen deposition was reported very prominently, for which the authors propose that SARS-CoV-2 promotes genetic programs associated with fibrosis in macrophages [11].
1.2. Activation of Fibroblasts and its Differentiation to Myofibroblasts
Fibroblasts are mesenchymal cells whose function is homeostasis and production of ECM [12]. In the lung, fibroblasts are found in the ECM of the interstitial space, where they participate in early and late wound repair [13]. For the progression of PF, a key factor is the proliferation and differentiation of fibroblasts into myofibroblasts [14]. TGF-β 1 participates in stimulating the differentiation of fibroblasts into myofibroblasts [15]. A study found that miR-21 expression is involved in collagen production by fibroblasts or fibroblast-like cells through negative regulation of Smad7. This suggests that Smad7 could be a potential therapeutic target in PF [16].
In IPF, type 1 collagen is the most abundant and relevant collagen, as it is deposited in greater quantities in the ECM [17][18]. Fibroblasts and myofibroblasts mainly secrete this type of collagen due to the action of several cytokines and factors, such as TGF-β, in addition to the hypoxia pathway [19]. In a study conducted by Philp, C. J et al., the inhibition of the formation of crosslinks through transglutaminase 2 modified the behavior of fibroblasts by reducing adhesion and proliferation of these; it also improved the renewal of ECM, which would mean that it could be a potential therapy for IPF by reducing excess ECM at the site of injury [20]. Figure 1 displays antifibrotic drugs with evidence of their use in PF related to COVID-19 and their possible mechanisms of antifibrotic action. For instance, Nimotuzumab inhibits EGFR, thereby inhibiting the JAK/STAT pathway and preventing fibrosis from progressing or occurring [21].
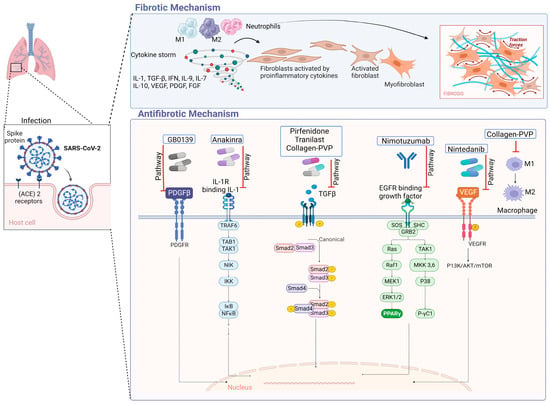
Figure 1. Antifibrotic drugs. Figure shows a possible antifibrotic mechanism involved in the treatment of pulmonary fibrosis induced by COVID-19. Pirfenidone, Nintedanib, and collagen-polyvinylpyrrolidone have different mechanisms that may regulate PF, while Nimotuzumab, Anakinra, Tranilast, and GB0139 have more specific mechanisms of action [22][23][24][25][26][27][28].
2. Signaling Pathways Involved in the Development of IPF
The pathological process by which IPF develops is very complex and involves the activation of alveolar epithelial cells as well as the proliferation of fibroblasts and differentiation into myofibroblasts; likewise, various growth factors such as TGF-β intervene, leading to irreversible damage to the alveolar architecture.
2.1. TGF-β Signaling
TGF-β is a member of a family of polypeptides that modulate various cellular functions, the most important of which are cell proliferation, differentiation, and apoptosis [29]. There are three major inactive isoforms of TGF-β (TGF-β1, TGF-β2, TGF-β3), which are synthesized and secreted in association with latency-associated peptide (LAP) [30]. When there are normal conditions, TGF-β in its inactive conformation binds to LAP, which, through disulfide bonds, binds to latent TGF-β-binding proteins, crosslinked with the ECM [31]. TGF-β can be activated within the large latent complex (CLL) in various ways, for example, acidification, temperature variations, oxidation, proteolytic cleavage, or through interaction with integrins, which will cause TGF-β to interact with TGF-β receptors (TGF-βR), and the consequent action is to mediate downstream effects [32]. TGF-β plays an important role in the development of IPF [33][34]. Upregulation and activation of all TGF-β isoforms and receptors have been reported to be involved in the pathogenesis of IPF [35].
It is postulated that TGF-β signaling, which is a profibrotic inducer, is stimulated by the protein N of SARS-CoV. Since this protein is 90% similar to that of SARS-CoV-2, it is considered to be one of the possible mechanisms of pulmonary fibrosis associated with SARS-CoV-2 infection [36]. Another possible mechanism is that coronaviruses can induce the lowering of ACE2; therefore, there is a reduction of angiotensin II (Ang II) in the lungs. In this way, Ang II could upregulate TGF-β and connective tissue growth factor [37]. Once the lung injury occurs, TGF-β, in addition to being released by the injured epithelial and endothelial cells, is released by macrophages, fibroblasts, T cells, and monocytes, which results in a self-sustained process [32]. The canonical TGF-β signaling pathway will involve the activation of SMAD proteins that will activate transcription [38]. TGF-β, in its non-canonical pathway, can act on mitogen-activated protein kinase (MAPK), extracellular signal-regulated kinase (ERK), and N-terminal Jun N kinase (JNK), as well as RHO-associated kinase (ROCK) and AKT pathways [39]. Some of these pathways are involved in the induction of the epithelial–mesenchymal transition (EMT), which promotes the exacerbation of fibrotic lesions [40].
2.2. Activation of Integrins
Integrins belong to a family of proteins composed of one of 18 unique α subunits and eight β subunits that pair to create one of the 24 identified integrins [41]. They play a crucial role in maintaining cell–cell junctions, cell–EM adhesion, and signal transduction from the ECM to the intracellular compartment [42]. Integrins have been found to participate in various disease processes, including sarcoidosis and PF [43]. Recent research has demonstrated that the S protein of SARS-CoV-2 binds to integrin ⍺5β1 in vascular endothelial cells (ECs) through the RGD domain. This binding activates the expression of nuclear factor kappa β (NF-κβ), which is responsible for vascular leakage and leukocyte adhesion. These findings suggest a new mechanism by which SARS-CoV-2 affects ECs and identify integrin ⍺5β1 as a potential therapeutic target for treating COVID-19-related inflammation [44]. αVβ6 is another integrin that recognizes RGD (Arg-Gly-Asp). It is overexpressed in many aggressive epithelial tumors, as well as in pulmonary and hepatic fibrosis [45]. A study found that high levels of αVβ6 suggest the presence of fibroblast foci and can also identify a specific endotype of progressive fibrotic lung disease [46].
3. Growth Factors Involved in Idiopathic Pulmonary Fibrosis
Several growth factors, including PDGF, CTGF, and FGF, have been reported to play a role in the development of IPF by regulating the activity of fibroblasts.
- -
-
PDGF is a mitogenic molecule that affects various connective tissue cells and other cell types [47]. This factor is composed of polypeptide chains A and B linked by a disulfide bond, which can form homo- and heterodimers [48]. PDGF is produced by several cell types, including macrophages, platelets, endothelial cells, and fibroblasts [49]. It has four isoforms that bind to two receptor tyrosine kinases (PDGFR α and β). These receptors are expressed in higher quantities in cells such as fibroblasts and myofibroblasts, where they participate in survival, proliferation, and migration [50]. PDGF works together with TGF-β to promote the release of activated alveolar macrophages and epithelial cells, which are directly involved in the self-activation cycle of fibrosis [51]. High levels of PDGF have been associated with FP, both in lung tissue and in bronchoalveolar lavage fluid, as demonstrated in various animal studies with models of PF [52]. PDGF-A and PDGF-C have also been found to be overexpressed in various cell types in a mouse model of injured lungs [53]. During pulmonary fibrosis, alveolar macrophages promote the release of PDGF, which contributes to the proliferation of alveolar myofibroblasts and fibrogenesis [54].
- -
-
Fibroblast growth factors (FGFs) belong to a family of 22 members [55][56]. These can be divided into hormone-like FGFs (FGF 15/19, 21 and 23), canonical FGFs (FGF 1–10, 16–18, 20 and 22), and intracellular FGFs (FGF 11–14) [57]. FGFs are involved in several biological responses, interacting with heparan sulfate glycosaminoglycans (HPSGs). Released FGFs interact with cell surface receptor (FGFR) domains [58]; a complex of FGF, FGFR, and HPSG is formed, and thus, signaling is carried out [59]. Abnormal FGF signaling is implicated in various pathologies and is known to participate in the pathogenesis of PF [60]. The inhibition of FGF signaling by using FGFR2c decreased lung fibroblast proliferation and differentiation in vitro and in vivo in a murine model [61]. The alteration in FGFR1 and FGF1 signaling is critical for fibroblast migration in PF [62]. In SARS-CoV infection, the EGFR signaling pathway remains active even after the virus is eliminated. This pathway is believed to be responsible for the effect of FGF in the conduction to PF [63].
- -
-
Connective tissue growth factor (CTGF) belongs to the IGFBP family, which is known as insulin-like growth factor-binding protein 8 (IGFBP-8) [64][65]. This 38 kDa mitogenic peptide is involved in fibrotic processes and stimulates migration, fibroblast proliferation, and ECM production [66]. CTGF is a cytokine that participates in the activation of fibroblasts; it was observed that in alveolar epithelial cells, the expression levels of CTGF were drastically reduced by inhibiting the Rho pathway. Additionally, the epithelial cells of the lung in the mice indicators of CTGF increased the activity of the promoter of CTGF when the authors treated them with bleomycin, an inducer of PF [67]. CTGF can interact with other molecules to develop its fibrotic effects, thus contributing to the generation of fibrosis; among the molecules with which it interacts, there are cytokines and growth factors such as IGF1, BMP4, BMP7, TGF-β and VEGF. This interaction with other molecules may positively or negatively alter the signaling pathways of which they are participants, resulting in changes in cell adhesion, migration, differentiation, angiogenesis, myofibroblast activation, deposition, and remodeling of ECM, which causes changes in structure and alters tissue remodeling [68].
- -
-
Metalloproteinases (MMPs) belong to the family of extracellular endopeptidases, whose primary function is the degradation of ECM components, while tissue inhibitors of metalloproteinases (TIMPs) block the activity of MMPs [69]. The participation of proinflammatory cytokines affects the overexpression of MMPs, which increases its activity and thus participates in the remodeling of the airways [70]. The level of TIMPs is increased in the presence of a fibrotic process such as PF [71]. MMP-2 and MMP-9 are downregulated in severe COVID-19 patients [72]. MMP-2 has shown a correlation with mortality in patients with COVID-19, so it could be a potential prognostic predictor for patients with COVID-19 [73]. One study reported that MMP-7 correlates with clinical and functional predictors of disease severity and mortality, so it can be used to distinguish IPF from other chronic diseases [74].
References
- Wen, J.H.; Li, D.Y.; Liang, S.; Yang, C.; Tang, J.X.; Liu, H.F. Macrophage autophagy in macrophage polarization, chronic inflammation and organ fibrosis. Front. Immunol. 2022, 13, 946832.
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795.
- Perciani, C.T.; MacParland, S.A. Lifting the veil on macrophage diversity in tissue regeneration and fibrosis. Sci. Immunol. 2019, 4, eaaz0749.
- Sica, A.; Bronte, V. Altered macrophage differentiation and immune dysfunction in tumor development. J. Clin. Investig. 2007, 117, 1155–1166.
- Krausgruber, T.; Blazek, K.; Smallie, T.; Alzabin, S.; Lockstone, H.; Sahgal, N.; Hussell, T.; Feldmann, M.; Udalova, I.A. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 2011, 12, 231–238.
- Lang, R.; Patel, D.; Morris, J.J.; Rutschman, R.L.; Murray, P.J. Shaping gene expression in activated and resting primary macrophages by IL-10. J. Immunol. 2002, 169, 2253–2263.
- Byrne, A.J.; Maher, T.M.; Lloyd, C.M. Pulmonary Macrophages: A New Therapeutic Pathway in Fibrosing Lung Disease? Trends Mol. Med. 2016, 22, 303–316.
- Zhu, L.; Fu, X.; Chen, X.; Han, X.; Dong, P. M2 macrophages induce EMT through the TGF-beta/Smad2 signaling pathway. Cell Biol. Int. 2017, 41, 960–968.
- Cao, H.; Wang, C.; Chen, X.; Hou, J.; Xiang, Z.; Shen, Y.; Han, X. Inhibition of Wnt/beta-catenin signaling suppresses myofibroblast differentiation of lung resident mesenchymal stem cells and pulmonary fibrosis. Sci. Rep. 2018, 8, 13644.
- Guo, X.; Li, T.; Xu, Y.; Xu, X.; Zhu, Z.; Zhang, Y.; Xu, J.; Xu, K.; Cheng, H.; Zhang, X.; et al. Increased levels of Gab1 and Gab2 adaptor proteins skew interleukin-4 (IL-4) signaling toward M2 macrophage-driven pulmonary fibrosis in mice. J. Biol. Chem. 2017, 292, 14003–14015.
- Wendisch, D.; Dietrich, O.; Mari, T.; von Stillfried, S.; Ibarra, I.L.; Mittermaier, M.; Mache, C.; Chua, R.L.; Knoll, R.; Timm, S.; et al. SARS-CoV-2 infection triggers profibrotic macrophage responses and lung fibrosis. Cell 2021, 184, 6243–6261.e6227.
- Plikus, M.V.; Wang, X.; Sinha, S.; Forte, E.; Thompson, S.M.; Herzog, E.L.; Driskell, R.R.; Rosenthal, N.; Biernaskie, J.; Horsley, V. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 2021, 184, 3852–3872.
- White, E.S. Lung extracellular matrix and fibroblast function. Ann. Am. Thorac. Soc. 2015, 12 (Suppl. S1), S30–S33.
- Khalil, N.; O’Connor, R. Idiopathic pulmonary fibrosis: Current understanding of the pathogenesis and the status of treatment. CMAJ 2004, 171, 153–160.
- Li, X.; Bi, Z.; Liu, S.; Gao, S.; Cui, Y.; Huang, K.; Huang, M.; Mao, J.; Li, L.; Gao, J.; et al. Antifibrotic Mechanism of Cinobufagin in Bleomycin-Induced Pulmonary Fibrosis in Mice. Front. Pharmacol. 2019, 10, 1021.
- Kwon, O.S.; Kim, K.T.; Lee, E.; Kim, M.; Choi, S.H.; Li, H.; Fornace, A.J., Jr.; Cho, J.H.; Lee, Y.S.; Lee, J.S.; et al. Induction of MiR-21 by Stereotactic Body Radiotherapy Contributes to the Pulmonary Fibrotic Response. PLoS ONE 2016, 11, e0154942.
- Hewlett, J.C.; Kropski, J.A.; Blackwell, T.S. Idiopathic pulmonary fibrosis: Epithelial-mesenchymal interactions and emerging therapeutic targets. Matrix Biol. 2018, 71–72, 112–127.
- Raghu, G.; Striker, L.J.; Hudson, L.D.; Striker, G.E. Extracellular matrix in normal and fibrotic human lungs. Am. Rev. Respir. Dis. 1985, 131, 281–289.
- Bellaye, P.S.; Shimbori, C.; Upagupta, C.; Sato, S.; Shi, W.; Gauldie, J.; Ask, K.; Kolb, M. Lysyl Oxidase-Like 1 Protein Deficiency Protects Mice from Adenoviral Transforming Growth Factor-beta1-induced Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 461–470.
- Philp, C.J.; Siebeke, I.; Clements, D.; Miller, S.; Habgood, A.; John, A.E.; Navaratnam, V.; Hubbard, R.B.; Jenkins, G.; Johnson, S.R. Extracellular Matrix Cross-Linking Enhances Fibroblast Growth and Protects against Matrix Proteolysis in Lung Fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 594–603.
- Abdo Cuza, A.A.; Avila, J.P.; Martinez, R.M.; Gonzalez, J.J.; Aspuro, G.P.; Gutierrez Martinez, J.A.; Suzarte, M.R.; Hernandez, D.S.; Ane-Kouri, A.L.; Ramos, T.C. Nimotuzumab for COVID-19: Case series. Immunotherapy 2021, 14, 185–193.
- Saiphoklang, N.; Patanayindee, P.; Ruchiwit, P. The Effect of Nintedanib in Post-COVID-19 Lung Fibrosis: An Observational Study. Crit. Care Res. Pr. 2022, 2022, 9972846.
- Londres, H.D.; Armada, J.J.; Martinez, A.H.; Abdo Cuza, A.A.; Sanchez, Y.H.; Rodriguez, A.G.; Figueroa, S.S.; Llanez Gregorich, E.M.; Torres Lahera, M.L.; Peire, F.G.; et al. Blocking EGFR with nimotuzumab: A novel strategy for COVID-19 treatment. Immunotherapy 2022, 14, 521–530.
- Acat, M.; Yildiz Gulhan, P.; Oner, S.; Turan, M.K. Comparison of pirfenidone and corticosteroid treatments at the COVID-19 pneumonia with the guide of artificial intelligence supported thoracic computed tomography. Int. J. Clin. Pr. 2021, 75, e14961.
- Gaughan, E.E.; Quinn, T.M.; Mills, A.; Bruce, A.M.; Antonelli, J.; MacKinnon, A.C.; Aslanis, V.; Li, F.; O’Connor, R.; Boz, C.; et al. An Inhaled Galectin-3 Inhibitor in COVID-19 Pneumonitis: A Phase Ib/IIa Randomized Controlled Clinical Trial (DEFINE). Am. J. Respir. Crit. Care Med. 2023, 207, 138–149.
- Mendez-Flores, S.; Priego-Ranero, A.; Azamar-Llamas, D.; Olvera-Prado, H.; Rivas-Redonda, K.I.; Ochoa-Hein, E.; Perez-Ortiz, A.; Rendon-Macias, M.E.; Rojas-Castaneda, E.; Urbina-Teran, S.; et al. Effect of polymerised type I collagen on hyperinflammation of adult outpatients with symptomatic COVID-19. Clin. Transl. Med. 2022, 12, e763.
- Oshitani, N.; Watanabe, K.; Sakuma, T.; Matsuda, M.; Oyama, Y. Tranilast, an antifibrotic agent and COVID-19-induced pulmonary fibrosis. QJM 2022, 115, 249–250.
- Nan, D.; Abraira-Meriel, C.; de la Roz-Fernandez, S.; Maestre-Orozco, T.; Hernandez, J.L.; Fernandez-Ayala, M. Delayed Use of the Recombinant Human IL-1 Receptor Antagonist Anakinra in Five COVID-19 Patients with Pulmonary Fibrosis and Persistent Hypoxaemia: A Preliminary Report. Eur. J. Case Rep. Intern. Med. 2021, 8, 002821.
- Hyytiainen, M.; Penttinen, C.; Keski-Oja, J. Latent TGF-beta binding proteins: Extracellular matrix association and roles in TGF-beta activation. Crit. Rev. Clin. Lab. Sci. 2004, 41, 233–264.
- Saharinen, J.; Taipale, J.; Keski-Oja, J. Association of the small latent transforming growth factor-beta with an eight cysteine repeat of its binding protein LTBP-1. EMBO J. 1996, 15, 245–253.
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGFbeta activation. J. Cell Sci. 2003, 116, 217–224.
- Aschner, Y.; Downey, G.P. Transforming Growth Factor-beta: Master Regulator of the Respiratory System in Health and Disease. Am. J. Respir. Cell Mol. Biol. 2016, 54, 647–655.
- Sheppard, D. Transforming growth factor beta: A central modulator of pulmonary and airway inflammation and fibrosis. Proc. Am. Thorac. Soc. 2006, 3, 413–417.
- Xu, Y.D.; Hua, J.; Mui, A.; O’Connor, R.; Grotendorst, G.; Khalil, N. Release of biologically active TGF-beta1 by alveolar epithelial cells results in pulmonary fibrosis. Am. J. Physiol. Lung. Cell Mol. Physiol. 2003, 285, L527–L539.
- Bartram, U.; Speer, C.P. The role of transforming growth factor beta in lung development and disease. Chest 2004, 125, 754–765.
- Gentile, F.; Aimo, A.; Forfori, F.; Catapano, G.; Clemente, A.; Cademartiri, F.; Emdin, M.; Giannoni, A. COVID-19 and risk of pulmonary fibrosis: The importance of planning ahead. Eur. J. Prev. Cardiol. 2020, 27, 1442–1446.
- Mohammadi, A.; Balan, I.; Yadav, S.; Matos, W.F.; Kharawala, A.; Gaddam, M.; Sarabia, N.; Koneru, S.C.; Suddapalli, S.K.; Marzban, S. Post-COVID-19 Pulmonary Fibrosis. Cureus 2022, 14, e22770.
- Ye, Z.; Hu, Y. TGFbeta1: Gentlemanly orchestrator in idiopathic pulmonary fibrosis (Review). Int. J. Mol. Med. 2021, 48, 132.
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584.
- Giacomelli, C.; Piccarducci, R.; Marchetti, L.; Romei, C.; Martini, C. Pulmonary fibrosis from molecular mechanisms to therapeutic interventions: Lessons from post-COVID-19 patients. Biochem. Pharmacol. 2021, 193, 114812.
- Barczyk, M.; Carracedo, S.; Gullberg, D. Integrins. Cell Tissue Res. 2010, 339, 269–280.
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687.
- Teoh, C.M.; Tan, S.S.; Tran, T. Integrins as Therapeutic Targets for Respiratory Diseases. Curr. Mol. Med. 2015, 15, 714–734.
- Robles, J.P.; Zamora, M.; Adan-Castro, E.; Siqueiros-Marquez, L.; Martinez de la Escalera, G.; Clapp, C. The spike protein of SARS-CoV-2 induces endothelial inflammation through integrin alpha5beta1 and NF-kappaB signaling. J. Biol. Chem. 2022, 298, 101695.
- Bugatti, K. alphaV beta6 Integrin: An Intriguing Target for COVID-19 and Related Diseases. Chembiochem 2021, 22, 2516–2520.
- Saini, G.; Porte, J.; Weinreb, P.H.; Violette, S.M.; Wallace, W.A.; McKeever, T.M.; Jenkins, G. alphavbeta6 integrin may be a potential prognostic biomarker in interstitial lung disease. Eur. Respir. J. 2015, 46, 486–494.
- Raica, M.; Cimpean, A.M. Platelet-Derived Growth Factor (PDGF)/PDGF Receptors (PDGFR) Axis as Target for Antitumor and Antiangiogenic Therapy. Pharmaceuticals 2010, 3, 572–599.
- Heldin, C.H.; Eriksson, U.; Ostman, A. New members of the platelet-derived growth factor family of mitogens. Arch. Biochem. Biophys. 2002, 398, 284–290.
- Heldin, C.H.; Westermark, B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 1999, 79, 1283–1316.
- Betsholtz, C. Biology of platelet-derived growth factors in development. Birth Defects Res. C Embryo Today 2003, 69, 272–285.
- Abdollahi, A.; Li, M.; Ping, G.; Plathow, C.; Domhan, S.; Kiessling, F.; Lee, L.B.; McMahon, G.; Grone, H.J.; Lipson, K.E.; et al. Inhibition of platelet-derived growth factor signaling attenuates pulmonary fibrosis. J. Exp. Med. 2005, 201, 925–935.
- Bonner, J.C. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev. 2004, 15, 255–273.
- Zhuo, Y.; Zhang, J.; Laboy, M.; Lasky, J.A. Modulation of PDGF-C and PDGF-D expression during bleomycin-induced lung fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 286, L182–L188.
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 22, 1276–1312.
- Prudovsky, I. Cellular Mechanisms of FGF-Stimulated Tissue Repair. Cells 2021, 10, 1830.
- Jones, S.A. Physiology of FGF15/19. Adv. Exp. Med. Biol. 2012, 728, 171–182.
- Brooks, A.N.; Kilgour, E.; Smith, P.D. Molecular pathways: Fibroblast growth factor signaling: A new therapeutic opportunity in cancer. Clin. Cancer Res. 2012, 18, 1855–1862.
- Olsen, S.K.; Garbi, M.; Zampieri, N.; Eliseenkova, A.V.; Ornitz, D.M.; Goldfarb, M.; Mohammadi, M. Fibroblast growth factor (FGF) homologous factors share structural but not functional homology with FGFs. J. Biol. Chem. 2003, 278, 34226–34236.
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253.
- Guzy, R.D.; Li, L.; Smith, C.; Dorry, S.J.; Koo, H.Y.; Chen, L.; Ornitz, D.M. Pulmonary fibrosis requires cell-autonomous mesenchymal fibroblast growth factor (FGF) signaling. J. Biol. Chem. 2017, 292, 10364–10378.
- Yu, Z.H.; Wang, D.D.; Zhou, Z.Y.; He, S.L.; Chen, A.A.; Wang, J. Mutant soluble ectodomain of fibroblast growth factor receptor-2 IIIc attenuates bleomycin-induced pulmonary fibrosis in mice. Biol. Pharm. Bull. 2012, 35, 731–736.
- MacKenzie, B.; Korfei, M.; Henneke, I.; Sibinska, Z.; Tian, X.; Hezel, S.; Dilai, S.; Wasnick, R.; Schneider, B.; Wilhelm, J.; et al. Increased FGF1-FGFRc expression in idiopathic pulmonary fibrosis. Respir. Res. 2015, 16, 83.
- Venkataraman, T.; Coleman, C.M.; Frieman, M.B. Overactive Epidermal Growth Factor Receptor Signaling Leads to Increased Fibrosis after Severe Acute Respiratory Syndrome Coronavirus Infection. J. Virol. 2017, 91, e00182-17.
- Kim, H.S.; Nagalla, S.R.; Oh, Y.; Wilson, E.; Roberts, C.T., Jr.; Rosenfeld, R.G. Identification of a family of low-affinity insulin-like growth factor binding proteins (IGFBPs): Characterization of connective tissue growth factor as a member of the IGFBP superfamily. Proc. Natl. Acad. Sci. USA 1997, 94, 12981–12986.
- Allen, J.T.; Knight, R.A.; Bloor, C.A.; Spiteri, M.A. Enhanced insulin-like growth factor binding protein-related protein 2 (Connective tissue growth factor) expression in patients with idiopathic pulmonary fibrosis and pulmonary sarcoidosis. Am. J. Respir. Cell Mol. Biol. 1999, 21, 693–700.
- Saito, S.; Alkhatib, A.; Kolls, J.K.; Kondoh, Y.; Lasky, J.A. Pharmacotherapy and adjunctive treatment for idiopathic pulmonary fibrosis (IPF). J. Thorac. Dis. 2019, 11, S1740–S1754.
- Yang, J.; Velikoff, M.; Canalis, E.; Horowitz, J.C.; Kim, K.K. Activated alveolar epithelial cells initiate fibrosis through autocrine and paracrine secretion of connective tissue growth factor. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L786–L796.
- Lipson, K.E.; Wong, C.; Teng, Y.; Spong, S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair. 2012, 5, S24.
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73.
- Amalinei, C.; Caruntu, I.D.; Balan, R.A. Biology of metalloproteinases. Rom. J. Morphol. Embryol. 2007, 48, 323–334.
- Selman, M.; Ruiz, V.; Cabrera, S.; Segura, L.; Ramirez, R.; Barrios, R.; Pardo, A. TIMP-1, -2, -3, and -4 in idiopathic pulmonary fibrosis. A prevailing nondegradative lung microenvironment? Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L562–L574.
- Ghezelbash, B.; Rostami, M.; Heidarvand, M.; Mafi, A.; Chegni, H.; Eskandari, N. Correlation of Expression of MMP-2, ACE2, and TMPRSS2 Genes with Lymphopenia for Mild and Severity of COVID-19. Iran J. Allergy Asthma Immunol. 2023, 22, 91–98.
- Avila-Mesquita, C.D.; Couto, A.E.S.; Campos, L.C.B.; Vasconcelos, T.F.; Michelon-Barbosa, J.; Corsi, C.A.C.; Mestriner, F.; Petroski-Moraes, B.C.; Garbellini-Diab, M.J.; Couto, D.M.S.; et al. MMP-2 and MMP-9 levels in plasma are altered and associated with mortality in COVID-19 patients. Biomed. Pharmacother. 2021, 142, 112067.
- Tzouvelekis, A.; Herazo-Maya, J.D.; Slade, M.; Chu, J.H.; Deiuliis, G.; Ryu, C.; Li, Q.; Sakamoto, K.; Ibarra, G.; Pan, H.; et al. Validation of the prognostic value of MMP-7 in idiopathic pulmonary fibrosis. Respirology 2017, 22, 486–493.
More
Information
Subjects:
Respiratory System
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
749
Revisions:
2 times
(View History)
Update Date:
04 Feb 2024
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No