Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | seetharamsing balamkundu | -- | 2363 | 2024-01-30 10:42:50 | | | |
| 2 | Rita Xu | -3 word(s) | 2360 | 2024-01-30 10:57:31 | | | | |
| 3 | Rita Xu | -1 word(s) | 2359 | 2024-02-02 06:57:52 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Balamkundu, S.; Liu, C. Lysosomal-Cleavable Peptide Linkers in Antibody–Drug Conjugates. Encyclopedia. Available online: https://encyclopedia.pub/entry/54520 (accessed on 23 July 2026).
Balamkundu S, Liu C. Lysosomal-Cleavable Peptide Linkers in Antibody–Drug Conjugates. Encyclopedia. Available at: https://encyclopedia.pub/entry/54520. Accessed July 23, 2026.
Balamkundu, Seetharamsing, Chuan-Fa Liu. "Lysosomal-Cleavable Peptide Linkers in Antibody–Drug Conjugates" Encyclopedia, https://encyclopedia.pub/entry/54520 (accessed July 23, 2026).
Balamkundu, S., & Liu, C. (2024, January 30). Lysosomal-Cleavable Peptide Linkers in Antibody–Drug Conjugates. In Encyclopedia. https://encyclopedia.pub/entry/54520
Balamkundu, Seetharamsing and Chuan-Fa Liu. "Lysosomal-Cleavable Peptide Linkers in Antibody–Drug Conjugates." Encyclopedia. Web. 30 January, 2024.
Copy Citation
Antibody–drug Conjugates (ADCs) are a powerful therapeutic modality for cancer treatment. ADCs are multi-functional biologics in which a disease-targeting antibody is conjugated to an effector payload molecule via a linker. The success of currently used ADCs has been largely attributed to the development of linker systems, which allow for the targeted release of cytocidal payload drugs inside cancer cells.
antibody–drug conjugate
lysosome
cathepsin
legumain
1. Introduction
Clinical approvals of Antibody–Drug Conjugates (ADCs) have seen a big leap in the past several years. In fact, 12 out of 15 ADCs on the market were approved from 2017 to 2022 [1][2]. More than 100 new ADCs are currently at various stages of clinical development, which reflects the huge potential of this class of medicines in cancer treatment. The mechanism of action of ADCs involves antibody binding to a specific antigen on cancerous cells and subsequent internalization via receptor-mediated endocytosis. Once inside the cancer cell, degradation of the antibody and/or cleavage of the linker in the endosomal–lysosomal compartments would release the drug payload, which then exerts its cytocidal effects in the cytoplasm or nucleus. Researchers have taken advantage of two important endosomal–lysosomal features in designing linker systems for drug release: (i) the acidic environment inside lysosomes for the design of acid-labile linkers, and (ii) the over-expression of specific lysosomal proteases for the design of protease-cleavable linkers (Figure 1A) [3][4][5][6][7][8][9]. Eight out of the fifteen approved ADCs have protease-recognizable peptide sequences in the linkers (Table 1), which are further attached to a self-immolated moiety. Upon cleavage of the peptide sequence by the protease, the self-immolated moiety readily undergoes elimination to release the free drug, which then defuses out of the lysosomal compartment. Linker–payload optimization is one of the most critical tasks in ADC development. The lysosomal protease-cleavable Valine-Citrulline-PABC (ValCitPABC) linker system is used in many of the approved ADCs [10][11], which is readily cleaved at the amide bond linking Cit and PABC, leading to self-immolate payload release. Initially, it was thought that only cathepsin B was responsible for the cleavage of ValCitPABC; however, later gene knockout studies have shown that other cathepsins, like cathepsin S, cathepsin L, and cathepsin F, are also involved in the cleavage mechanism [12]. It has also been revealed that a variety of dipeptide sequences can act as substrates for these lysosomal enzymes. After the initial development of the ValCitPABC linker, researchers have tested a plethora of peptide/peptidomimetic sequences to further improve the linker system [2][3][4][5][7][13] for faster release of payloads and to discover more enzyme-specific peptide sequences, which led to the identification of new cathepsin-sensitive dipeptide sequences such as ValAla, AlaAla, and cBuCit (cBu: cyclobutane-1,1-dicarboxamide).
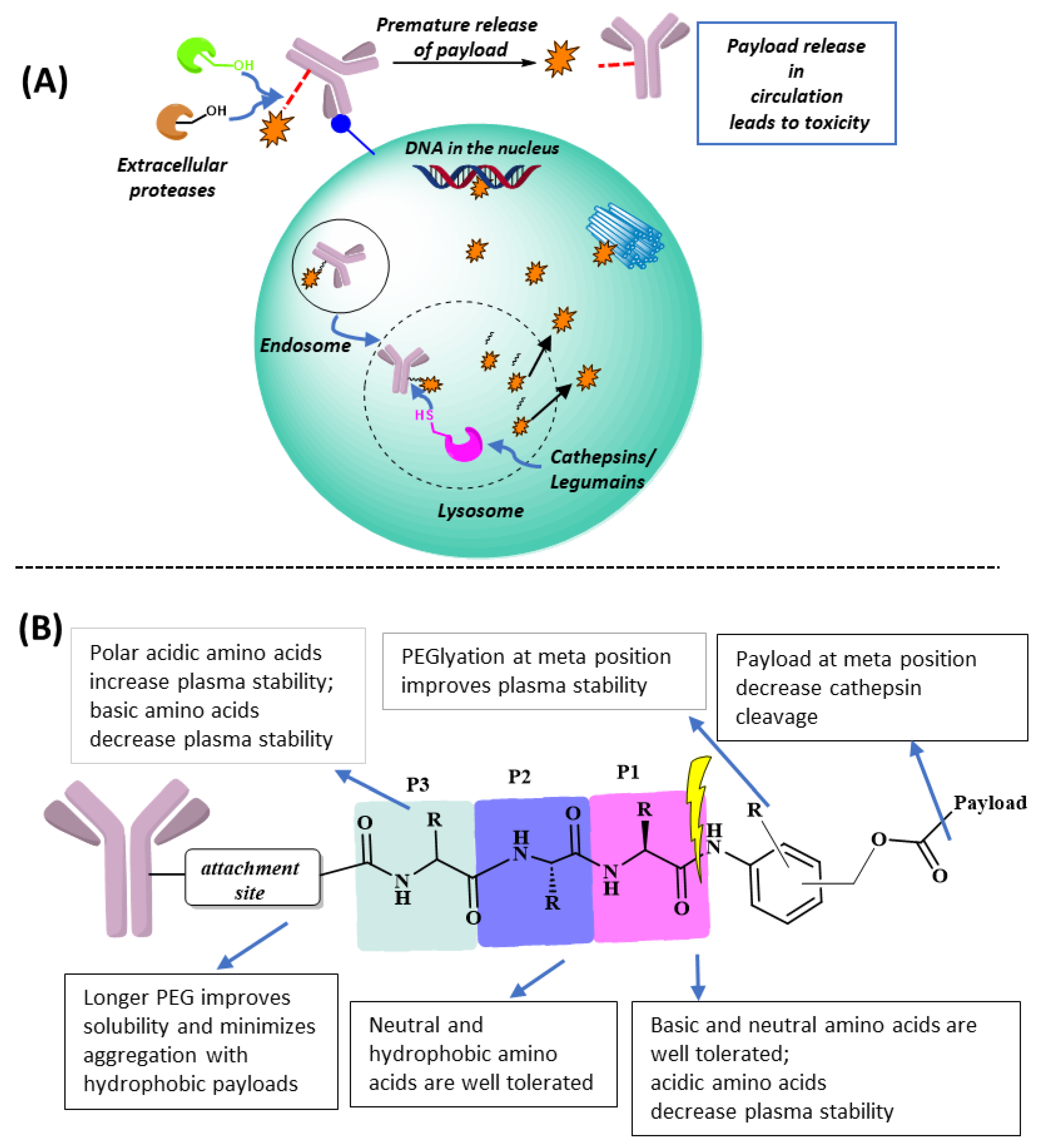
Figure 1. (A) Fate of an ADC before and after internalization. Premature cleavage of the linker in extracellular matrix is often associated with off-target toxicity. Antigen-mediated endocytosis delivers ADC in the endosomal–lysosomal system and lysosomal linker cleavage releases the drug, which acts to exert its cytotoxicity. (B) Effect of amino acid composition in the linker peptide and substitution on the benzene ring of PABC on linker stability in mouse plasma.
Table 1. Clinically approved ADCs.
| S. No. | ADC | Linker System | Cleavage Mechanism | Payload | Company |
|---|---|---|---|---|---|
| 1 | Gemtuzumab ozogamicin (Mylotarg) |
4-(4-acetylphenoxy) butanoic acid | pH sensitive | Calicheamicin | Pfizer/Wyeth |
| 2 | Brentuximab vedotin (Adcetris) |
mc-ValCitPABC | Lysosomal | MMAE | Seattle/Takeda |
| 3 | Trastuzumab emtansine (Kadcyla) |
MCC | Non-cleavable | Maytansine DM1 | Genentech Roche |
| 4 | Inotuzumab ozogamicin (Besponsa) |
Hydrazone | pH sensitive | Calicheamicin | Pfizer/Wyeth |
| 5 | Polatuzumab vedotin (Polivy) |
mc-ValCitPABC | Lysosomal degradation | MMAE | Genentech Roche |
| 6 | Enfortumab vedotin (Padcev) |
mc-ValCitPABC | Lysosomal degradation | MMAE | Astellas/Seattle Genetics |
| 7 | Trastuzumab deruxtecan (Enhertu) |
mc-GGFG-aminomethoxy | Lysosomal degradation | Deruxtecan, Dxd | Daiichi-Sankyo/AstraZeneca |
| 8 | Sacituzumab govitecan (Trodelvy) |
mc-PEG-carbonate | pH | SN-98 | Immunomedics |
| 9 | Belantamab mafodotin (Blenerp) * | mc-MMAF | Non-cleavable | MMAF | GSK |
| 10 | Loncastuximab tesirine (Zynlonta) |
mc-ValCitPABC | Lysosomal degradation | SG3199, PDB dimer | ADC Therapeutics |
| 11 | Tisotumab vedotin (Tivdak) |
mc-ValCitPABC | Lysosomal degradation | MMAE | Genmab and Seattle Genetics |
| 12 | Disitamab Vedotin (Aidixi) |
mc-ValCitPABC | Lysosomal degradation | MMAE | RemeGen |
| 13 | Moxetumomab pasudotox (Lumoxiti) | mc-ValCitPABC | Lysosomal degradation | PE38 | AstraZeneca |
| 14 | Cetuximab sarotalocan (Akalux) | NA | NA | IRDye700DX | Rakuten Medical |
| 15 | Mirvetuximab Soravtansine (ELAHERE) |
Disulfide-containing cleavable linker sulfo-SPDB | Glutathione cleavable | Maytansinoid DM4 | ImmunoGen |
The ValCitPABC linker system has proven to have good stability in human serum, which is the utmost criterion for ADCs to sustain enzymatic degradation in systemic circulation. However, many studies have shown that ValCitPABC is susceptible in mouse plasma and that the degradation is caused by a protease called Carboxylesterase C1 (Ces1C) [14]. This instability in mouse plasma hampers the preclinical evaluation of the ADCs. Therefore, the preclinical studies have to be conducted in transgenic mice with knock-out Ces1C. Target-independent uptake toxicity [15], dose-limiting neutropenia, and thrombocytopenia [3][4][5][6][8][16] are major causes of concern in ADC development. It has been shown that ADC treatment often leads to neutropenia in cancer patients [17], especially when employing ValCitPABC-MMAE. Zhao et al. [17] employed purified neutrophil elastase, a serine protease, to evaluate the in vitro stability of ADCs with a cleavable valine-citrulline linker (vc-MMAE), or a non-cleavable maleimidocaproyl linker (mc-MMAF), and showed that the ValCit linker was readily cleaved by elastase to release free MMAE, whereas the MC linker was not.
2. Adding a P3 Polar Acidic Residue to the ValCitPABC Linker Increases Plasma Stability
The ValCitPABC linker is designed to be cleaved by cathepsin B at the amide bond between the P1 residue citrulline and P1′ PABC (Figure 1B) [10]. Cathepsin B, a cysteine protease primarily found in lysosomes, is first synthesized as a pre-proenzyme in the endoplasmic reticulum. Upon removal of the N-terminal signal peptide, the proenzyme is delivered to the Golgi apparatus, where it is modified via glycosylation. The glycosylated proenzyme then translocates from the trans-Golgi network to endo/lysosome, where auto-proteolytic processing converts it into the mature form composed of two disulfide-linked polypeptide chains [18]. Like other lysosomal cathepsins, cathepsin B is often upregulated in cancer cells [19][20], which makes the ValCitPABC linker particularly attractive for ADC payload release. However, it has been shown that ValCitPABC can be cleaved by Ces1C in mouse plasma, as verified with the purified Ces1C enzyme and the Ces1C knockout mouse as the positive and negative controls, respectively [14]. In the same study, the ValCit linker was found to be stable when co-incubated with Ces1C inhibitors. After clearly establishing the Ces1C-mediated cleavage mechanism, the authors synthesized several novel linker–payload molecules with a substitution at the P3 position and tested their stability (Figure 1B). Interestingly, they observed increased mouse plasma stability when certain hydrophilic groups were introduced at the P3 position; in particular, a 2-hydroxy acetamide group greatly increased the plasma stability. Inspired by these results, Anami et al. introduced hydrophilic amino acids at the P3 position [21] and found that, while SerValCit showed little improvement in stability, linkers with an acidic amino acid at the P3 position, GluValCit and AspValCit, showed excellent stability in mouse plasma. In contrast, having a basic amino acid Lys at P3 made the LysValCit linker more labile than the parent ValCit linker. This indicates that an acidic amino acid at the P3 position effectively blocks the access by Ces1C, whereas a basic amino acid enhances the interaction between Ces1C and the linker. Importantly, all the linker–payload designs discussed above were very stable in human serum and they remained susceptible to the lysosomal enzyme cathepsin B, which is crucial for drug release inside the cancer cell, suggesting that lysosomal cleavage enzymes do not have stringent sequence specificity. It is hypothesized that the human homolog of the Ces1C enzyme in human liver has the active-site serine residue deep inside the substrate binding cleft [14], whereas the active site of human cathepsin B is shallow; thus, substituents at the P3 site are well tolerable.
3. Polar Basic Residue Substitution at the P1 Position of the ValCit-PABC Linker Improves Lysosomal Cleavage Activity
Since its initial discovery, the ValCit-PABC linker system has been used in many ADC constructs with a variety of antibodies and payloads. Researchers have designed a plethora of P2-P1 dipeptides via the substitution of P2-Val and P1-Cit, aiming to increase the linker’s lysosomal cleavability and improve its stability in mouse plasma. Poudel et al. synthesized various linkers with uncialamycin as the payload [22]. When the polar citrulline was replaced with alanine, the stability in plasma was further decreased, although payload release by cathepsin B was not affected. When the polar negatively charged aspartic acid was introduced at the P1 position, there was no significant change in mouse plasma stability; however, it showed a decreased release of the payload by cathepsin B. On the contrary, another study on the effect of the P1 amino acid on cathepsin-mediated cleavage showed that an arginine residue at the P1 position improved the cleavage by nine fold [23]. These results suggest that a polar or basic P1 amino acid is preferable for efficient payload release, whereas an acidic residue decreases the cleavage efficiency.
4. Peptidomimetic Substitution on the Cathepsin-Specific Dipeptide-PABC Linker
In a structure-based study of novel peptidomimetic substitutions at the P2 position of the ValCit-PABC linker, Wei et al. [23] took the key assumption that reducing the number of hydrolyzable peptide bonds would yield cathepsin B-specific linkers with improved extracellular stability [23]. They used simplified constructs where linkers were attached to norfloxacin via PABC and the N-terminal was protected as benzyl carbamate. Cleavage efficiency was evaluated using the Michaelis–Menten steady state Vmax and the Km data by keeping the cathepsin B concentration constant. When they replaced the P2-P1 peptide bond with fluoroolefin and triazole, the cleavage activity by cathespsin B was greatly reduced, which suggests hydrogen bond interactions are crucial for enzyme binding. Based on computational shape similarity search, they identified cyclobutane-1,1-dicarboxamide as a suitable P2 residue. This novel structural unit is able to provide three hydrogen bonding interactions and the cyclobutyl group has an optimum size to fit in the S2 binding pocket. They synthesized various linkers with varying sizes of cycloalkyl rings and side chains at the P1 site; however, none of these showed increased cleavage activity compared to the ValCit counterpart. To address the discrepancies between the computational model and the observed cleavage results, they obtained a crystal structure of human cathepsin B complexed with thiophen-cBuCit-CN (Figure 2A), where the electrophilic CN group was introduced to capture the Cys thiol group at the active site. Indeed, the crystal structure showed expected hydrogen bonding interactions and the key thioimidate bond formed between the cysteine and C≡N and the cyclobutyl group perfectly sitting in the S2 binding pocket. Encouraged from the crystal structure results, they went on to synthesize the ADCs, anti-Her2-mc-cBuCitPABC-MMAE, and anti-Her2-mc-ValCitPABC-MMAE (Figure 3), using an engineered cysteine strategy with DAR between 1.8 and 2.0. When these ADCs were evaluated against Her-2-expressing SKBR-3 cells, both ADCs showed almost similar antigen dependent anti-cancer activity and were equally stable in the in vivo mouse models. When incubating the ValCit and cBuCit ADCs with inhibitors of cathepsin B, they found that cBuCit cleavage activity was reduced by 90% vs. 50% for ValCit, demonstrating the high specificity of the cBuCit linker towards cathepsin B.
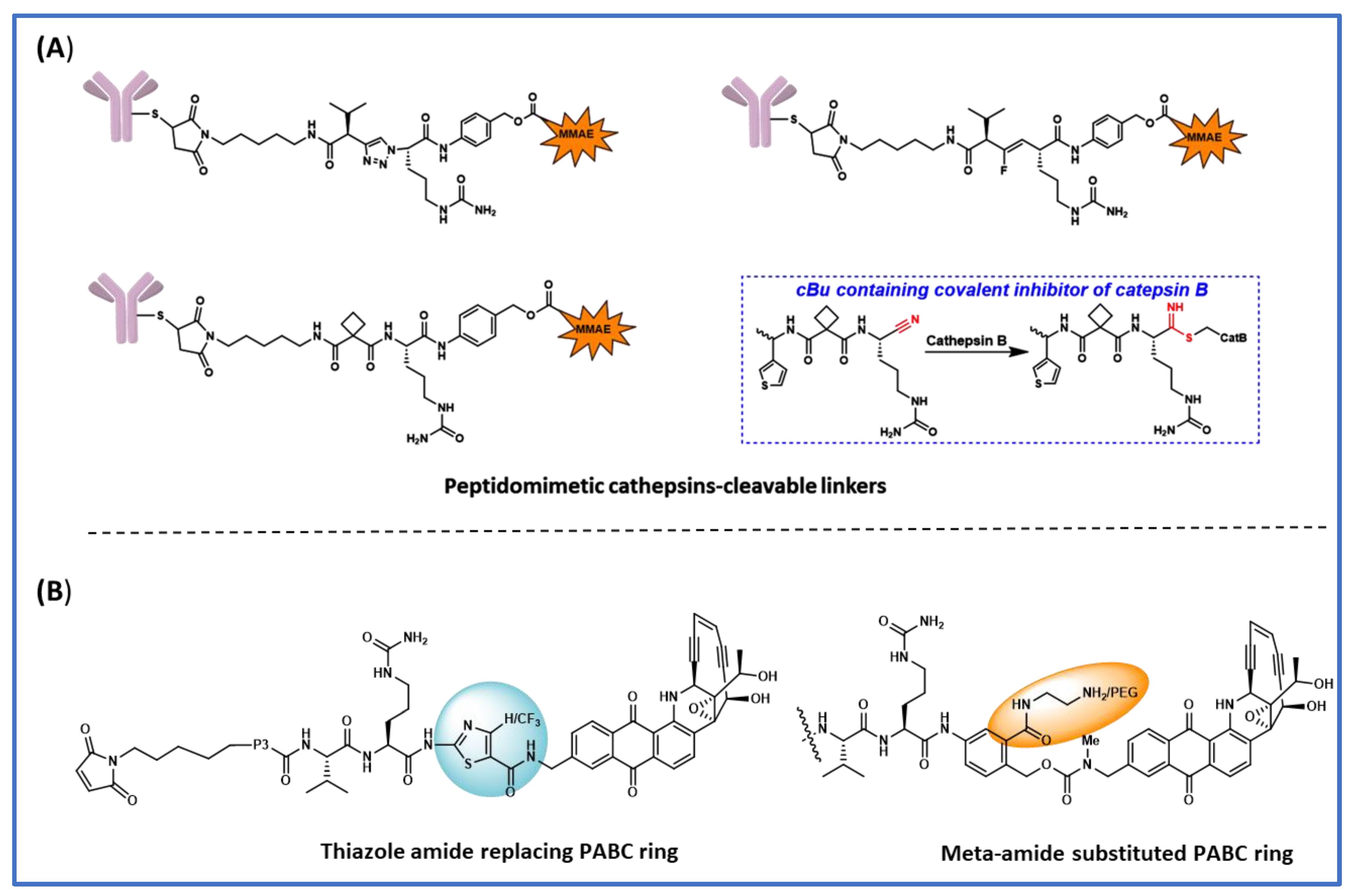
Figure 2. Various Linker payload designs for faster lysosomal cleavage and improved plasma stability. (A) Peptidomimetic linkers specifically cleaved by cathepsin B; (B) modifications to central PABC ring.
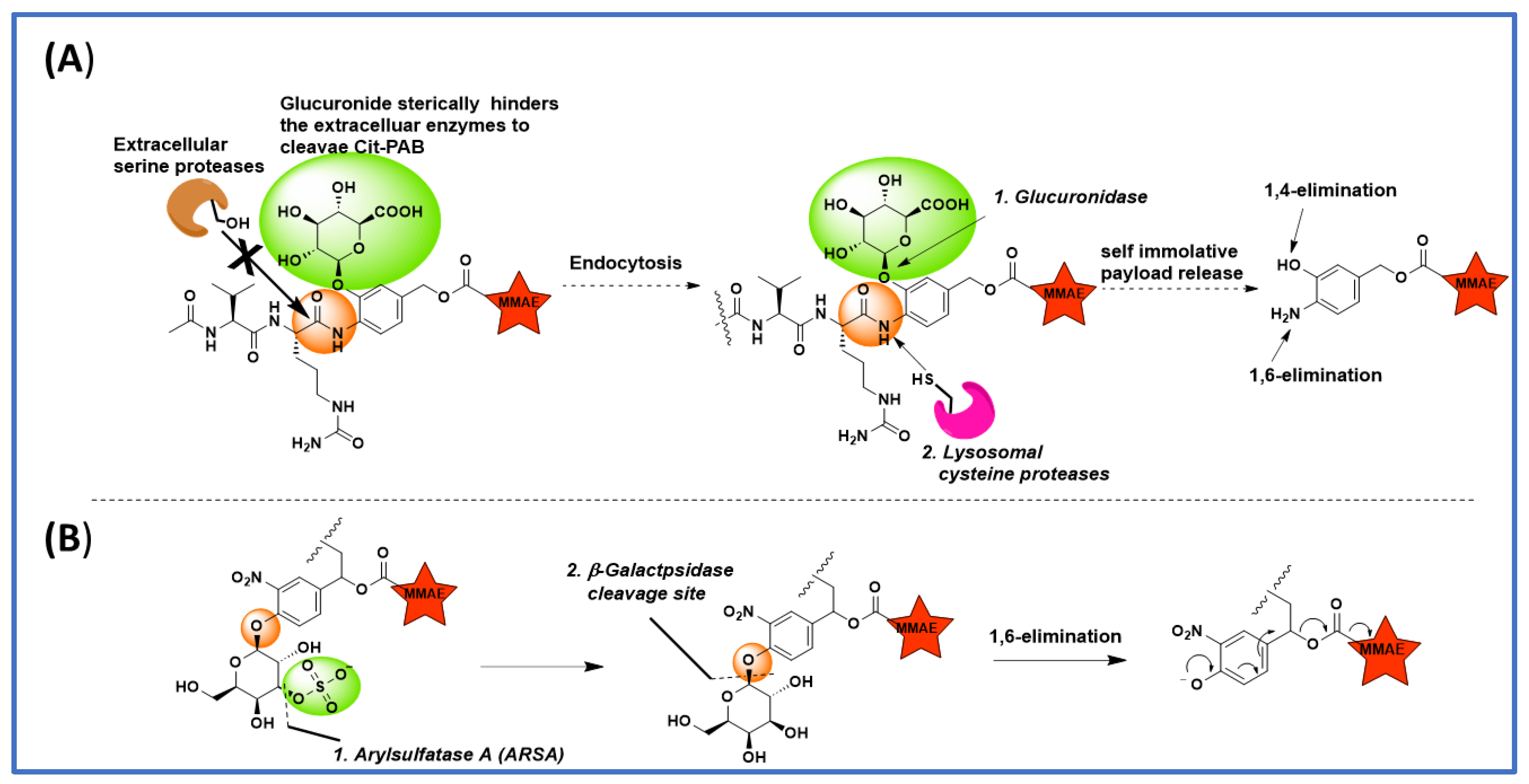
Figure 3. Various Linker payload designs for faster lysosomal cleavage and improved plasma stability. (A) Tandem cleavable linkers’ glucuronide group masks the linker system to maintain stability in extracellular environment; (B) Aryalsulfatase A (ARSA) and β-galactosidase dual cleavable 3-O-sulfo-β-galactose linker.
5. The Effect of Substitution on the PABC Benzene Ring
Directly attaching Val-Cit to the payload leads to a less efficient payload release due to the steric factors of the payload, which inhibits cathepsin binding to the ValCit dipeptide. This problem is partially solved when a spacer is added between the payload and ValCit. PABC (para-aminobenzyl carbamate) not only improves cathepsin binding, but it also undergoes self-immolative 1,6-elimination to release the payload in the unmodified form. Currently, PABC is used with a variety of payloads and peptide linker systems. To improve the stability of the ValCit-PABC linker system toward Ces1C in mouse plasma, Podule et al. synthesized several uncialamycin-linker conjugates [22][24] with substitutions on or the replacement of PABC, including replacement by heterocycles like thiazole. These conjugates were prepared from the reaction of MC-peptide-PABC-uncialamycin linkers and N-acetyl cysteine. They determined the percentage of the drug released after 24 h incubation with cathepsin B, human serum, and mouse serum. Introducing a thiazole amide group in place of PABC decreased the cleavage in mouse serum; however, cleavage in human serum was also observed. When an electron-withdrawing CF3 group was introduced on the thiazole ring, the cleavage in mouse serum was further decreased, and so was the cleavage in human serum. Combining the CF3-substituted thiazole with an aspartic acid at the P3 position generated a linker with only 6% and 4% cleavage in mouse and human serum after 24 h incubation. It is noteworthy that all of these PABC linker analogues were cleaved 100% by cathepsin B, which indicates that the modifications are well tolerated by cathepsin B. Next, they tried to see the impact of substitutions on the PABC benzene ring. When ValCit was attached via ortho allylation to benzyl alcohol, another possible position for self-immolation, cathepsin cleavage activity was completely lost due to the high steric hindrance of the ortho substitution. Furthermore, the linker did not survive in mouse serum. Encouraging results were obtained when an N-methyl carboxyamide group was introduced at the meta position. The resultant MA-PABC was only 3% cleaved in mouse serum after 24 h incubation and its cathepsin-mediated release was not affected. When a glutamic acid was added at the P3 position to the same MA-PABC, the mouse serum cleavage was further reduced to 7% over 24 h. Adding a glutamic acid not only improves the stability towards Ces1C but also the solubility, which is especially important when hydrophobic payloads are used. The authors went further to increase the hydrophilicity of linkers by replacing the methyl group with 2-aminoethyl and its amino-PEGylated form (Figure 2B). These new modifications provided the linkers with excellent mouse and human serum stability without compromising the cathepsin B-mediated cleavage. The ADCs generated from the above PEG-linker–payload using bacterial transglutaminase chemistry showed antigen-dependent activity without problems of linker hydrolysis in mouse serum. The above study clearly demonstrates that combining various attributes from structure–activity relationship studies can yield ideal linker–payload systems with high stability in plasma while maintaining cathepsin susceptibility.
References
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: The “biological missile” for targeted cancer therapy. Signal Transduct Target. Ther. 2022, 7, 93.
- Tong, J.T.; Harris, P.W.; Brimble, M.A.; Kavianinia, I. An insight into FDA approved antibody-drug conjugates for cancer therapy. Molecules 2021, 26, 5847.
- Lyon, R. Drawing lessons from the clinical development of antibody-drug conjugates. Drug Discov. Today Technol. 2018, 30, 105–109.
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody–drug conjugates for cancer. Lancet 2019, 394, 793–804.
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337.
- Hughes, B. Antibody-drug conjugates for cancer: Poised to deliver? Highlighted by Genentech’s recent US regulatory submission for trastuzumab-DM1, antibody-drug conjugation technology could be heading for the mainstream in anticancer drug development. Nat. Rev. Drug Discov. 2010, 9, 665–668.
- Carter, P.J.; Senter, P.D. Antibody-drug conjugates for cancer therapy. Cancer J. 2008, 14, 154–169.
- Donaghy, H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. MAbs 2016, 8, 659–671.
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344.
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B.; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotech. 2003, 21, 778–784.
- Doronina, S.O.; Bovee, T.D.; Meyer, D.W.; Miyamoto, J.B.; Anderson, M.E.; Morris-Tilden, C.A.; Senter, P.D. Novel peptide linkers for highly potent antibody—Auristatin conjugate. Bioconjug. Chem. 2008, 19, 1960–1963.
- Caculitan, N.G.; Chuh, J.D.C.; Ma, Y.; Zhang, D.; Kozak, K.R.; Liu, Y.; Pillow, T.H.; Sadowsky, J.; Cheung, T.K.; Phung, Q.; et al. Cathepsin B is dispensable for cellular processing of cathepsin B-cleavable antibody-drug conjugates. Cancer Res. 2017, 77, 7027–7037.
- Salomon, P.L.; Reid, E.E.; Archer, K.E.; Harris, L.; Maloney, E.K.; Wilhelm, A.J.; Miller, M.L.; Chari, R.V.; Keating, T.A.; Singh, R. Optimizing Lysosomal Activation of Antibody–Drug Conjugates (ADCs) by Incorporation of Novel Cleavable Dipeptide Linkers. Mol. Pharm. 2019, 16, 4817–4825.
- Dorywalska, M.; Dushin, R.; Moine, L.; Farias, S.E.; Zhou, D.; Navaratnam, T.; Lui, V.; Hasa-Moreno, A.; Casas, M.G.; Tran, T.-T. Molecular basis of valine-citrulline-pabc linker instability in site-specific ADCs and its mitigation by linker designmolecular basis of VC-PABC linker instability. Mol. Cancer Ther. 2016, 15, 958–970.
- Mahalingaiah, P.K.; Ciurlionis, R.; Durbin, K.R.; Yeager, R.L.; Philip, B.K.; Bawa, B.; Mantena, S.R.; Enright, B.P.; Liguori, M.J.; Van Vleet, T.R. Potential mechanisms of target-independent uptake and toxicity of antibody-drug conjugates. Pharmacol. Ther. 2019, 200, 110–125.
- Younes, A.; Gopal, A.K.; Smith, S.E.; Ansell, S.M.; Rosenblatt, J.D.; Savage, K.J.; Ramchandren, R.; Bartlett, N.L.; Cheson, B.D.; De Vos, S. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J. Clin. Oncol. 2012, 30, 2183.
- Zhao, H.; Gulesserian, S.; Malinao, M.C.; Ganesan, S.K.; Song, J.; Chang, M.S.; Williams, M.M.; Zeng, Z.; Mattie, M.; Mendelsohn, B.A. A potential mechanism for ADC-induced neutropenia: Role of neutrophils in their own demisemechanism for ADC-induced neutropenia. Mol. Cancer Ther. 2017, 16, 1866–1876.
- Mort, J.S.; Buttle, D.J. Cathepsin b. Int. J. Biochem. Cell Biol. 1997, 29, 715–720.
- Xie, Z.; Zhao, M.; Yan, C.; Kong, W.; Lan, F.; Narengaowa; Zhao, S.; Yang, Q.; Bai, Z.; Qing, H. Cathepsin B in programmed cell death machinery: Mechanisms of execution and regulatory pathways. Cell Death Dis. 2023, 14, 255.
- Mohamed, M.M.; Sloane, B.F. Cysteine cathepsins: Multifunctional enzymes in cancer. Nat. Rev. Cancer 2006, 6, 764–775.
- Anami, Y.; Yamazaki, C.M.; Xiong, W.; Gui, X.; Zhang, N.; An, Z.; Tsuchikama, K. Glutamic acid–valine–citrulline linkers ensure stability and efficacy of antibody–drug conjugates in mice. Nat. Commun. 2018, 9, 2512.
- Poudel, Y.B.; Chowdari, N.S.; Cheng, H.; Iwuagwu, C.I.; King, H.D.; Kotapati, S.; Passmore, D.; Rampulla, R.; Mathur, A.; Vite, G. Chemical modification of linkers provides stable linker–payloads for the generation of antibody–drug conjugates. ACS Med. Chem. Lett. 2020, 11, 2190–2194.
- Wei, B.; Gunzner-Toste, J.; Yao, H.; Wang, T.; Wang, J.; Xu, Z.; Chen, J.; Wai, J.; Nonomiya, J.; Tsai, S.P. Discovery of peptidomimetic antibody–drug conjugate linkers with enhanced protease specificity. J. Med. Chem. 2018, 61, 989–1000.
- Poudel, Y.B.; Rao, C.; Kotapati, S.; Deshpande, M.; Thevanayagam, L.; Pan, C.; Cardarelli, J.; Chowdari, N.; Kaspady, M.; Samikannu, R. Design, synthesis and biological evaluation of phenol-linked uncialamycin antibody-drug conjugates. Bioorg. Med. Chem. Lett. 2020, 30, 126782.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
3.9K
Revisions:
3 times
(View History)
Update Date:
02 Feb 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No