Anaplastic lymphoma kinase (ALK)+ Non-small cell lung cancer (NSCLC), affecting about 5% of cases, is characterized by a mutation in the ALK gene, leading to poor life expectancy and a high risk of brain metastases. Unmet needs in metastatic NSCLC include the development of treatments that improve survival, reduce toxicity, and effectively address brain metastases. Evaluating Food and Drug Administration (FDA)-approved ALK inhibitors in advanced NSCLC highlights their unique effectiveness and safety profiles. Crizotinib exhibits notable benefits regarding progression-free survival (PFS) and objective response rate (ORR); however, multiple studies consistently position alectinib as the superior option. Alectinib distinguishes itself with extended PFS, increased central nervous system (CNS) activity, and excellent patient-reported outcomes.
1. Introduction
Lung cancer comprises non-small cell lung cancer (NSCLC) (81% of cases) and small cell lung cancer (SCLC) (14% of cases). In the US, NSCLC dominates and is projected to affect around 238,340 adults by 2023, resulting in 127,070 deaths. Globally, 2,206,771 people were diagnosed with lung cancer in 2020, encompassing both NSCLC and SCLC cases
[1][2].
Anaplastic lymphoma kinase (ALK), belonging to the insulin receptor superfamily, plays a significant role in various cancers. The
ALK gene is in the 2p23.2-p23.1 chromosomal region, encoding a protein of 26 exons and 1620 amino acid residues. In 1994, a groundbreaking discovery revealed the fusion between nucleolar phosphoprotein (NPM)-1 and ALK in anaplastic large cell non-Hodgkin’s lymphoma (ALCL), emphasizing ALK’s importance. Understanding the t(2;5)(p23;q35) chromosomal translocation in ALCL led to identifying ALK and the resulting NPM-ALK oncogenic protein
[3][4]. On the other hand, different changes in the
ALK gene—such as alternative splicing, amplification, and mutations—are linked to various tumors, such as inflammatory myofibroblastoma and NSCLC
[4][5][6][7].
Crizotinib has demonstrated significant efficacy in reducing tumor size by approximately 50% to 60% in patients with ALK protein alterations, even among those previously treated with chemotherapy. Typical side effects encompass nausea, vomiting, diarrhea, constipation, bloating, fatigue, edema, and eye issues. At the same time, more severe outcomes may involve reduced leukocyte count and detected changes in the lungs and heart. Moreover, clinical investigations highlight the robust efficacy of a co-targeting approach, combining epidermal growth factor (EGF) receptor (EGFR)-TKIs with crizotinib as targeted therapies, especially in metastatic NSCLC
[8]. Furthermore, combining crizotinib with immune checkpoint inhibitors (ICIs) targeting programmed cell death protein 1 (PD-1), programmed cell death-1 ligand-1 (PD-L1), and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) show potential for NSCLC treatment. Still, their efficacy in oncogenic mutated proteins such as EGFR or ALK remains uncertain. ALK alterations have been linked to increased immune checkpoint expression, raising questions about the effectiveness of immunotherapy alone or combined with targeted therapies in this subset of patients. However, trials evaluating immunotherapy in NSCLC often need more representation of ALK-rearranged patients, limiting robust conclusions about their clinical benefit in this population
[9][10].
However, second-generation ALK-TKIs have demonstrated superior clinical activity in terms of median progression-free survival (PFS), objective response rate (ORR), intracranial disease control, and duration of response when compared to crizotinib. The second-generation ALK-TKIs are the gold-standard first-line treatment for ALK-rearranged metastatic NSCLC. Among these options, alectinib is considered to have the most favorable profile of clinical activity and safety, making it a preferred choice for upfront therapy. Ongoing trials and biomarker analyses will provide further insights into the optimal treatment approach
[11]. Additionally, second-generation ALK-TKIs (alectinib, ceritinib, brigatinib) were developed to combat resistance emerging with crizotinib. Initially, these TKIs showed promising effectiveness, validated in several phase 3 trials as the primary treatment for newly diagnosed ALK+ NSCLC
[12][13][14][15][16].
2. The ALK Structural Biology
2.1. ALK Extracellular Side
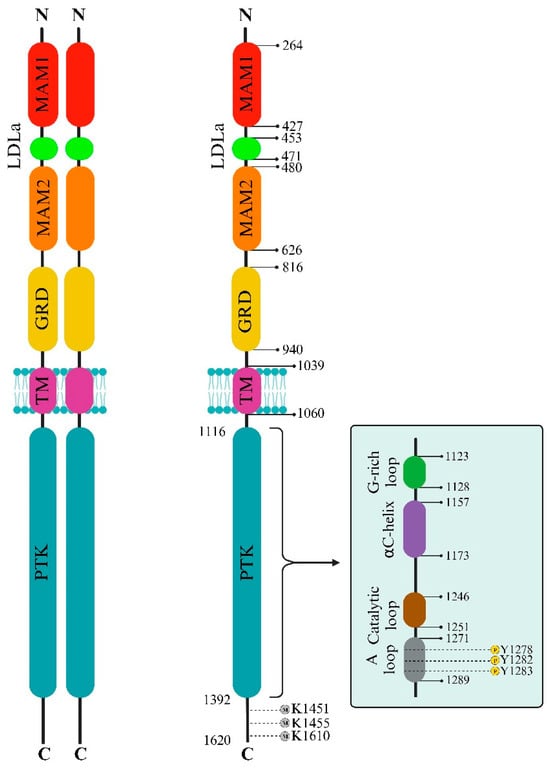
The ALK extracellular domain (ECD) consists of distinct segments believed to serve specific roles such as binding ligands, interacting with potential co-receptors and secreted regulatory proteins, and facilitating dimerization. These functions could trigger structural changes initiating activation within the intracellular protein tyrosine kinase (PTK) domain. The ECD of ALK stands out among receptor tyrosine kinases (RTKs) due to its distinct glycine-rich section. At the same time, ALK includes an additional low-density lipoprotein receptor class A (LDLa) and two meprin, A5 protein, and receptor protein tyrosine phosphatase mu (MAM) domains (
Figure 1). Pleiotrophin (PTN) and midkine (MK) are recognized as triggers for mammalian ALK, playing pivotal roles in neural development, survival, and tumorigenesis
[17]. These growth factors, binding to heparin, can activate various receptors, including receptor protein tyrosine phosphatase-β (RPTPβ), RPTPζ, N-syndecan, low-density lipoprotein receptor-related protein (LRP), and integrins. PTN can specifically engage RPTPβ and RPTPζ phosphatases to initiate ALK signaling.
Figure 1. ALK Domain Structure and Regulatory Elements. This comprehensive depiction illustrates ALK’s extracellular and intracellular aspects, emphasizing key structural domains and functional motifs. The extracellular domain comprises sections crucial for ligand binding and potential activation, including MAMs and LDLa domains. Within the intracellular protein kinase domain (PTK), essential regulatory segments governing ALK’s active and inactive states are highlighted, shedding light on its allosteric control and potential therapeutic targeting. Notably, the A-loop, housing pivotal amino acid residues Y1278, Y1282, and Y1283, drives ALK activation and downstream signaling. In contrast, specific C-terminal lysine residues serve as targets for methylation, contributing to regulatory functions. Furthermore, this figure delineates the duality of ALK functionality: ligand-dependent dimerization (Left) and ligand-independent monomeric activity (Right).
The structure of ALK deviates from the typical architecture due to its unique domain composition. One distinctive element is the glycine-rich domain (GRD) near the membrane. This GRD, which contains a cysteine-rich area resembling the fold of EGF, is an unconventional and less defined region. Interestingly, this peculiar GRD is a shared feature between ALK and LTK. Despite its high glycine content, often associated with structural disorders, the GRD alone can drive receptor activity regulated by its ligands. Vertebrate ALKs’ ligands, known as ALKALs (ALK and LTK Activating Ligands), include Fam150A (AUGβ) and Fam150B (AUGα). These ligands, consisting of approximately 100 amino acids, contain a highly conserved domain termed the ALKAL domain, which stimulates ALK activity
[18][19][20].
2.2. ALK Intracellular Side
The activation loop (A-loop), a pivotal segment governing access to the active site, commences with a conserved Asp-Phe-Gly (DFG) sequence, significantly regulating ALK’s active and inactive states. ALK boasts two distinct hydrophobic spines named the “regulatory” and “catalytic”, contributing to vital allosteric control within and between the lobes. These spines, housing conserved hydrophobic amino acids, facilitate the transition between active and inactive states. The ALK regulatory spine, encompassing I1171, C1182, H1247, F1271, and D1311, assembles during kinase activation and disengages during inactivation
[21]. Researchers also explored the structure of the unphosphorylated human ALK kinase domain in tandem with ATP-competitive ligands such as PHA-E429 and NVP-TAE684. This analysis provided invaluable insights into the distinct attributes of the ALK active site, aiding in the quest for selective ALK inhibitors. Specifically, the ALK-PTK-PHA-E429 structure uncovered a potential regulatory mechanism, linking a brief helical segment after the DFG motif to a two-stranded beta-sheet at the N-terminal. The ALK structure begins with an initial 13-residue segment featuring two β-strands (β1′ and β2′) before the bilobal protein kinase fold. This configuration encompasses an N-terminal lobe housing a core five-stranded β-sheet and an α-helix.
In contrast, the C-terminal lobe is primarily α-helical and accommodates the critical activation loop pivotal for enzyme activation. The gaps in the ALK structures, particularly in the complexes with PHA-E429 and TAE684, indicate regions of structural disorder within the protein. Comparative analysis with other kinase structures highlighted discrepancies in lobe closure and αC helix positioning, suggesting potential inactivity in the ALK-PTK structure due to these structural deviations (Figure 1).
Furthermore, ALK exhibits a distinctive autophosphorylation motif, Y
XXXYY (Y
RASYY), within the A-loop. In instances of ALK fusions, the tyrosine at position Y1278 is the primary site for phosphorylation within this sequence. Notably, an inhibitory structural feature within the ALK kinase domain involves a short α-helix in the A-loop closely associated with the αC-helix. At the same time, a β-turn motif containing C1097 obstructs the region for substrate binding. This arrangement prevents Y1278 from being accessible for phosphorylation as it forms a bond with C1097 within the amino-terminal β-sheet
[22][23][24]. These insights suggest that the initial activation of ALK could potentially involve the regulation of Y1278 phosphorylation, thereby freeing ALK from inactive structural constraints (
Figure 2)
[25].
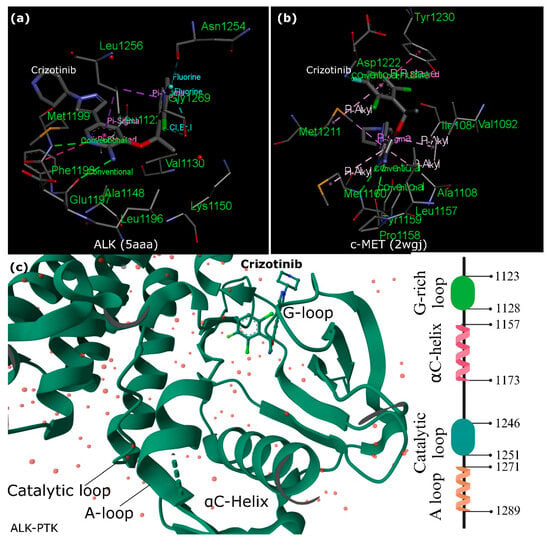
Figure 2. Crizotinib Interactions with ALK and c-MET. (
a,
b) Comparison of Crizotinib’s Interactions with (
a) ALK (PDB id: 5aaa
[26]) and (
b) c-MET (PDB id: 2wgj
[27]). The figure illustrates distinct binding sites of Crizotinib within unphosphorylated c-MET and ALK-PTK, highlighting crucial π interactions and conventional hydrogen bonds. Fundamental interactions, such as those with specific amino acids (e.g., M1211 in c-MET), contribute significantly to Crizotinib’s inhibitory effect. (
c) ALK-PTK (PDB id: 5aaa). The figure details the binding interactions of Crizotinib within the ALK-PTK domain, emphasizing critical regions such as the G-loop, A-loop, and catalytic loop. ALK’s activation loop (A-loop) with the conserved Asp-Phe-Gly (DFG) sequence plays a pivotal role in regulating ALK’s active and inactive states. Hydrophobic spines named the “regulatory” and “catalytic” contribute to vital allosteric control between lobes, impacting the transition between active and inactive states. This figure was designed using the BIOVIA Discovery Studio Visualizer (v.21.1).
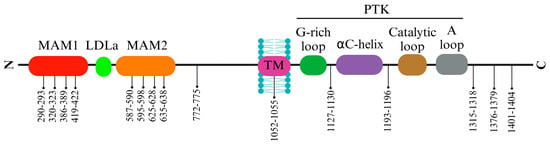
The ALK protein’s structural intricacies and regulatory mechanisms are governed by specific amino acid residues within its sequence. Residues within the range of 1095–1401 display intermittent gaps due to structural disorder, encapsulating crucial segments such as the glycine-rich loop (1123–1128) and the activation loop (1271–1288), essential for ALK’s functional modulation. Among these residues, 1150 (K1150) and 1167 (E1167) stand out for their involvement in pivotal hydrogen bond formations, contributing to ALK’s enzymatic activity. Residue 1245 (F1245) notably interacts, potentially impeding the initiation of a critical structural element termed β9.
2.3. Crizotinib and ALK Inhibition versus c-MET
Crizotinib, a drug with diverse effects on various kinases, demonstrates different inhibitory patterns in enzyme and cellular assays. While it affects multiple kinases in enzymatic tests, its cellular actions show potent inhibition, specifically on mesenchymal-epithelial transition factor (c-MET) and ALK (
Figure 2). This selectivity is linked to distinct binding sites shaped by unphosphorylated c-MET’s unique conformation. However, in the ALK-PTK, crizotinib shows similarities in binding to c-MET but lacks a crucial interaction in ALK, possibly explaining its weaker potency against ALK. Critical interactions with specific amino acids (M1211 in c-MET) play a vital role in maintaining crizotinib’s inhibitory effect and are found similarly in RON (a c-MET-related receptor) and ALK
[27]. This drug demonstrates promising potential in inhibiting c-MET and ALK phosphorylation, curbing tumor cell growth, exhibiting antiangiogenic properties, and inducing apoptosis in specific cancer cells.
Overall, crizotinib presents itself as a promising targeted therapy across diverse cancer types by selectively inhibiting c-MET and ALK, leading to crizotinib’s accelerated approval by the US Food and Drug Administration (FDA) on 26 August 2011, for treating ALK+ locally advanced or metastatic NSCLC. The approval relied on two single-arm trials, showcasing ORRs of 50% and 61%, along with median response durations lasting 42 and 48 weeks
[28].
Furthermore, the co-crystal structure of the ALK kinase domain complexed with crizotinib has a binding configuration like c-MET. However, unlike c-MET, the interaction involving tyrosine π–π stacking is absent in the ALK co-crystal structure, potentially contributing to a slight decrease in potency against ALK compared to c-MET.
Additionally, interactions with the gatekeeper residue and other critical elements of the inhibitor were consistent across both structures, maintaining similar distances and positions. Although the overall protein conformations between the two crystal structures were broadly comparable, a notable difference was observed in the gatekeeper residue (L1196 to M1196).
In addition, ALK possesses multiple LC3-interacting region (LIR) motifs across different domains, hinting at a direct connection between ALK and autophagy. This suggests a complex interplay between inhibiting ALK kinase activity and activating autophagy, potentially complicating targeted therapies for NSCLC and other conditions. Understanding the dual roles of autophagy in cancer—serving as both an immune response facilitator and a tumor growth promoter—underscores the need to categorize ALK + NSCLC based on hepatocyte growth factor (HGF)/c-MET signaling or autophagy-related subtypes to guide treatment decisions for optimal patient outcomes (
Figure 3)
[29].
Figure 3. ALK Sequences as LC3-Interacting Targets. LC3, a mammalian counterpart to yeast Atg8, is a precise marker for autophagy monitoring. The diagram highlights various LC3-interacting region (LIR) motifs across ALK’s diverse domains, indicating a direct link between ALK and autophagy. This connection suggests a complex interplay between suppressing ALK kinase activity and activating autophagy, potentially complicating therapeutic approaches for NSCLC and similar conditions. Understanding autophagy’s dual role in cancer, both as an immune response facilitator and a promoter of tumor growth, underscores the need to classify ALK+ NSCLC based on HGF/c-MET signaling or autophagy-related subtypes. This stratification enables customized treatment strategies for optimizing patient outcomes.
3. ALK Cleavage and Modifications
ALK protein manifests diverse sizes and variations, encompassing an 8.0 kb message detected in rhabdomyosarcoma, small intestine, and brain, alongside transcripts of around 6.5 kb, presumed as typical cDNA. Additionally, various ALK messages—approximately 6.0 kb in the human testis, placenta, and fetal liver, and a distinct 4.4 kb transcript found solely in the testis—highlight tissue-specific isoforms likely arising from alternative transcriptional start sites or polyadenylation signals. These distinct sizes potentially signify tissue-specific functions generated via alternative splicing, leading to diverse receptor forms with varying ligand-binding capabilities and biological activities. Investigations have striven to unravel the precise roles of these diverse ALK isoforms in specific tissues, shedding light on their significance in mammalian development and growth, particularly in neural signaling pathways and development
[30].
A study delved into the expression of ALK in a specific subset of neurons associated with nociception and explored factors influencing its cleavage, shedding light on the potential roles of ALK in sensory neuron development and pain perception
[31]. The study identified that 73% of these sensory neurons expressed ALK, with a significant portion also expressing markers for nociception. This suggests that ALK might be a marker for neurons sensing pain.
The
ALK gene has emerged as a significant player in neuroblastoma development, making it an attractive target for therapeutic interventions. Studies identified specific mutations at F1174 and R1275 in neuroblastoma tumor cells that activate ALK, establishing its role in the disease. Researchers clarified distinct behaviors between the standard and mutated forms of the ALK receptor. They identified that the altered ALK receptors are primarily inside the cell, notably in the reticulum/Golgi structures. This internal retention was particularly noticeable in the F1174L mutation compared to the R1275Q variation
[32].
Treatments inhibiting ALK kinase activity resulted in the translocation of mutated receptors to the cell membrane. This sheds light on potential therapeutic avenues, suggesting that targeting ALK with kinase inhibitors or specific antibodies could hold promise in neuroblastoma treatment, especially considering the possibility of these antibodies inducing receptor internalization and downregulation. These findings open avenues for therapeutic approaches targeting both the wild-type and mutated ALK receptors in neuroblastoma treatment, offering potential complementary strategies to kinase inhibitors
[32].
Furthermore, a recent study introduced a potentially groundbreaking therapeutic approach targeting ALK using a peptide derived from neuronal growth regulator 1 (Negr1)
[33]. Negr1 has been linked to regulating various RTKs, and the researchers observed that acute treatment with soluble Negr1 reduced ALK protein levels, suggesting it prompts ALK protein degradation
[34]. The study proposed that the Negr1-derived peptide might influence ALK levels and downstream signaling pathways impacting cell proliferation. The peptides derived from Negr1 may interact differently with ALK compared to the full-length protein, potentially leading to ALK degradation. This could be a promising strategy as ALK activation is known to drive neuroblastoma growth, and therapies targeting ALK have shown efficacy but are prone to resistance and adverse effects. The Negr1-derived peptide demonstrated the ability to degrade ALK and slow tumor growth both in vitro and in vivo, presenting a promising avenue for treating aggressive neuroblastoma resistant to current ALK inhibitors
[33].
On the flip side, although cleaving ALK’s intracellular domain may help ALK-targeted therapy, cleavage of its extracellular side fosters ALK-related tumor formation and the movement of cells in neuroblastoma. Another potential regulatory process involves the proteolytic breakdown of the full-length ALK receptor ECD
[35], releasing an ECD fragment approximately 80 kDa in size alongside a significantly tyrosine-phosphorylated 140 kDa truncated receptor. The precise physiological significance and the molecular mechanisms driving this cleavage event remain ambiguous; it is uncertain whether cleaved ALK might be more stable or active than intact ALK and whether this cleavage plays a role in ligand-mediated activation
[25].
Researchers noted that blocking this cleavage in neuroblastoma cells reduced migration and invasion. Intriguingly, introducing the cleavable form of ALK in cells with minimal ALK expression significantly boosted their migration, whereas mutations preventing cleavage did not have the same impact
[36]. This indicates the critical role of this cleavage process in driving cell movement, supported by changes in gene activity linked to cell motion. Grasping this process’s developmental role is vital, as abnormal expression in neuroblastoma cells might heighten tumor spread. The study suggests this cleavage could affect a protein called β-catenin, regulating cell motion. When ALK undergoes cleavage at the N654-L655 site, it might release β-catenin, enabling its movement into the cell nucleus to activate genes related to cell motion.
On the other hand, N-linked glycosylation impacts ALK function in neuroblastoma cells. However, it is essential to note that the N654 cleavage site targeted by MMP-9 activates β-catenin signaling. A substantial decrease in the binding of β-catenin to the truncated membrane-bound ALK 655-1604 receptor indicated that the cleavage of the ECD releases β-catenin from ALK, allowing its transportation to the nucleus
[36]. Nonetheless, previous findings showed that N-linked glycosylations impact ALK
[37] and other RTKs
[38].
4. ALK Signaling and TKI Resistance
The majority of cases in NSCLC include a subset of two to seven percent of patients exhibiting gene rearrangements of the
ALK gene or chromosomal fusions of ALK with echinoderm microtubule-associated protein-like 4 (EML4)
[39][40]. Using ALK-TKIs has significantly improved the outcomes for NSCLC patients with these specific genetic abnormalities. Nevertheless, emerging evidence underscores the clinical challenge of primary or secondary resistance to ALK inhibitors during treatment, necessitating a shift to second- or third-generation ALK-TKIs and the meticulous monitoring of NSCLC patients on ALK-TKIs through repeated molecular testing
[41]. The latest generation of ALK-TKIs offers benefits for most individuals with EML4-ALK fusions.
While several ALK inhibitors, such as crizotinib, alectinib, and ceritinib, have been utilized clinically for ALK+ NSCLC treatment, resistance commonly develops against these inhibitors. The mutated forms of ALK, along with ALK fusion proteins such as NPM-ALK, can activate various signaling pathways that contribute to cell transformation and the maintenance of a cancerous state. This persistent activation triggers the recruitment of several adaptors, initiating multiple signaling pathways. Mutated ALK and ALK chimeras induce mitogenic signaling, predominantly through the RAS/mitogen-activated protein (MAP) kinase pathway, facilitated by the direct binding of IRS1, SHC, and SRC to specific tyrosine residues within ALK’s intracytoplasmic segment. The SHP2/growth factor receptor-bound protein 2 (GRB2) complex interaction with p130Cas alters cytoskeletal organization. Activation of the phosphatidylinositol 3 kinase (PI3K) pathway by ALK results in a significant anti-apoptotic signal, mainly mediated by pAKT1/2 and its downstream molecules that inhibit BAD and FOXO3a-mediated transcription, while regulating cell cycle progression.
Further, in neuroendocrine prostate cancer (NEPC), a severe form of prostate cancer, a mutation (ALK F1174C) in the
ALK gene responded well to alectinib. An experimental model combining ALK F1174C and N-Myc led to aggressive NEPC, mirroring poor outcomes seen in human datasets. This combination also activated the wnt/β-catenin pathway
[42]; however, as mentioned earlier, ALK cleavage by MMP-9 in neuroblastoma results in β-catenin release from ALK
[36].
Further investigation revealed that ALK controls MYC’s transcriptional expression and activates c-MYC’s regulation of target genes in NSCLC. Silencing MYC, either through RNAi or small molecules, sensitizes ALK+ cells to crizotinib. These findings illuminate a dual oncogenic mechanism whereby ALK stimulates the MYC signaling axis, suggesting that targeting MYC could potentially prevent or overcome crizotinib resistance
[43].
In addition, findings suggested that targeting Src signaling could be a promising therapeutic strategy for ALK+ NSCLC cases that have developed resistance to ALK-TKIs. Researchers discovered that Src signaling is a key resistance mechanism to alectinib, and combining ALK and Src inhibitors effectively halted the growth of ALK-TKI-resistant cells. Further, blocking Src in alectinib-resistant cells effectively countered the activation of phospho-receptor tyrosine kinases and downstream PI3K/AKT signaling. This combined inhibition of ALK and Src also displayed effectiveness against other ALK+ NSCLC cell lines resistant to ceritinib or lorlatinib
[44].
On the other hand, in NSCLC, where ALK genes are rearranged, resistance to ALK-TKIs remains a challenge despite their success. Research into resistance mechanisms uncovered a new adaptive resistance mechanism linked to JNK/c-Jun signaling. This pathway contributes to the survival of cells tolerant to alectinib and brigatinib. Blocking JNK/c-Jun improved the effectiveness of ALK-TKI treatment in curbing cell growth and promoting cell death. Combining the inhibition of JNK with ALK-TKIs increased cell death by suppressing Bcl-xL proteins, surpassing the effects observed with ALK-TKI treatment alone. Targeting both JNK signaling and ALK might be a promising method to improve outcomes for ALK-rearranged NSCLC
[45].
Additionally, treatment advancements for NSCLC with the EML4-ALK fusion gene have been made with ALK-TKIs. In ALK-TKI-resistant cells, the expression of EML4-ALK decreased at the transcriptional level, while the phosphorylation of EGFR, HER2, and HER3 increased compared to parental-sensitive cells. This increase in the activation of HER family proteins coincided with a higher secretion of EGF. Treatment with an EGFR-TKI induced apoptosis in ALK-TKI resistant cells but not in sensitive cells. In the parental cells, the inhibition of extracellular signal-regulated kinase (ERK) and STAT3 phosphorylation by the selective ALK-TKI TAE684 was disrupted when these cells were exposed to exogenous EGF, leading to reduced sensitivity in cell growth to TAE684
[46]. However, resistance, notably the G1202R mutation in ALK, limits their effectiveness.
In addition, research exploring protein methylation, notably SET and MYND domain containing 2 (SMYD2) methyltransferases, discovered their role in methylating specific lysine residues (1451, 1455, and 1610) in the ALK protein. Lowering SMYD2 levels or using an SMYD2 inhibitor reduced EML4-ALK protein phosphorylation in NSCLC cell lines. Modification of these lysine residues hindered ALK methylation and inhibited downstream AKT phosphorylation, impeding cell growth. Combining SMYD2 and ALK inhibitors demonstrated enhanced efficacy in restraining NSCLC cell growth. Hence, this SMYD2-mediated ALK methylation is suggested as a novel treatment avenue for ALK fusion gene-related tumors
[47].
5. FDA-Approved ALK Inhibitors
5.1. Main Clinical Trials
Studies comparing various ALK inhibitors for advanced NSCLC have highlighted distinct efficacy profiles and safety concerns associated with each medication. Examining the efficacy of crizotinib, alectinib, brigatinib, ceritinib, and lorlatinib in treating ALK+ NSCLC has provided valuable insights into their performance, safety, and unique adverse event profiles
[48].
Clinical studies
[49] highlighted crizotinib’s superior performance over chemotherapy, emphasizing extended PFS and higher ORR. Similarly, another research
[50] reinforced these findings in PROFILE 1029 (NCT01639001), underlining the significant advantages of crizotinib in terms of PFS, ORR, and prompt response time among East Asian patients despite negligible differences in overall survival (OS). Conversely, several studies have consistently drawn attention to alectinib’s advantages compared to crizotinib.
The global phase III ALEX study showcased notable enhancements in PFS and OS when comparing alectinib to crizotinib in treatment-naive individuals with ALK + NSCLC. Subsequent phase III trials in Japanese and Asian populations (J-ALEX and ALESIA) affirmed the clinical advantages of alectinib over crizotinib as a first-line therapy.
On the other hand, in the ALTA-1L trial (NCT02737501), brigatinib’s effectiveness was evaluated against crizotinib among patients with locally advanced or metastatic ALK+ NSCLC who had not previously received ALK inhibitors
[51]. Brigatinib demonstrated significant superiority over crizotinib in terms of PFS and ORR. The trial highlighted brigatinib’s substantial increase in response duration compared to crizotinib, with an OS rate of 86% for crizotinib and 85% for brigatinib. Notably, the adverse effect profiles differed between the two treatments, with distinct patterns of side effects observed in each group.
In the multicenter, randomized ASCEND-4 trial (NCT01828099) comparing ceritinib to platinum-based chemotherapy, the efficacy and safety of ceritinib in ALK-rearranged nonsquamous NSCLC were assessed
[52]. Ceritinib demonstrated significantly longer PFS than chemotherapy, with substantial, rapid, and durable responses observed in the ceritinib group. However, adverse events, particularly diarrhea, nausea, and vomiting, were more familiar with ceritinib, including higher-grade events such as elevated liver enzymes. The effectiveness and tolerability of ceritinib were further analyzed in a Japanese subgroup of patients from the ASCEND-5 (NCT01828112) study
[12][53]. The ceritinib group exhibited prolonged PFS compared to chemotherapy, though it came with a higher incidence of suspected drug-related adverse events.
In the CROWN clinical trial (NCT03052608), a phase 3, open-label, multicenter, randomized study, researchers investigated the efficacy of lorlatinib as a first-line treatment for advanced ALK+ NSCLC in comparison to crizotinib
[54]. The trial enrolled 296 patients without previously received systemic therapy for their metastatic disease. Patients were randomized to receive either lorlatinib (100 mg daily) or crizotinib (250 mg twice daily) for 28 days. The study revealed significant advantages in favor of lorlatinib over crizotinib. The proportion of patients alive at 12 months without disease progression was notably higher in the lorlatinib arm compared to the crizotinib arm (78% versus 39%,
p < 0.001).
5.2. Efficacy and Tolerability Profiles
In a clinical study involving 72 Chinese patients diagnosed with ALK+ NSCLC, crizotinib demonstrated favorable effectiveness and was well-tolerated. Administered orally at 250 mg twice daily, the patients, primarily characterized as young, non/light smokers with adenocarcinoma histology, achieved an ORR of 52.2% and a disease control rate of 64.2%. Common adverse effects included mild visual disturbances, nausea, vomiting, diarrhea, and constipation. The findings suggest that crizotinib is well-tolerated and effective in this patient cohort, highlighting the need for further prospective, multicenter studies with larger sample sizes to validate these results
[55].
Furthermore, lorlatinib, identified as a potent and highly active ALK inhibitor with favorable outcomes in later-line settings, aligns with clinical trial data. In a real-world evaluation of 38 heavily pretreated patients with ALK + NSCLC, lorlatinib demonstrated notable efficacy and tolerability. The overall response rate was 44%, and the disease control rate was 81%, indicating substantial antitumor activity. Lorlatinib dose adjustments, including reduction (18%), interruption (16%), and discontinuation (3%), were consistent with the trial experience. Median OS from advanced ALK+ diagnosis ranged from 45.0 to 69.9 months, while median PFS from lorlatinib initiation varied from 7.3 to 27.7 months.
In conclusion, examining various ALK inhibitors for advanced NSCLC has unveiled their distinct efficacy profiles and safety considerations. While crizotinib showcased notable advantages regarding PFS and ORRs, alectinib consistently emerged as a superior performer across multiple studies. Alectinib’s prolonged PFS heightened CNS activity, and excellent patient-reported outcomes stood out prominently. Despite its effectiveness in extending PFS, brigatinib exhibited more adverse effects than crizotinib. Ceritinib’s ability to improve PFS and reduce the risk of disease progression or mortality underscored its clinical significance. As a first-line treatment, lorlatinib demonstrated promising outcomes, displaying superior PFS and a higher proportion of patients without disease progression at the 12-month than crizotinib. However, it is essential to note that lorlatinib showed varying adverse effect profiles and impacts on different facets of patients’ quality of life. These findings provide clinicians with invaluable insights to tailor therapies according to individual patient needs and tolerability, ultimately advancing the development of personalized treatment strategies for ALK+ NSCLC.
6. Advancements in ALK-Targeted Therapy and Future Horizons
6.1. ALK and Immunotherapy
Changes in the structure of the
ALK gene significantly contribute to the onset of different human cancers, and therapies aimed at this gene have revolutionized how we treat these tumors driven by this specific oncogene. However, overcoming inherent or acquired resistance remains a significant hurdle. Variations in the
ALK gene, such as gene rearrangements or mutations, also influence the immune environment within tumors. Harnessing immunotherapy to target the
ALK gene holds promise in clinical settings
[56]. The association between ALK rearrangement and immune cells is complex and contingent on the specific characteristics of the tumor microenvironment.
In ALK + NSCLC, the proportion of tumors expressing PD-L1 was lower compared to KRAS + NSCLC. Furthermore, T cells expressing immune checkpoint proteins, including T-cell immunoglobulin and mucin-domain containing-3 (TIM-3), CTLA4, Lymphocyte activation gene 3 (LAG3), and PD-1, were less prevalent in ALK+ NSCLC than in EGFR/KRAS + NSCLC. Additionally, the levels of CD3, CD8 T cells, and CD20 B cells were lower in ALK+ NSCLC compared to KRAS+ NSCLC, while CD4 helper T cell levels were higher in ALK+ NSCLC than in EGFR/KRAS+ NSCLC. TIM3 repression was higher in ALK+ NSCLC than in KRAS+ NSCLC. Notably, high expression of PD-L1 and CTLA4 was associated with lower OS in advanced-stage ALK-rearranged NSCLC patients treated with ALK-TKIs. These findings suggest an immunosuppressive tumor microenvironment in ALK + NSCLC, emphasizing the need for further exploration and validation of immunotherapy in this patient population through clinical trials
[57]. Additionally, individuals with EGFR mutations or ALK rearrangements exhibited the lowest proportion of tumors expressing both PD-L1 and CD8 (PD-L1+/CD8+), at 5.0%. In contrast, at 63.5%, the highest proportion was observed in tumors lacking both PD-L1 and CD8 expression (PD-L1-/CD8-). Conversely, those with wild-type EGFR and ALK presented 14.2% of tumors showing PD-L1+/CD8+ and 50.3% with PD-L1-/CD8-. Consequently, patients harboring EGFR mutations or ALK rearrangements demonstrated a diminished PD-L1 and CD8 co-expression level in the tumor microenvironment, potentially contributing to an inadequate response to ICIs. The co-expression of PD-L1 and CD8 in EGFR-mutated or ALK-rearranged lung cancer serves as a biomarker for poor prognosis, correlating with a shorter OS
[58].
On the other hand, interleukin (IL)-6, IL-8, and IL-10 have been associated with disease progression in NSCLC, specifically in ALK+ patients. The interactions between TLRs and various interleukins underscore their involvement in lung cancer pathogenesis, progression, and potential prognostic value
[59]. Moreover, serum-soluble IL-2R levels are a reliable marker for disease activity in hairy cell leukemia and adult T-cell leukemia/lymphoma patients. In addition, ALCL patients often display CD30 and CD25 expression in malignant cells.
The occurrence of any mutation in ALK results in the promotion of PD-L1 expression. Increasing the expression of immunosuppressive molecules such as PD-L1 may lead to tolerance and immune evasion in patients with tumors and cancers. Tian et al. have shown that upregulation of PD-L1 can be identified as a biomarker for ALK-rearrange NSCLC. In addition, it has been recognized that the TME in the presence of upregulated expression of PD-L1 encompasses an immunosuppressive condition
[60][61]. ICIs have shown significant promise in various cancers
[62][63]. In ALK+ NSCLC, these inhibitors have been explored, mainly due to the upregulation of PD-L1 expression in ALK+ tumors
[64][65]. However, studies on the prognosis of ALK+ patients using ICIs have yielded conflicting results, necessitating further investigation
[64]. Initial data from randomized studies suggested lower effectiveness of immunotherapies in ALK+ tumors compared to wild-type tumors.
In addition, another study studied how ALK fusion proteins regulate PD-L1 expression and immune function in ALK+ NSCLC. Researchers observed a correlation between PD-L1 expression, EGFR mutations, and ALK fusion genes in NSCLC cell lines. Elevating ALK fusion protein levels boosted PD-L1 expression, leading to T-cell apoptosis in co-culture systems. Blocking ALK with TKIs amplified IFN-γ production. Anti-PD-1 antibodies were effective in both crizotinib-sensitive and -resistant NSCLC cells. However, combining ALK-TKIs with anti-PD-1 antibodies did not benefit co-culture systems. ALK-TKIs suppressed tumor growth and indirectly bolstered antitumor immunity by reducing PD-L1 expression.
6.2. Molecular Diagnosis of ALK: Insights from Next-Generation Sequencing
Molecular analyses, mainly focusing on genetic rearrangements in genes such as ALK, ROS1, RET, and NTRK
[66], have become standard practices in patients with advanced NSCLC—immunohistochemistry (IHC) functions as the primary screening method, valued for its ease of implementation and interpretation. Fluorescence in situ hybridization (FISH) confirms rearrangements, especially in cases with ambiguous immunostainings. Although FISH is acknowledged as the most sensitive method for detecting ALK and ROS1 rearrangements, it requires comprehensive guidelines for result interpretation
[67].
On the other hand, advanced genomic analyses, such as next-generation sequencing (NGS), meticulously scrutinize the genetic composition of NSCLC. The pivotal roles of ALK, ROS1, and RET genes in NSCLC development make their fusion events crucial for targeted therapies. Researchers characterize these fusion events by employing cutting-edge techniques, aiming for a profound understanding of the molecular intricacies driving NSCLC progression. The expanding coverage of genetic testing has led to the discovery of numerous ALK fusion subtypes and partners, with over 90 rare ALK fusion subtypes identified in NSCLC. While common fusions such as EML4-ALK have established clinical data, rare fusions such as striatin (STRN)-ALK and huntingtin interacting protein 1 (HIP1)-ALK lack substantial clinical evidence. ALK-TKIs are clinically applied based on ALK gene positivity, irrespective of the fusion partner
[68][69].
The research utilized target-capture DNA NGS to identify ALK, ROS1, and RET fusions in NSCLC, examining genomic breakpoints as predictors of targeted therapy efficacy. Categorizing canonical and uncommon fusions among 3787 samples based on breakpoint positions, RNA sequencing revealed 12.8% of uncommon fusions as nonproductive. The study stressed unreliable efficacy prediction for uncommon genomic breakpoints, recommending RNA or protein validation
[70]. In another investigation, a hybridization-based NGS approach on 302 NSCLC tumors identified three non-EML4-ALK fusions and additional fusions through RNA sequencing, emphasizing NGS as promising for ambiguous cases and novel fusion detection
[71].
6.3. ALK and Co-Targeting Approaches
The continuous evolution of ALK inhibitors has significantly improved PFS in NSCLC. Notably, second- and third-generation inhibitors such as brigatinib and lorlatinib exhibit remarkable efficacy in controlling brain metastases. The shift toward personalized medicine, involving genetic panels for diagnosis and tailored targeted therapies, represents a new paradigm. Adopting broad molecular panels as the standard of care will facilitate the detection of resistance mechanisms. This prolonged PFS is anticipated to transform the disease into a manageable, chronic condition. Effective treatment sequencing will be vital for patient survival, and the potential replacement of tissue biopsies with liquid biopsies is on the horizon
[72]. In addition, clinical trials have shown that ALK inhibitors exhibit excellent efficacy against brain metastases. Consequently, initiating treatment with these specific inhibitors is considered reasonable in asymptomatic patients. Radiotherapy can then be employed during tumor progression or when symptoms arise, ensuring the best possible quality of life for patients
[73][74].
Lineage transformation, recognized as a resistance mechanism to ALK-TKIs, occurs at a low frequency, less than 5%, and is primarily attributed to changes in transcriptional patterns rather than acquiring new genomic mutations in the cells
[75]. In cases of resistance to second-generation ALK-TKIs, treatment strategies should be personalized according to the identified resistance mechanisms. Lorlatinib is the preferred option for patients with ALK mutations resistant to these TKIs, providing comprehensive coverage, including mutations such as G1202R and L1196M. For situations without specific resistance mutations, alternative options such as atezolizumab, bevacizumab, and platinum-based chemotherapy may be explored
[76]. In cases of oligo-progression, the approach may involve maintaining the existing systemic treatment despite progression, accompanied by adding local therapies to address advancing lesions. Strategies to counteract on-target resistance mechanisms in ALK-TKI resistance include developing 4th generation TKIs (such as TPX-0131 and NVL-655) and proteolysis-targeting chimeras (PROTACs). In off-target (ALK-independent) resistance cases, potential options include combination therapies targeting ALK along with other downstream or parallel pathways, novel antibody-drug conjugates, or combining ALK inhibitors with chemotherapy and immunotherapy
[77].
Crizotinib exhibits notable efficacy in ALK+ lung cancers, but variable responses and acquired resistance pose challenges. Clinical observations of an exceptional response to an insulin-like growth factor 1 receptor (IGF-1R)-specific antibody in an ALK+ patient led to the identification of therapeutic synergism between ALK and IGF-1R inhibitors. ALK fusion proteins bind to insulin receptor substrate 1 (IRS-1), and inhibiting IRS-1 enhances ALK inhibitors’ antitumor effects. In models of ALK-TKI resistance, activation of the IGF-1R pathway is observed, and combined ALK and IGF-1R inhibition improves therapeutic efficacy. Biopsy samples from patients progressing on crizotinib monotherapy show increased levels of IGF-1R and IRS-1, suggesting a role for the IGF-1R-IRS-1 pathway in both ALK-TKI-sensitive and ALK-TKI-resistant states, supporting further clinical development of dual ALK and IGF-1R inhibitors
[78].
An extraordinary responder in a trial employing erlotinib and IGF-1R antibody unveiled a synergistic impact between ALK and IGF-1R inhibitors. Despite the initial unresponsiveness of the patient’s tumor to erlotinib alone, a remarkable 17-month response emerged with the combination. As subsequent molecular profiling identified an ALK rearrangement, the study proposed the IGF-1R–IRS-1 signaling axis as a potential therapeutic focus in ALK+ lung cancer, providing insights for upcoming clinical trials
[79].
Furthermore, co-targeting primary anticancer targets and corresponding drug escape pathways might enhance anticancer therapeutics. The clinical status and targets of 23 approved and 136 clinical trial multi-target anticancer drugs, focusing on co-targeting ALK, EGFR, HER2, Abl, VEGFR2, mTOR, PI3K, MEK, KIT, and DNA topoisomerase, demonstrated that the majority of approved (73.9%) and phase 3 (75.0%) drugs, as well as a significant portion of phase 2 (62.8%) and phase 1 (53.6%) drugs, co-targeted cancer drug escape pathways, suggesting a potential clinical advantage in co-targeting anticancer targets and drug escape pathways, encouraging further exploration of this strategy
[80].
6.4. Other ALK-Innovative Approaches
In ALK+ cancer models, DNA vaccines directed against the ALK gene exhibited notable effectiveness
[81]. These vaccines prompted specific immune reactions against ALK, fostering CD8+ T cell-mediated cytotoxicity and provoking IFN-γ responses. When combined with chemotherapy, ALK-DNA vaccination significantly extended the survival of mice afflicted with ALK+ lymphomas. In the context of ALK+ NSCLC models, the ALK-DNA vaccine triggered robust immune responses, curtailing tumor growth and elongating survival. However, lung tumors with ALK rearrangements establish an immunosuppressive setting, diminishing the efficacy of the ALK vaccine by upregulating PD-L1 expression.
ALK vaccine pairing with ALK-TKIs notably delayed tumor relapse post-TKI treatment. Further research explored the treatment of ALK-rearranged NSCLC with ALK-TKI and ICIs. The findings revealed that ICIs were ineffective in prompting the rejection of ALK+ lung tumors. However, a vaccination with a single ALK peptide successfully reinstated the activation of ALK-specific CD8+ T cells. When coupled with ALK-TKIs, this vaccination eradicated lung tumors and impeded metastatic spread to the brain. The research additionally pinpointed human ALK peptides suitable for vaccination, demonstrating their immunogenicity in mice and their recognition by CD8+ T cells in individuals with NSCLC. This breakthrough implies the potential development of a clinical vaccine for treating ALK+ NSCLC
[82].
Additionally, alternative ALK vaccines utilizing peptides or lipid vesicles encapsulating ALK antigens showcased potential in restraining tumor advancement in preclinical models. Further, the application of anti-EGF-VacAbs targeting EGF in ALK+ NSCLC cell lines amplified the effectiveness of ALK-TKIs, impeding the emergence of resistance and intercepting downstream oncogenic pathways
[83].
On the other hand, scientists developed a highly sensitive NanoBiT LATS bioluminescent biosensor (BS) to track LATS kinase activity in the Hippo signaling pathway in lab settings and living organisms. This new biosensor showed greater sensitivity and stability than previous versions, even when expressed at significantly lower levels. Using this advanced biosensor, they could monitor LATS activity in live cells at physiologically relevant levels and simplify kinase activity analysis in vitro.
Alternatively, researchers recently investigated the role of the Nuclear Interaction Partner of ALK (NIPA) in a specific type of lymphoma induced by the NPM-ALK gene. Previous studies highlighted NIPA’s significance in cell division control and bone marrow failure but had yet to explore its involvement in NPM-ALK-driven lymphomas
[84]. Researchers demonstrated that NIPA interacts with NPM-ALK, and its absence or downregulation led to significant impairment in the growth and transformation of cells associated with this lymphoma in lab tests.
Furthermore, T-LAK cell-oriented protein kinase (TOPK), recognized as a potential therapeutic target in cancer, has been scrutinized in ALK+ NSCLC. The study identified ALK as an upstream kinase of TOPK, phosphorylating it specifically at Y74. This phosphorylation notably promotes tumor growth in ALK+ lung cancer cells, a finding supported by a phosphoproteomic analysis delineating downstream pathway involvement
[85]. Comparatively, TOPK emerges as a superior target for cancer therapy compared to other direct downstream molecules of ALK, including Smad4, STAT3, PI3K, and PLC-γ. Clinical studies have consistently associated TOPK with a marker of poor prognosis in various cancers and an independent predictor for OS
[86][87][88][89]. Encouragingly, inhibitors such as HI-032 and SKLB-C05, which target TOPK, have demonstrated promising potential.
Moreover, combining TOPK inhibition with alectinib, an ALK inhibitor, has shown remarkable synergy in impeding cell proliferation and promoting apoptosis. This combined approach proposes a promising strategy to counter drug resistance in ALK+ NSCLC
[85]. These research findings advance our understanding of ALK’s oncogenic signaling network and suggest the potential efficacy of co-inhibition of ALK and TOPK as a novel therapeutic strategy to treat ALK+ NSCLC and potentially delay the onset of drug resistance.
 +1 credit
+1 credit