 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Andrey Shumega | -- | 4071 | 2024-01-25 09:25:23 | | | |
| 2 | Mona Zou | Meta information modification | 4071 | 2024-01-26 10:00:53 | | |
Video Upload Options
The discovery of the CRISPR/Cas9 microbial adaptive immune system has revolutionized the field of genetics, by greatly enhancing the capacity for genome editing. CRISPR/Cas9-based editing starts with DNA breaks (or other lesions) predominantly at target sites and, unfortunately, at off-target genome sites. DNA repair systems differing in accuracy participate in establishing desired genetic changes but also introduce unwanted mutations, that may lead to hereditary, oncological, and other diseases. New approaches to alleviate the risks associated with genome editing include attenuating the off-target activity of editing complex through the use of modified forms of Cas9 nuclease and single guide RNA (sgRNA), improving delivery methods for sgRNA/Cas9 complex, and directing DNA lesions caused by the sgRNA/Cas9 to non-mutagenic repair pathways.
1. Structure, Mechanism of Action and DNA Lesions Caused by CRISPR/Cas9 Editing Tools
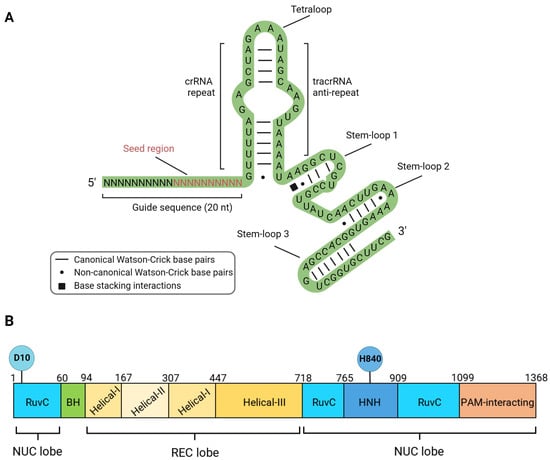
2. The Mechanism of Inaccurate Repair of Cas9-Induced DNA Lesions
3. On-Target and Off-Target Activity: Hot Points of the sgRNA/Cas9-Dependent Mutagenesis
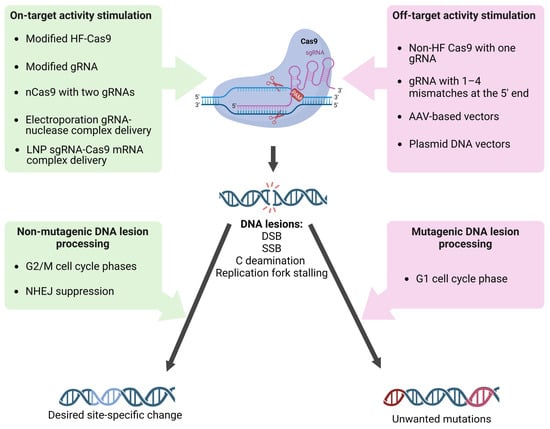
References
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR Provides Acquired Resistance against Viruses in Prokaryotes. Science 2007, 315, 1709–1712.
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821.
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; et al. Evolutionary Classification of CRISPR-Cas Systems: A Burst of Class 2 and Derived Variants. Nat. Rev. Microbiol. 2020, 18, 67–83.
- Barrangou, R. Diversity of CRISPR-Cas Immune Systems and Molecular Machines. Genome Biol. 2015, 16, 247.
- Wong, N.; Liu, W.; Wang, X. WU-CRISPR: Characteristics of Functional Guide RNAs for the CRISPR/Cas9 System. Genome Biol. 2015, 16, 218.
- Nishida, K.; Arazoe, T.; Yachie, N.; Banno, S.; Kakimoto, M.; Tabata, M.; Mochizuki, M.; Miyabe, A.; Araki, M.; Hara, K.Y.; et al. Targeted Nucleotide Editing Using Hybrid Prokaryotic and Vertebrate Adaptive Immune Systems. Science 2016, 353, aaf8729.
- Skrekas, C.; Limeta, A.; Siewers, V.; David, F. Targeted In Vivo Mutagenesis in Yeast Using CRISPR/Cas9 and Hyperactive Cytidine and Adenine Deaminases. ACS Synth. Biol. 2023, 12, 2278–2289.
- Chen, L.; Zhu, B.; Ru, G.; Meng, H.; Yan, Y.; Hong, M.; Zhang, D.; Luan, C.; Zhang, S.; Wu, H.; et al. Re-Engineering the Adenine Deaminase TadA-8e for Efficient and Specific CRISPR-Based Cytosine Base Editing. Nat. Biotechnol. 2023, 41, 663–672.
- Zhang, Y.; Zhang, H.; Wang, Z.; Wu, Z.; Wang, Y.; Tang, N.; Xu, X.; Zhao, S.; Chen, W.; Ji, Q. Programmable Adenine Deamination in Bacteria Using a Cas9-Adenine-Deaminase fusion. Chem. Sci. 2020, 11, 1657–1664.
- Wei, Y.; Zhang, X.H.; Li, D.L. The “New Favorite” of Gene Editing Technology-Single Base Editors. Yi Chuan 2017, 39, 1115–1121.
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable Editing of a Target Base in Genomic DNA without Double-Stranded DNA Cleavage. Nature 2016, 533, 420–424.
- Komor, A.C.; Zhao, K.T.; Packer, M.S.; Gaudelli, N.M.; Waterbury, A.L.; Koblan, L.W.; Kim, Y.B.; Badran, A.H.; Liu, D.R. Improved Base Excision Repair Inhibition and Bacteriophage Mu Gam Protein Yields C:G-to-T:A Base Editors with Higher Efficiency and Product Purity. Sci. Adv. 2017, 3, eaao4774.
- Grunewald, J.; Zhou, R.; Lareau, C.A.; Garcia, S.P.; Iyer, S.; Miller, B.R.; Langner, L.M.; Hsu, J.Y.; Aryee, M.J.; Joung, J.K. A Dual-Deaminase CRISPR Base Editor Enables Concurrent Adenine and Cytosine Editing. Nat. Biotechnol. 2020, 38, 861–864.
- Zhao, Y.; Li, M.; Liu, J.; Xue, X.; Zhong, J.; Lin, J.; Ye, B.; Chen, J.; Qiao, Y. Dual Guide RNA-Mediated Concurrent C&G-to-T&A and A&T-to-G&C Conversions Using CRISPR Base Editors. Comput. Struct. Biotechnol. J. 2023, 21, 856–868.
- Heyer, W.D.; Ehmsen, K.T.; Liu, J. Regulation of Homologous Recombination in Eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139.
- Marini, F.; Rawal, C.C.; Liberi, G.; Pellicioli, A. Regulation of DNA Double Strand Breaks Processing: Focus on Barriers. Front. Mol. Biosci. 2019, 6, 55.
- Lieber, M.R. The mechanism of double-strand DNA Break Repair by the Nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211.
- Sfeir, A.; Symington, L.S. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem. Sci. 2015, 40, 701–714.
- Liu, L.; Malkova, A. Break-Induced Replication: Unraveling Each Step. Trends Genet. 2022, 38, 752–765.
- Zhuk, A.S.; Shiriaeva, A.A.; Andreychuk, Y.V.; Kochenova, O.V.; Tarakhovskaya, E.R.; Bure, V.M.; Pavlov, Y.I.; Inge-Vechtomov, S.G.; Stepchenkova, E.I. Detection of Primary DNA Lesions by Transient Changes in Mating Behavior in Yeast Saccharomyces cerevisiae Using the Alpha-Test. Int. J. Mol. Sci. 2023, 24, 12163.
- Geisinger, J.M.; Turan, S.; Hernandez, S.; Spector, L.P.; Calos, M.P. In Vivo Blunt-End Cloning through CRISPR/Cas9-Facilitated Non-Homologous End-Joining. Nucleic Acids Res. 2016, 44, e76.
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the End Game: DNA Double-Strand Break Repair Pathway Choice. Mol. Cell 2012, 47, 497–510.
- Li, X.; Heyer, W.D. Homologous Recombination in DNA Repair and DNA Damage Tolerance. Cell Res. 2008, 18, 99–113.
- Abuetabh, Y.; Wu, H.H.; Chai, C.; Al Yousef, H.; Persad, S.; Sergi, C.M.; Leng, R. DNA Damage Response Revisited: The p53 Family and Its Regulators Provide Endless Cancer Therapy Opportunities. Exp. Mol. Med. 2022, 54, 1658–1669.
- Hussmann, J.A.; Ling, J.; Ravisankar, P.; Yan, J.; Cirincione, A.; Xu, A.; Simpson, D.; Yang, D.; Bothmer, A.; Cotta-Ramusino, C.; et al. Mapping the Genetic Landscape of DNA Double-Strand Break Repair. Cell 2021, 184, 5653–5669.e25.
- Xue, C.; Greene, E.C. DNA Repair Pathway Choices in CRISPR-Cas9-Mediated Genome Editing. Trends Genet. 2021, 37, 639–656.
- Alkan, F.; Wenzel, A.; Anthon, C.; Havgaard, J.H.; Gorodkin, J. CRISPR-Cas9 off-Targeting Assessment with Nucleic Acid Duplex Energy Parameters. Genome Biol. 2018, 19, 177.
- Mathiasen, D.P.; Lisby, M. Cell Cycle Regulation of Homologous Recombination in Saccharomyces cerevisiae. FEMS Microbiol. Rev. 2014, 38, 172–184.
- Liu, X.; Homma, A.; Sayadi, J.; Yang, S.; Ohashi, J.; Takumi, T. Sequence Features Associated with the Cleavage Efficiency of CRISPR/Cas9 System. Sci. Rep. 2016, 6, 19675.
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kuhn, R. Increasing the Efficiency of Homology-Directed Repair for CRISPR-Cas9-Induced Precise Gene Editing in Mammalian Cells. Nat. Biotechnol. 2015, 33, 543–548.
- Maruyama, T.; Dougan, S.K.; Truttmann, M.C.; Bilate, A.M.; Ingram, J.R.; Ploegh, H.L. Increasing the Efficiency of Precise Genome Editing with CRISPR-Cas9 by Inhibition of Nonhomologous End Joining. Nat. Biotechnol. 2015, 33, 538–542.
- Robert, F.; Barbeau, M.; Ethier, S.; Dostie, J.; Pelletier, J. Pharmacological Inhibition of DNA-PK Stimulates Cas9-Mediated Genome Editing. Genome Med. 2015, 7, 93.
- Pinder, J.; Salsman, J.; Dellaire, G. Nuclear Domain ‘Knock-in’ Screen for the Evaluation and Identification of Small Molecule Enhancers of CRISPR-Based Genome Editing. Nucleic Acids Res. 2015, 43, 9379–9392.
- Yang, D.; Scavuzzo, M.A.; Chmielowiec, J.; Sharp, R.; Bajic, A.; Borowiak, M. Enrichment of G2/M Cell Cycle Phase in Human Pluripotent Stem Cells Enhances HDR-Mediated Gene Repair with Customizable Endonucleases. Sci. Rep. 2016, 6, 21264.
- Caldecott, K.W. Single-Strand Break Repair and Genetic Disease. Nat. Rev. Genet. 2008, 9, 619–631.
- Park, S.; Beal, P.A. Off-Target Editing by CRISPR-Guided DNA Base Editors. Biochemistry 2019, 58, 3727–3734.
- Kouzminova, E.A.; Kuzminov, A. Fragmentation of Replicating Chromosomes Triggered by Uracil in DNA. J. Mol. Biol. 2006, 355, 20–33.
- Kuzminov, A. Single-Strand Interruptions in Replicating Chromosomes Cause Double-Strand Breaks. Proc. Natl. Acad. Sci. USA 2001, 98, 8241–8246.
- Krokan, H.E.; Bjoras, M. Base Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583.
- Northam, M.R.; Robinson, H.A.; Kochenova, O.V.; Shcherbakova, P.V. Participation of DNA Polymerase Zeta in Replication of Undamaged DNA in Saccharomyces cerevisiae. Genetics 2010, 184, 27–42.
- Northam, M.R.; Moore, E.A.; Mertz, T.M.; Binz, S.K.; Stith, C.M.; Stepchenkova, E.I.; Wendt, K.L.; Burgers, P.M.; Shcherbakova, P.V. DNA Polymerases Zeta and Rev1 Mediate Error-Prone bypass of Non-B DNA Structures. Nucleic Acids Res. 2014, 42, 290–306.
- Stepchenkova, E.I.; Zadorsky, S.P.; Shumega, A.R.; Aksenova, A.Y. Practical Approaches for the Yeast Saccharomyces cerevisiae Genome Modification. Int. J. Mol. Sci. 2023, 24, 11960.
- Wang, S.; Zong, Y.; Lin, Q.; Zhang, H.; Chai, Z.; Zhang, D.; Chen, K.; Qiu, J.L.; Gao, C. Precise, Predictable Multi-Nucleotide Deletions in Rice and Wheat Using APOBEC-Cas9. Nat. Biotechnol. 2020, 38, 1460–1465.
- Corsi, G.I.; Qu, K.; Alkan, F.; Pan, X.; Luo, Y.; Gorodkin, J. CRISPR/Cas9 gRNA Activity Depends on Free Energy Changes and on the Target PAM Context. Nat. Commun. 2022, 13, 3006.
- Daer, R.M.; Cutts, J.P.; Brafman, D.A.; Haynes, K.A. The Impact of Chromatin Dynamics on Cas9-Mediated Genome Editing in Human Cells. ACS Synth. Biol. 2017, 6, 428–438.
- Chen, E.; Lin-Shiao, E.; Trinidad, M.; Saffari Doost, M.; Colognori, D.; Doudna, J.A. Decorating Chromatin for Enhanced Genome Editing Using CRISPR-Cas9. Proc. Natl. Acad. Sci. USA 2022, 119, e2204259119.
- Schep, R.; Brinkman, E.K.; Leemans, C.; Vergara, X.; van der Weide, R.H.; Morris, B.; van Schaik, T.; Manzo, S.G.; Peric-Hupkes, D.; van den Berg, J.; et al. Impact of Chromatin Context on Cas9-Induced DNA Double-Strand Break Repair Pathway Balance. Mol. Cell 2021, 81, 2216–2230.e10.
- Nambiar, T.S.; Baudrier, L.; Billon, P.; Ciccia, A. CRISPR-Based Genome Editing through the Lens of DNA Repair. Mol. Cell 2022, 82, 348–388.
- Lohia, A.; Sahel, D.K.; Salman, M.; Singh, V.; Mariappan, I.; Mittal, A.; Chitkara, D. Delivery Strategies for CRISPR/Cas Genome Editing Tool for Retinal Dystrophies: Challenges and Opportunities. Asian J. Pharm. Sci. 2022, 17, 153–176.
- Carpenter, M.A.; Law, E.K.; Serebrenik, A.; Brown, W.L.; Harris, R.S. A Lentivirus-Based System for Cas9/gRNA Expression and Subsequent Removal by Cre-Mediated Recombination. Methods 2019, 156, 79–84.
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.S. Highly Efficient RNA-Guided Genome Editing in Human Cells via Delivery of Purified Cas9 Ribonucleoproteins. Genome Res. 2014, 24, 1012–1019.
- Ramakrishna, S.; Kwaku Dad, A.B.; Beloor, J.; Gopalappa, R.; Lee, S.K.; Kim, H. Gene Disruption by Cell-Penetrating Peptide-Mediated Delivery of Cas9 Protein and Guide RNA. Genome Res. 2014, 24, 1020–1027.
- Qiu, M.; Glass, Z.; Chen, J.; Haas, M.; Jin, X.; Zhao, X.; Rui, X.; Ye, Z.; Li, Y.; Zhang, F.; et al. Lipid Nanoparticle-Mediated Codelivery of Cas9 mRNA and Single-Guide RNA Achieves Liver-Specific In Vivo Genome Editing of Angptl3. Proc. Natl. Acad. Sci. USA 2021, 118, e2020401118.
- Taha, E.A.; Lee, J.; Hotta, A. Delivery of CRISPR-Cas Tools for In Vivo Genome Editing Therapy: Trends and Challenges. J. Control. Release 2022, 342, 345–361.
- Sinclair, F.; Begum, A.A.; Dai, C.C.; Toth, I.; Moyle, P.M. Recent Advances in the Delivery and Applications of Nonviral CRISPR/Cas9 Gene Editing. Drug Deliv. Transl. Res. 2023, 13, 1500–1519.
- Yu, X.; Yang, Z.; Zhang, Y.; Xia, J.; Zhang, J.; Han, Q.; Yu, H.; Wu, C.; Xu, Y.; Xu, W.; et al. Lipid Nanoparticle Delivery of Chemically Modified NGF(R100W) mRNA Alleviates Peripheral Neuropathy. Adv. Healthc. Mater. 2023, 12, e2202127.
- Guo, C.; Ma, X.; Gao, F.; Guo, Y. Off-Target Effects in CRISPR/Cas9 Gene Editing. Front. Bioeng. Biotechnol. 2023, 11, 1143157.
- Evers, M.J.W.; Du, W.; Yang, Q.; Kooijmans, S.A.A.; Vink, A.; van Steenbergen, M.; Vader, P.; de Jager, S.C.A.; Fuchs, S.A.; Mastrobattista, E.; et al. Delivery of Modified mRNA to Damaged Myocardium by Systemic Administration of Lipid Nanoparticles. J. Control. Release 2022, 343, 207–216.
- Chen, F.; Alphonse, M.; Liu, Q. Strategies for Nonviral Nanoparticle-Based Delivery of CRISPR/Cas9 Therapeutics. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020, 12, e1609.
- Greig, J.A.; Martins, K.M.; Breton, C.; Lamontagne, R.J.; Zhu, Y.; He, Z.; White, J.; Zhu, J.X.; Chichester, J.A.; Zheng, Q.; et al. Integrated Vector Genomes May Contribute to Long-Term Expression in Primate Liver after AAV Administration. Nat. Biotechnol. 2023.
- Nathwani, A.C.; Reiss, U.M.; Tuddenham, E.G.; Rosales, C.; Chowdary, P.; McIntosh, J.; Della Peruta, M.; Lheriteau, E.; Patel, N.; Raj, D.; et al. Long-Term Safety and Efficacy of Factor IX Gene Therapy in Hemophilia B. N. Engl. J. Med. 2014, 371, 1994–2004.
- Hanlon, K.S.; Kleinstiver, B.P.; Garcia, S.P.; Zaborowski, M.P.; Volak, A.; Spirig, S.E.; Muller, A.; Sousa, A.A.; Tsai, S.Q.; Bengtsson, N.E.; et al. High Levels of AAV Vector Integration into CRISPR-Induced DNA breaks. Nat. Commun. 2019, 10, 4439.
- Kim, D.; Luk, K.; Wolfe, S.A.; Kim, J.S. Evaluating and Enhancing Target Specificity of Gene-Editing Nucleases and Deaminases. Annu. Rev. Biochem. 2019, 88, 191–220.
- Ghani, M.W.; Iqbal, A.; Ghani, H.; Bibi, S.; Wang, Z.; Pei, R. Recent Advances in Nanocomposite-Based Delivery Systems for Targeted CRISPR/Cas Delivery and Therapeutic Genetic Manipulation. J. Mater. Chem. B 2023, 11, 5251–5271.
- Kulishova, L.M.; Vokhtantsev, I.P.; Kim, D.V.; Zharkov, D.O. Mechanisms of the Specificity of the CRISPR/Cas9 System in Genome Editing. Mol. Biol. 2023, 57, 269–284.
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell 2013, 154, 1380–1389.
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally Engineered Cas9 Nucleases with Improved Specificity. Science 2016, 351, 84–88.
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-Fidelity CRISPR-Cas9 Nucleases with No Detectable Genome-Wide off-Target Effects. Nature 2016, 529, 490–495.
- Chen, J.S.; Dagdas, Y.S.; Kleinstiver, B.P.; Welch, M.M.; Sousa, A.A.; Harrington, L.B.; Sternberg, S.H.; Joung, J.K.; Yildiz, A.; Doudna, J.A. Enhanced Proofreading Governs CRISPR-Cas9 Targeting Accuracy. Nature 2017, 550, 407–410.
- Bravo, J.P.K.; Liu, M.S.; Hibshman, G.N.; Dangerfield, T.L.; Jung, K.; McCool, R.S.; Johnson, K.A.; Taylor, D.W. Structural Basis for Mismatch Surveillance by CRISPR-Cas9. Nature 2022, 603, 343–347.
- Casini, A.; Olivieri, M.; Petris, G.; Montagna, C.; Reginato, G.; Maule, G.; Lorenzin, F.; Prandi, D.; Romanel, A.; Demichelis, F.; et al. A Highly Specific SpCas9 Variant is Identified by In Vivo Screening in Yeast. Nat. Biotechnol. 2018, 36, 265–271.
- Lee, J.K.; Jeong, E.; Lee, J.; Jung, M.; Shin, E.; Kim, Y.H.; Lee, K.; Jung, I.; Kim, D.; Kim, S.; et al. Directed Evolution of CRISPR-Cas9 to Increase Its Specificity. Nat. Commun. 2018, 9, 3048.
- Vakulskas, C.A.; Dever, D.P.; Rettig, G.R.; Turk, R.; Jacobi, A.M.; Collingwood, M.A.; Bode, N.M.; McNeill, M.S.; Yan, S.; Camarena, J.; et al. A High-Fidelity Cas9 Mutant Delivered as a Ribonucleoprotein Complex Enables Efficient Gene Editing in Human Hematopoietic Stem and Progenitor Cells. Nat. Med. 2018, 24, 1216–1224.
- Hu, J.H.; Miller, S.M.; Geurts, M.H.; Tang, W.; Chen, L.; Sun, N.; Zeina, C.M.; Gao, X.; Rees, H.A.; Lin, Z.; et al. Evolved Cas9 Variants with Broad PAM Compatibility and High DNA Specificity. Nature 2018, 556, 57–63.
- Pattanayak, V.; Lin, S.; Guilinger, J.P.; Ma, E.; Doudna, J.A.; Liu, D.R. High-throughput Profiling of off-Target DNA Cleavage Reveals RNA-Programmed Cas9 Nuclease Specificity. Nat. Biotechnol. 2013, 31, 839–843.
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823.
- Anders, C.; Niewoehner, O.; Duerst, A.; Jinek, M. Structural Basis of PAM-Dependent Target DNA Recognition by the Cas9 Endonuclease. Nature 2014, 513, 569–573.
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.S. Analysis of off-Target Effects of CRISPR/Cas-Derived RNA-Guided Endonucleases and Nickases. Genome Res. 2014, 24, 132–141.
- Kawamata, M.; Suzuki, H.I.; Kimura, R.; Suzuki, A. Optimization of Cas9 Activity through the Addition of Cytosine Extensions to Single-Guide RNAs. Nat. Biomed. Eng. 2023, 7, 672–691.
- Wyvekens, N.; Topkar, V.V.; Khayter, C.; Joung, J.K.; Tsai, S.Q. Dimeric CRISPR RNA-Guided FokI-dCas9 Nucleases Directed by Truncated gRNAs for Highly Specific Genome Editing. Hum. Gene Ther. 2015, 26, 425–431.
- Rozners, E. Chemical Modifications of CRISPR RNAs to Improve Gene-Editing Activity and Specificity. J. Am. Chem. Soc. 2022, 144, 12584–12594.


