Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Andrei C Sposito | -- | 7608 | 2024-01-23 14:17:47 | | | |
| 2 | Camila Xu | Meta information modification | 7608 | 2024-01-24 02:01:07 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Carmo, H.R.P.; Bonilha, I.; Barreto, J.; Tognolini, M.; Zanotti, I.; Sposito, A.C. High-Density Lipoproteins, the NLRP3 Inflammasome and Myocardial Infarction. Encyclopedia. Available online: https://encyclopedia.pub/entry/54253 (accessed on 25 July 2026).
Carmo HRP, Bonilha I, Barreto J, Tognolini M, Zanotti I, Sposito AC. High-Density Lipoproteins, the NLRP3 Inflammasome and Myocardial Infarction. Encyclopedia. Available at: https://encyclopedia.pub/entry/54253. Accessed July 25, 2026.
Carmo, Helison R. P., Isabella Bonilha, Joaquim Barreto, Massimiliano Tognolini, Ilaria Zanotti, Andrei C. Sposito. "High-Density Lipoproteins, the NLRP3 Inflammasome and Myocardial Infarction" Encyclopedia, https://encyclopedia.pub/entry/54253 (accessed July 25, 2026).
Carmo, H.R.P., Bonilha, I., Barreto, J., Tognolini, M., Zanotti, I., & Sposito, A.C. (2024, January 23). High-Density Lipoproteins, the NLRP3 Inflammasome and Myocardial Infarction. In Encyclopedia. https://encyclopedia.pub/entry/54253
Carmo, Helison R. P., et al. "High-Density Lipoproteins, the NLRP3 Inflammasome and Myocardial Infarction." Encyclopedia. Web. 23 January, 2024.
Copy Citation
The NLRP3 inflammasome is an intracellular defender that acts against the cellular fragments and cytoplasmic contents of injured and/or dead cells, called damage-associated molecular patterns (DAMPs) (also known as alarmins). The ultimate consequence of prolonged cardiac ischemia is myocardial infarction (MI), a condition accountable for one-third of global fatalities. The extent of MI is a potent predictor of various adverse cardiovascular events, encompassing mortality, recurrent MI, arrhythmias, congestive heart failure, angina, and the need for revascularization, as substantiated by several studies.
HDL
NLRP3 inflammasome
myocardial infarction
ischemia-reperfusion injury
1. Introduction
Owing to its unique role, the myocardium perpetually engages in electromechanical and biochemical metabolic activities, leading to a significant oxygen consumption per unit of tissue [1]. Consequently, the myocardium encounters substantial hurdles when confronted with ischemia, a condition characterized by limited oxygen supply, with its severity influenced by various factors, including the duration and extent of the at-risk myocardial tissue [2]. Furthermore, the presence of cardiometabolic disorders can alter the accessibility of energy substrates, including fatty acids, glucose, and ketones, thereby exacerbating the ischemic injury [3].
The ultimate consequence of prolonged cardiac ischemia is myocardial infarction (MI), a condition accountable for one-third of global fatalities. The extent of MI is a potent predictor of various adverse cardiovascular events, encompassing mortality, recurrent MI, arrhythmias, congestive heart failure, angina, and the need for revascularization, as substantiated by several studies [4][5][6][7]. Consequently, there has been widespread adoption of abbreviated reperfusion therapies aimed at attenuating the extent of MI. In other words, reperfusion therapies aim to restore blood flow by unclogging an occlusive thrombus, mostly resulting from the rupture of an atherosclerotic plaque. This is achieved through two primary methods: non-invasive procedures, which involve the use of thrombolytic drugs, and invasive procedures, such as primary percutaneous coronary intervention (PCI) or coronary artery bypass graft (CABG) surgery. These methods allow for blood reperfusion and aid in the restoration of appropriate circulation. Notably, all existing research consistently demonstrates an inverse relationship between the duration of reperfusion therapy and both short-term and long-term mortality rates, underscoring the significance of prompt intervention [8].
Consequently, early and effective reperfusion therapies have become a cornerstone in reducing mortality and morbidity associated with MI. Yet, reperfusion itself triggers a cascade of molecular mechanisms that contribute to additional myocardial damage, collectively known as myocardial ischemic and reperfusion injury (IRI) [9][10]. From the mechanistic point of view, following reperfusion, there is a rapid intracellular pH shift, disruption of ionic balance, heightened oxidative stress, increased activity of proteolytic enzymes, initiation of inflammatory responses, and activation of several cell death pathways, encompassing apoptosis, necroptosis, and pyroptosis [11]. These metabolic changes activate the intracellular sensor responsible for mediating the local immune system response. This sensor is formed by nucleotide oligomerization domain (NOD)-like receptors, leucine-rich repeat (LRR)-containing receptors (NLRs), and the multiprotein signaling complex named the NLRP3 inflammasome, and is made of pyrin domain-containing 3 (NLRP3), adaptor apoptosis-associated speck-like proteins containing a CARD (ASC), and pro-caspase-1 [12]. The NLRP3 inflammasome is an intracellular defender that acts against the cellular fragments and cytoplasmic contents of injured and/or dead cells, called damage-associated molecular patterns (DAMPs) (also known as alarmins).
Indeed, MI activates the NLRP3 inflammasome through the DAMPs tissue dissemination, resulting in two deleterious mechanisms: (i) the activation of proinflammatory interleukins (pro-IL-1β and pro-IL18), (ii) and lytic cell death due to the pore formation on the cellular membrane, characterized by proinflammatory cell death or pyroptosis. Regarding the secretion of proinflammatory cytokines, the activation of the NLRP3 inflammasome leads to pathogenicity in various resident cardiac cells. The increased activity of the NLRP3 inflammasome leads to an exacerbated production of IL1β and IL18, responsible for an acute immune and inflammatory response. This effect can manifest locally as myocardial inflammation or systemically targeting the vascular endothelium and multiple organs. The presence of IL1 receptors in immune cells facilitates the paracrine and autocrine effects of these proteins, potentially inducing the increased transcription of proinflammatory genes, amplifying their synthesis and secretion [10]. Detrimental clinical effects such as fever, hypotension, myocardial contractility dysfunction, and increased ischemic events are mainly associated with elevated plasma IL1β levels. However, insights from in vitro experiments indicate that IL-1β secretion is primarily associated with cardiac fibroblasts, whereas IL-18 secretion is more closely linked to cardiomyocytes [13][14]. Other proinflammatory plasmatic markers such as high-mobility group protein B1 (HMGB1), leukotrienes, prostaglandins, chemokines, interferon gamma (IFN-γ), necrosis factor-alpha (TNF-α), interleukin 6 (IL-6), cyclooxygenase-2, intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1) have also been associated with pyroptosis damage at the systemic level [12][15][16]. A comprehensive overview of the suggested cellular mechanisms involved in these interactions is provided in Figure 1.
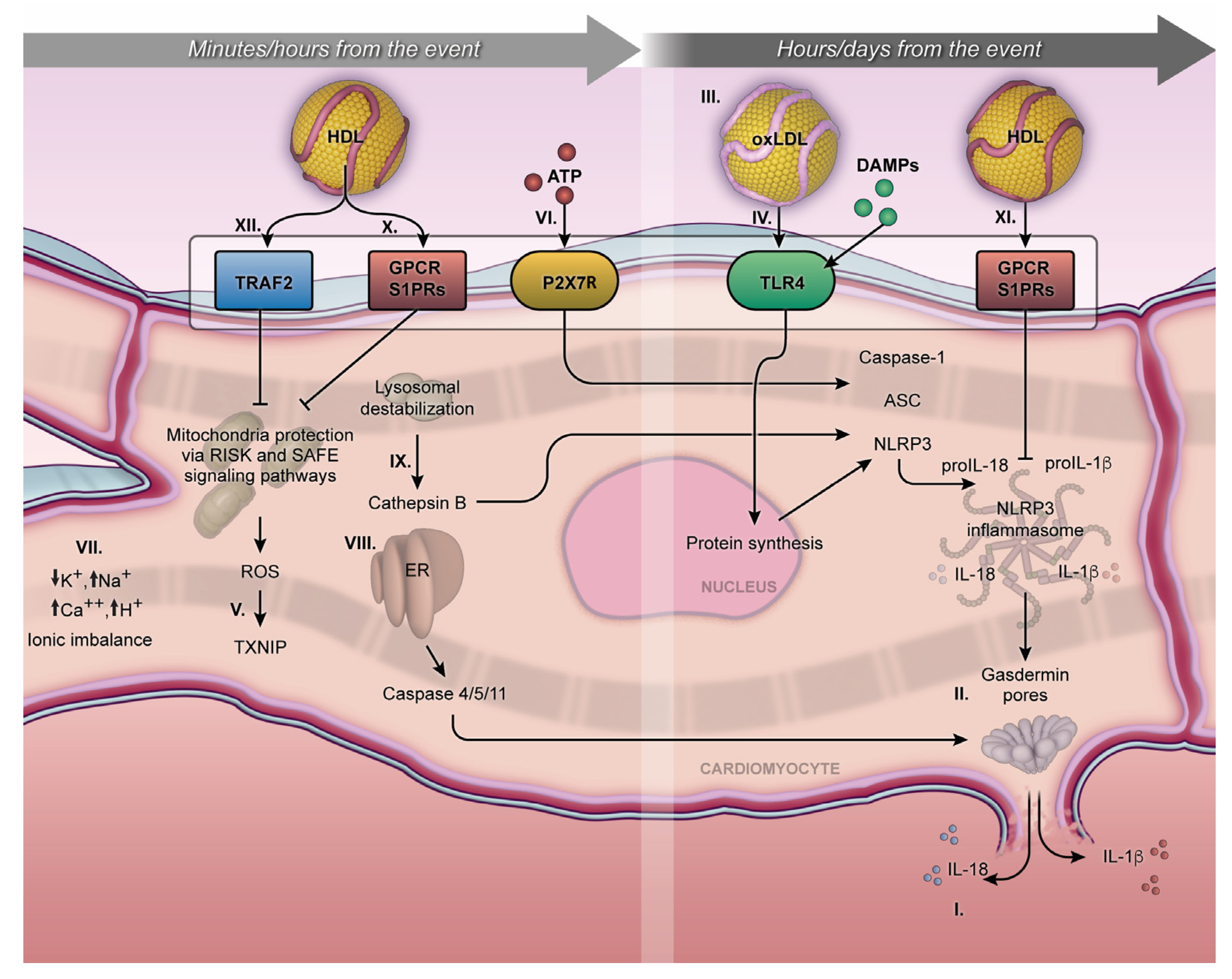
Figure 1. Minutes/hours from MI and hours/days from MI. The NLRP3 inflammasome cellular mechanism initiates during the first minutes of MI and can persist for days, exerting an influence on acute or chronic outcomes. Activation of pro-IL-1β and pro-IL18 and their respective release outside the membrane (I); gasdermin-D on the lytic cell death due to the pore formation on the cellular membrane, characterized by proinflammatory cell death or pyroptosis (II); proinflammatory oxidized LDL (III); TLR4 and NLRP3 inflammasome transcription pathway (IV); MI exacerbates the production of mitochondrial ROS in a process mediated by the TXNIP (V); P2X7R receptor under ATP stimulation promotes calcium and sodium influx, resulting in potassium efflux and classic signaling for NLRP3 inflammasome activation (VI); ionic imbalance (mainly by increase of Ca2+ mobilization and decrease of K+ efflux) (VII); endoplasmic reticulum stress, triggering caspase 4/5/11 activation (VIII); Cathepsin B, inducing NLRP3 activation (VIX); S1PRs play a crucial role in the interaction of S1P, facilitating cytoprotective mechanisms both short- and long-term, following MI (X and XI). The activation of the Tumor Necrosis Factor 2 Receptor (TRAF2) by HDL can influence the activity of sphingosine kinase-1 and consequently increase the production of sphingosine-1-phosphate (XII). The arrows mean the increase (↑) or decrease (↓) in ion concentration into the cytoplasmic milieu.
Studies conducted in animal models of MI estimate that IRI contributes to as much as 50% of the final infarct size (Figure 2) [9]. These ex vivo studies also shed light on diverse strategies with the potential to alleviate IRI, either by the inhibition of apoptosis or pyroptosis pathways [17][18]. However, the translation of their findings into clinical benefits has proven to be challenging. This difficulty hints at a complex interplay between IRI and both the multitude and the redundancy of systems at play in the MI scenario.
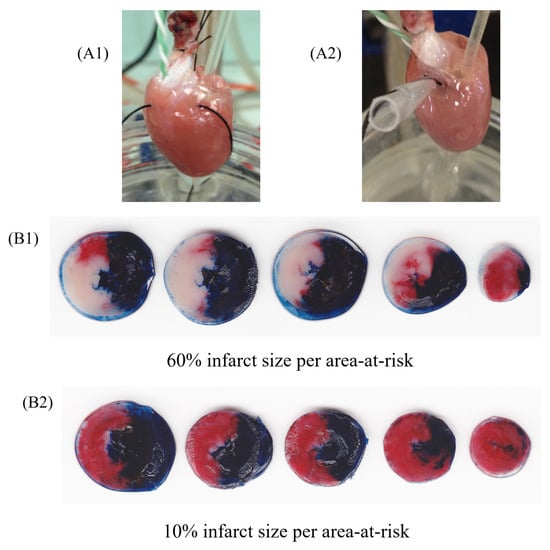
Figure 2. Representative image of isolated perfused hearts (ex vivo) using Langendorff apparatus undergoing ischemia and reperfusion protocol. Rat heart during regional ischemia (A1) and reperfusion (A2). Figures represent groups that received vehicle (B1) or HDL (200 µg/mL) infusion (B2), after the protocol of staining with 2,3,5-triphenyltetrazolium chloride staining. The heart sections (B1,B2) are presented in tree colors for histologic analysis: blue, the non-ischemic area (also known as non-risk area); red, the ischemic area (also known as area-at-risk); and white or pale, the infarct area. The infarct size was calculated as a percentage of the area-at-risk.
2. The Mechanisms Underlying the MI-Induced Activation of NLRP3 Inflammasome Signaling
The detection of DAMPs is a fundamental aspect of the innate immune system’s ancient defense mechanism against invaders. It not only counters threats but also notifies neighboring cells through proinflammatory cytokines. In MI patients, the initiation of proinflammatory signaling through DAMPs may occur as a consequence of IRI [10][19]. Additionally, the activation of this signaling could potentially pre-exist and be driven by modified lipoproteins like oxidized low-density lipoproteins (oxLDL), which have the potential to enhance proinflammatory responses [20]. Nevertheless, the antioxidant function of the paraoxonase family, notably paraxonase-1 (PON1) within HDL, significantly mitigates the inflammatory impact mediated by ox-LDL. Animal model experiments have indicated a direct correlation between the absence of the PON1 gene and inflammatory responses. In humans, plasma levels of PON1 exhibit an inverse correlation with the development of atherosclerosis [21].
Pattern-recognition receptors (PRRs) play a pivotal role in mediating signaling within the innate immune defense system, triggering proinflammatory responses upon detecting DAMPs. Notably, PRRs are not limited to immune cells but are also found in non-immune cardiac cells, including cardiac fibroblasts, cardiomyocytes, and endothelial cells [22][23][24][25]. Proinflammatory cytokines can act in a paracrine manner via PRRs, amplifying and intensifying signals along the same inflammasome pathway [26].
These receptors can be categorized into two subgroups: (i) transmembrane receptors, such as Toll-like receptors (TLRs) and C-type lectin receptors (CLRs) [27], and (ii) intracellular sensors, including cytoplasmic nucleotide-binding oligomerization domain-like receptors (NLRs), retinoic acid-inducible gene I-like receptors (RLRs), absent in melanoma 2-like receptors (ALRs), and cytosolic DNA receptors [28]. However, among these, the TLR and NLR families are most extensively characterized in cardiac cells, with particular focus on members like TLR4 and the NLRP3 inflammasome (Figure 1).
While the NLRP3 inflammasome serves a protective role against harmful stimuli, its dysregulated activity can paradoxically intensify proinflammatory signaling and exacerbate MI damage, ultimately leading to cell death [29]. In fact, NLRP3 inflammasome-mediated inflammation is initiated in the early stages of myocardial ischemia and can persist for days, exerting a profound influence on chronic outcomes like cardiac remodeling and congestive heart failure [30][31].
In preclinical studies modeling myocardial IRI, it has been observed that just minutes of reperfusion can trigger NLRP3 inflammasome activation [32]. Interestingly, the activation kinetics of the NLRP3 inflammasome in the heart after IRI display a gradual increase over a 6 to 24 h period during reperfusion [32]. This sustained activation contributes significantly to the inflammatory response and the severity of myocardial damage [32].
The most extensively studied pathways that lead to the activation of the NLRP3 inflammasome triggered by the MI include the following: (i) the production of reactive oxygen species (ROS) derived from the mitochondria, (ii) lysosomal destabilization, (iii) ionic imbalance (mainly presented by K+ efflux and Ca2+ mobilization), and, more recently, (iv) intracellular signaling through the adenosine triphosphate (ATP)-activated P2 purinergic receptor (P2X7R) [13].
Mitochondrial ROS production arises from a complex interplay of sources, including electron leakage from complex I and II, leading to the formation of superoxide, as well as the generation of hydrogen peroxide, which can induce the creation of other reactive species [33]. This cascade of ROS generation can trigger the activation of the NLRP3 inflammasome, a process facilitated by its interaction with thioredoxin-interacting protein (TXNIP), a member of the α-arrestin superfamily of proteins found ubiquitously in non-ischemic myocardial tissue [34] (refer to Figure 1). Notably, TXNIP itself becomes activated by ROS, setting in motion a positive feedback loop that further amplifies ROS production [35]. In agreement with these findings, studies using mice with a cardiomyocyte-specific knockout of the TXNIP gene have demonstrated a reduction in the extent of MI [36].
Lysosomal destabilization represents another crucial mechanism to consider within the context of MI [37]. This process involves the release of cathepsin B hydrolase, a lysosomal cysteine protease belonging to the papain family, from the lysosome into the cytosol. This release contributes significantly to the activation of the NLRP3 inflammasome and, consequently, exacerbates myocardial damage [38].
Mitochondrial Ca2+ overload has emerged as a contributor to the activation of the NLRP3 inflammasome during ischemia [39]. An excessive or sustained increase in mitochondrial Ca2+ levels can lead to mitochondrial damage and cell death [40]. During MI, damaged cardiac cells can release ATP from their membranes, initiating the activation of the P2X7R. This receptor, an ATP-gated extracellular ion channel known for its involvement in transmembrane ion migration [41][42], responds to ATP by promoting the influx of Ca2+ and Na+ while inducing K+ efflux. This classic signaling pattern triggers the activation of the NLRP3 inflammasome [35] (Figure 1). Consequently, extracellular ATP binding to P2X7R rapidly disrupts the cytosolic ion concentration gradient.
The details of K+ efflux in NLRP3 inflammasome activation remain less comprehensively understood and appear to be independent of Ca2+ mobilization. For instance, Katsnelson et al. [43] disrupted the cytosolic concentration gradient by employing BAPTA, a potent Ca2+ chelator and cytosolic Ca2+ buffer. Their findings demonstrated that cellular K+ efflux during NLRP3 activation operates independently of intracellular Ca2+ dynamics [41].
Furthermore, the P2X7R has been identified as a critical mediator of NLRP3 inflammasome activation through its interaction with the serine-threonine kinase NIMA-related Kinase 7 (NEK7), a member of the mammalian NIMA-related kinases (Nek protein) family [44]. Recently, using an in vivo animal model of MI, Yanqin Li and colleagues [45] revealed that an upregulation of P2X7R, NEK7, and NLRP3 inflammasome activation occurs after MI, and is accompanied by a deterioration in cardiac function and an increase in inflammatory markers.
3. The Mechanistic Basis of NLRP3 Inflammasome Priming and Activation
The NLRP3 inflammasome is expressed in various cardiovascular cells, such as endothelial cells and cardiomyocytes. Among several identified inflammasomes, including NLRP1, NLRC4, and AIM2, NLRP3 is the most extensively studied in MI pathogenesis. In a 2009 study by Iyer et al. [46] using a necrotic cell model, NLRP3 inflammasome activation was demonstrated, leading to IL-1β release. Subsequently, Kawaguchi et al. [47] emphasized the significance of NLRP3 inflammasome activity in cardiac fibroblasts during IRI.
The pro-inflammatory signaling triggered by DAMPs in MI involves two phases, termed signal 1 and signal 2 [35], both dependent on the NLRP3 inflammasome. Signal 1, also known as priming, initiates when DAMPs bind to TLRs, initiating downstream signaling [39]. In mammals, 13 TLRs are recognized [48][49], but it remains uncertain whether all are present in myocardial tissue. Most of them (TLR-2 to 7 and 9) have been identified through mRNA expression in murine cardiac cells in vitro and in vivo [50]. In humans, TLR1 to 10 have been identified thus far, with recent findings indicating increased expression in the Tlr2, Tlr3, Tlr4, and Tlr9 genes in chronic ischemic diseases [51].
To date, the most comprehensive signal description involves TLR4-mediated nuclear factor kappa B (NF-κB) signaling. DAMPs activate TLR4, initiating an intracellular signaling cascade that, in turn, activates the adapter protein myeloid differentiation primary response protein (MyD88). MyD88 subsequently triggers downstream signaling by binding to interleukin-1 receptor-associated kinase 4 (IRAK4), followed by IRAK1/2, resulting in the formation of a large complex called the Myddosome [27]. This Myddosome complex associates with the ubiquitin ligase E3 TNF receptor-associated factor 6 (TRAF6), leading to the activation of the kappa light polypeptide gene enhancer nuclear factor in the B-cell inhibitor alpha (IκBα) within the IKK complex, and subsequent translocation, along with NF-κB, into the cell nucleus. NF-κB serves as the principal transcription factor driving the proinflammatory priming response during MI [52]. Consequently, priming signaling targets gene transcription for NLRP3 inflammasome proteins (e.g., ASC and pro-caspase-1) and pro-inflammatory interleukins (pro-IL1-β and pro-IL18) [53][54][55][56] (Figure 1).
Furthermore, alternative pathways for the transcriptional activation of the priming effect may occur directly through IL-1β via the IL-1 receptor (IL1R), also coupled to MyD88, leading to transcriptional signaling via NF-κB. It is plausible that multiple non-canonical pathways exist to indicate pro-inflammatory priming, potentially involving enzymatic deubiquitination.
Signal 2, also referred to as activation, is a process initiated by DAMPs, leading to the assembly or oligomerization of the NLRP3 complex. This activation is driven by proximity-induced autocatalytic activity [12]. In the presence of DAMPs, NLRP3 interacts with ASC through PYD interactions and pro-caspase-1 through CARD interactions. It is worth noting that ASC serves as a crucial scaffold protein necessary for recruiting the pro-caspase-1 effector enzyme to form the NLRP3 inflammasome [57]. Consequently, the activation of the NLRP3 inflammasome leads to the mediation of caspase-1 activity, which is a cysteine protease with specificity for aspartic residues and is classified among the proinflammatory caspases, including caspases 1, 4, 5, 11, and 12 [58] (Figure 1).
The NLRP3 inflammasome exhibits a multi-target nature, attributed to its possession of three distinct domains: (i) an LRR domain, (ii) a NACHT domain, and (iii) an amino-terminal PYRIN (PYD) domain [7]. This unique feature renders NLRP3 susceptible to the stimulation of various DAMPs. Nevertheless, the precise mechanisms linking the NLRP3 inflammasome to these DAMPs remain unclear [59]. It is widely acknowledged that the primary mode of NLRP3 inflammasome activation involves the release of pro-inflammatory cytokines via the formation of membrane pores, a process termed pyroptosis [60] (Figure 1). Inhibiting NLRP3 inflammasome activity using a caspase-1-specific probe can significantly safeguard the myocardium against IRI, mitigating the harmful consequences of pyroptosis. Consequently, pyroptosis emerges as the principal outcome of NLRP3 inflammasome activation.
4. NLRP3 Inflammasome Activation and Pyroptosis after Coronary Reperfusion
Historically, MI treatment primarily focused on mitigating two forms of cell death. The first is necrosis, primarily stemming from traumatic events that disrupt metabolic homeostasis, activating independent cellular pathways [61][62]. The second form is apoptosis, characterized by programmed cell death driven by intrinsic or extrinsic molecular pathways. While apoptosis serves adaptive functions, its dysregulation can lead to lethal outcomes. Pyroptosis, a cell death triggered by NLRP3 inflammasome activation, emerges as an alternative cell death pathway, presenting a distinctive challenge in MI management. Coined by Cookson and Brennan in 2001, pyroptosis, derived from the Greek word’s “pyro” meaning fire and “ptosis” meaning fall or death, was initially observed in macrophages during bacterial infection processes [63][64]. Remarkably, pyroptosis exhibits characteristics of both apoptosis, such as nuclear condensation and DNA fragmentation, and necrosis, including plasma membrane rupture and an inflammatory response [65][66]. Pyroptosis underpins the systemic elevation of IL-1β. This cytokine is associated with a wide array of inflammatory conditions, including rheumatoid arthritis, osteoarthritis, chronic obstructive pulmonary disease, asthma, inflammatory bowel disease (including Crohn’s disease and ulcerative colitis), multiple sclerosis, Alzheimer’s disease, atherosclerosis, and type 2 diabetes [23][67].
Pyroptosis entails the activation of caspase-1 and the subsequent cleavage of gasdermin-D (GSDM-D), enabling the secretion of pro-inflammatory interleukins. Hence, pyroptosis is widely recognized as a caspase-1-dependent cell death program. The cleavage of GSDM-D leads to the release of its active N-terminal fragment, which relocates to the plasma membrane’s lipid region, instigating oligomerization and pore formation. These membrane pores facilitate the outward release of active interleukins and result in lytic cell death due to the leakage of cytosolic contents [68] (Figure 1).
Preclinical studies have convincingly demonstrated the molecular signaling pathway of pyroptosis mediated by NLRP3/ASC/caspase-1 in the context of myocardial damage following MI [57]. Consistently, Yang et al. [69] demonstrated that treatment with the same caspase-1 inhibitor mitigated myocardial damage in vivo. Likewise, several other inhibitors are also capable of mitigating or inhibiting the pyroptosis pathway by targeting the NLRP3 inflammasome, caspase-1, and interleukins (specifically IL-1β and IL-18). This intervention leads to myocardial protection, as confirmed by the assessment of biological markers, including proinflammatory cytokines and Troponin I [70]. These experimental protocols suggest that the NLRP3/ASC/caspase-1 molecular signaling pathway could serve as a promising therapeutic target for mitigating pyroptosis.
5. The Non-Canonical Activation of Pyroptosis during MI
The non-canonical mechanisms governing inflammasome activation and its connection to pyroptosis remain partially elucidated, yet they offer a potential alternative pathway to the previously described ones. Initial investigations, utilizing lipopolysaccharide (LPS) stimulation, have unveiled immune defense system activation through mechanisms under the control of other members of the inflammatory caspase family. Specifically, in humans, caspase-4 and 5 have been implicated, whereas in mice, caspase-11 is a key player, and in both species, caspase-12 contributes to this process [71][72] (Figure 1). There is a plausible notion that specific inflammatory caspases may assume an alternative function in the proteolysis of the GSDM-D protein, which was previously regarded as the exclusive role of caspase-1 [73].
Caspase-4 and 5 in humans exhibit similarities to caspase-11 in mice, thereby classifying them as orthologs [74]. Kayagaki et al. [75] demonstrated that murine macrophages, when exposed to LPS, activate caspase-11, leading to the proteolytic cleavage of GSDM-D. This constitutes the primary mechanism of pyroptosis, as GSDM-D releases an N-terminal fragment, instigating membrane pore formation, which may indirectly activate the NLRP3 inflammasome. Likewise, in an experimental model of endotoxic shock in mice, LPS stimulation directly triggers caspase-11 activation, which, in turn, facilitates the activation of IL-1β by cleaving the transmembrane protein pannexin-1 (Panx-1) and allowing its release [76]. In the context of MI, where the classic experimental protocol of coronary artery occlusion is employed to induce in vivo myocardial IRI, an observed elevation in caspase-4 activity and GSDM-D expression underscores the potential pathophysiological relevance of this non-canonical mechanism for myocardial damage [77].
Several studies have indicated the potential role of endoplasmic reticulum (ER) stress as a central hub for alternative caspase activation [78][79]. One plausible mechanism involves the signaling of the inositol 1,4,5-triphosphate receptor (IP3R1) within the endoplasmic reticulum (ER), a crucial regulator of Ca2+ transport. This signaling pathway has been implicated in modulating the NLRP3 inflammasome pathway during myocardial injury [80]. Excessive Ca2+ accumulation, functioning as a DAMP, may potentially serve as the link mediating this connection and triggering pyroptosis.
Moreover, it is conceivable that stress triggered by diverse DAMPs might activate alternative pathways that culminate in pyroptosis and cytokine release, bypassing the primary involvement of NLRP3 inflammasome activity [80]. Nevertheless, compelling evidence indicates that in instances of prolonged pathological events, the continued presence of DAMPs is likely to ultimately lead to the canonical activation of the NLRP3 inflammasome [81].
6. The Double-Edged Effects of HDL Particles in MI
The plasma concentration of HDL particles, measured by either their cholesterol content or apolipoprotein A1 levels, is a widely recognized robust predictor of cardiovascular events. This phenomenon can be attributed to multiple mechanisms, encompassing the antioxidant, anti-inflammatory, antithrombotic, and antiapoptotic properties associated with these particles [82].
HDL are endogenous particles, sizing approximately 7–12 nm, with prominent characteristics such as: (i) high diffusion capacity through the intravascular space; (ii) promotion of the reverse cholesterol transport from peripheral tissues to the liver; and (iii) cross-talking with endothelial cells by carrying several active metabolites, among which proteins, lipids, phospholipids, apoproteins and micro-RNAs (miRNAs) are in constant and intense hemodynamic traffic [83][84]. The structure of HDL consists of numerous proteins, lipids, and miRNA, organized with a hydrophobic core and a hydrophilic surface. Nevertheless, this structure undergoes continuous remodeling within the bloodstream, achieved through processes such as lipid transfer, hydrolysis, esterification, and the capture of proteins that exhibit high affinity for HDL.
Interestingly, there is evidence suggesting that HDL mimetic therapy may confer myocardial protection post-MI [85]. This effect should contribute to reducing the extent of MI in studies using ex vivo models that tested coronary reperfusion with HDL. HDL exhibits multiple cytoprotective actions that can engage various protective mechanisms against NLRP3 inflammasome activation and DAMP signaling [86]. These mechanisms can be initiated through the interaction between HDL and endothelial cells or cardiomyocytes. In essence, cardiomyocyte protection can be achieved directly via lipoprotein-to-cardiomyocyte contact, on indirectly via endothelium–cardiomyocyte contact or cross-talking [87].
While direct evidence of the interaction between HDL and the myocardium remains limited, the presence of scavenger receptor (SR-BI) expression in cardiomyocytes emerges as a significant factor contributing to the cardioprotective effect [88]. In general, the binding, internalization, and transport of HDL across endothelial cells involve three key proteins: (i) SR-BI, (ii) endothelial lipase, and (iii) the ATP-binding cassette (ABCG1). Additionally, lipid-free apoA-I undergoes lipidation by the ATP-binding cassette A1 (ABCA1) before being transported as part of the HDL complex [89]. Directly or indirectly, HDL’s cardioprotective effect, achieved by dampening inflammasome activation in the myocardium, may be attributed to various mechanisms.
7. The Protective Mechanisms of HDL on Cardiomyocyte Pyroptosis
HDL plays a crucial role in preserving mitochondrial function by activating intracellular protein kinase signaling through various receptors. In addition, HDL can also engage with intracellular G protein-coupled receptors (GPCRs), both trimeric and small [90]. Upon binding to SR-BI, ABCA1, or GPCRs, these receptors initiate downstream signaling cascades, influencing cellular processes like proliferation, metabolic regulation, and cell survival. In the context of reperfusion following MI, these activated pathways are collectively referred to as the reperfusion salvage kinase (RISK) [91]. The RISK pathway comprises two signaling routes mediated by the phosphoinositide 3-kinases (PI3Ks) family plus protein B (Akt) and mitogen-activated protein kinase 1, plus extracellular signal-regulated kinase 1/2 (MEK1-ERK1/2) [92]. Both are triggered by stimuli to transmembrane GPCRs.
PI3K-Akt inhibits the mitochondrial permeability transition pore (mPTP) by reducing glycogen synthase kinase 3 beta (GSK3β) activity, preserving mitochondrial integrity [93][94]. Additionally, it activates hexokinase II (HKII), essential for glycolysis during ischemic or hypoxic conditions [95][96][97][98]. MEK1-ERK1/2 stimulates anti-apoptotic mechanisms and desensitizes mPTP via the cyclophilin D pathway [99]. Furthermore, in non-cardiac cells, PI3K-Akt signaling has shown potential in regulating NLRP3 inflammasome and caspase-1 activity [100][101][102]. HDL can activate the RISK pathway during MI, thereby modulating mitochondrial DAMPs (mtDAMPs) in the cytoplasm and reducing NLRP3 inflammasome activation [103][104]. In addition, recent findings also suggest that Akt’s phosphorylation of NLRP3 on serine 5 inhibits its assembly and activity [105] (Figure 3). The phosphorylation of AKT at serine 570 may exert an inhibitory effect on peroxisome proliferator-activated receptor coactivator 1α (PGC-1α), a transcription factor responsible for mediating the dynamics of mitochondrial metabolism [106]. Possibly, a negative regulation of PGC-1α would result in increased expression of the NLRP3 inflammasome, mtDNA release, and oxidative stress [107]. Additionally, this downregulation could decrease the expression of tumor necrosis factor α-induced protein 3 (TNFAIP3), an intermediate in TLR signaling that inhibits the NLRP3 inflammasome pathway [108]. Hence, this specific AKT phosphorylation appears to have detrimental effects, which is intriguingly contradictory considering that the phosphoinositide 3-kinases (PI3Ks) family plus protein B (Akt) pathway (PI3K-Akt pathway) typically promotes cardio protection by facilitating a shift in glycolytic metabolism during ischemic events [109][110].
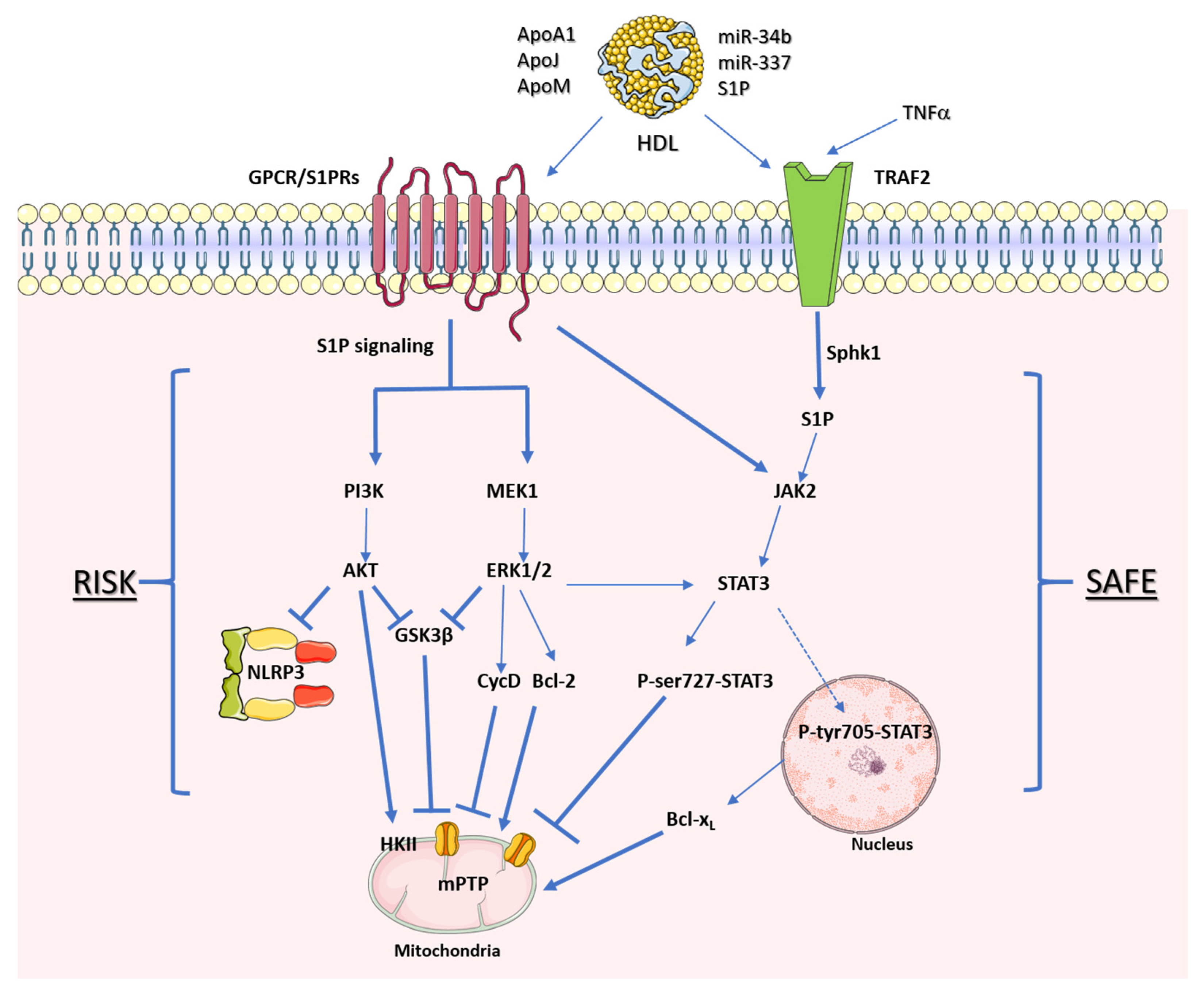
Figure 3. HDL receptors of cardioprotective protein kinase pathways. Representative signaling mediated by HDL through the reperfusion salvage kinase (RISK) pathway and the survivor-activating factor enhancement (SAFE) pathway. The arrows represent stimulation, while the hammerheads represent inhibition.
Another pathway activated during reperfusion, known as survivor activating factor enhancement (SAFE), is pertinent to myocardial protection. In SAFE, the key player is the signal transducer and activator of transcription 3 (STAT3) protein kinase. Typically activated via phosphorylation, tyrosine phosphorylated STAT3 can translocate to the nucleus to regulate gene expression or the mitochondrial membrane [111]. Like the RISK pathway, HDL particles can activate the SAFE pathway, illustrating their crosstalk in myocardial pro-survival mechanisms [112][113].
Serine phosphorylation of STAT3 (Ser-727 residue) is implicated in preserving respiratory complex I and inhibiting mPTP opening following MI [93][94][112]. While the STAT3 pathway is extensively studied for its role in myocardial IR protection, its direct correlation with NLRP3 inflammasome modulation remains unclear. Nevertheless, the activation of the SAFE pathway may indirectly contribute to reducing mtDAMPs by preventing NLRP3 inflammasome activation [114]. Experimental evidence with other cell types supports the feasibility of STAT3-mediated NLRP3 inflammasome signaling [115][116][117].
The SAFE pathway involves two signaling routes driven by TNF-alpha and Janus kinase 2 (JAK2)-STAT3, eventually converging downstream in the cytoplasm. TNF-alpha activates via its release and binding with the Tumor Necrosis Factor 2 Receptor (TRAF2). HDL’s interaction with TNF-alpha signaling during reperfusion, while less explored, influences TRAF2 activation, enhancing sphingosine kinase-1 (Sphk1) activity and sphingosine-1-phosphate (S1P) production, a well-studied bioactive sphingolipid, in the cytoplasm [111].
Increased S1P levels trigger intracellular TRAF2 activation, leading to further S1P production via Sphk-1 activity, and ultimately instigating JAK2-STAT3 activation through an ‘inside-out’ signaling pathway [118][119]. Inside-out signaling is believed to facilitate JAK2 activation through S1P receptors (S1PRs), which may induce phosphorylation and docking sites for STAT3 proteins, enabling their subsequent phosphorylation at serine or tyrosine residues [111].
HDL particles can activate two additional kinase pathways that require further examination in the context of MI. First, HDL may target the positive regulatory phosphorylation of NLRP3 at serine 198 in humans and serine 194 in mice by c-Jun terminal kinase (JNK) [120]. HDL has demonstrated its potential to reduce this signaling pathway’s activity, as seen in human pancreatic beta cells and endothelial cells [121][122]. Second, the protein kinase A (PKA) signaling pathway, which negatively regulates NLRP3 assembly through serine 295 phosphorylation in humans and serine 291 phosphorylation in mice [123], is associated primarily with HDL/ApoA-I via ABCA-1 and SR-BI receptors [124].
HDL particles transport sphingolipids that regulate Ca2+ dynamics through various mechanisms. Among these, S1P can modulate intracellular Ca2+ levels via S1P1–5 receptors coupled to G proteins [125]. Specifically, S1P2 and S1P3 receptors are thought to activate phospholipase C and inhibit adenylyl cyclase, facilitating intracellular Ca2+ mobilization [125]. Ke et al. [126] used confocal microscopy to demonstrate that S1P, in cardiomyocytes subjected to hypoxia and reoxygenation, mitigated mitochondrial Ca2+ overload. This effect preserved mitochondrial membrane potential by preventing the opening of the mitochondrial permeability transition pore (mPTP).
While further investigations are required to confirm these effects in the context of MI, promising results have emerged from ApoA-I infusion therapies [127]. These efforts date back to the 1980s when reconstituted HDL (rHDL), with ApoA-I as the primary protein component, was utilized [128][129]. More recently, the anti-inflammatory effects of rHDL were documented in preclinical [130][131] and translational (human ex vivo and in vivo) models, with challenges with LPS in healthy human volunteers [132]. Among these studies, the noteworthy impact of CSL-112, a recombinant HDL (rHDL), in reducing IL-1β secretion, a robust indicator of NLRP3 inflammasome pathway activation, deserves attention [133]. A comprehensive review of these studies was conducted by Morin et al. [134].
The use of rHDL therapy for patients in the acute phase of MI was evaluated in a randomized clinical trial, which showed a significant reduction in coronary atheroma volume [135]. Currently, an ongoing Phase 3 multicenter double-blind randomized placebo-controlled trial, titled ‘Study to Investigate CSL-112 in Subjects with Acute Coronary Syndrome’ (AEGIS-II; NCT03473223), holds the promise of offering further evidence regarding the therapeutic efficacy of rHDL in enhancing post-MI outcomes. While this trial was not specifically designed to assess the extent of MI, the data related to cardiovascular mortality and the incidence of heart failure could indicate the potential relevance of this mechanism.
ApoM is found not only in HDL particles but also in other lipoproteins like LDL, very low-density lipoprotein (VLDL), and chylomicrons. Its protective effect on mitochondrial function is linked to its ability to bind with S1P, forming the ApoM–S1P complex, which facilitates intravascular transport. It is noteworthy that roughly 65% of intravascular S1P is carried by HDL particles through their interaction with ApoM [136].
S1P is naturally produced intracellularly through the catalytic activity of sphingosine kinase isoforms 1 and 2 (SphK1 and SphK2), regulated by phosphorylation reactions [137]. S1P can indeed initiate signaling cascades in vascular endothelium and cardiomyocytes, offering various protective functions. Notably, this sphingolipid plays a role in a range of biological processes, encompassing cell motility, proliferation, differentiation, cytoskeleton organization, cell growth, and survival. It achieves this through paracrine and autocrine cell communication, particularly during acute pathological conditions and responses to metabolic stress, such as MI [138][139][140][141].
Exploring S1P metabolism during MI poses challenges. The transcription rate of cardiac SphK1, responsible for S1P synthesis, increases during MI [142]. Correspondingly, plasma S1P levels remain stable during the initial 2 h following MI onset, rise over the subsequent 12 h, and then revert to levels comparable to those seen in patients with stable coronary artery disease (sCAD) [143]. However, HDL-bound plasma S1P levels are lower in both MI and sCAD patients compared to controls, indicating a reduced capacity to retain S1P within the HDL of these patients [143]. The observed decline in apolipoprotein M production during the acute inflammatory response could potentially explain this discovery [144]. Nevertheless, further research is necessary to establish a definitive understanding of the interplay between MI, HDL-bound S1P and apolipoprotein M.
Preclinical studies have showcased myocardial protection against IRI by augmenting S1P levels through diverse strategies, including the inhibition of the endogenous catabolic enzyme S1P lyase [138], S1P supplementation using native or recombinant HDL particles [145], and elevating S1P availability via genetic modifications [146]. The crucial role played by S1P’s interaction with myocardial cells via S1PR1–5, G protein-coupled receptors belonging to the class A family [147], underpins these cytoprotective mechanisms. While the precise effects of S1PR1–5 are not fully elucidated, the interconnections represented by complexes like S1P–S1PR1, S1P–S1PR2, and S1P–S1PR3 are associated with shielding against acute myocardial IRI [139]. An illustrative example involves the interplay between the inhibitory regulator of G proteins and β-arrestin within the S1P1 signaling pathway. HDL particles binding to S1P exert a regulatory influence, leading to anti-inflammatory effects by inhibiting the NF-κB pathway via ERK-1/2. This ultimately holds the potential to reduce the synthesis of NLRP3 inflammasome proteins [99][148].
The connection between the apoM-S1P complex and the NLRP3 inflammasome is still a subject of ongoing research. Weigert et al. [149] shed light on another avenue of interaction between S1P metabolism and NLRP3 inflammasome activity in an in vitro study. They demonstrated that caspase-1’s proteolytic activity hinders SphK2 function by cleaving its N-terminal portion, thereby disrupting S1P metabolism. However, the in–out signaling whose interactions with the NLRP3 inflammasome are best demonstrated are due to the synthesis of S1P via SphK1. So far, it remains uncertain whether SphK1 might also play a role in this interaction.
ApoJ, also known as clusterin, is a chaperone composed of heterodimeric glycoproteins. It is primarily found in smaller, denser HDL particles, and plays a vital role in cholesterol transport and the prevention of oxidative stress caused by Reactive Oxygen Species (ROS) [150]. In a porcine model of myocardial IRI induced by the percutaneous occlusion of the left anterior coronary artery, there was an increase in the plasma concentration of ApoJ, suggesting a potential connection between ApoJ and acute cardiac injury [151]. The precise mechanism underlying this effect remains unclear. However, ApoJ exhibits antioxidant properties and can activate the RISK pathway, which implies it might be a compensatory metabolic response during the acute post-injury phase [152]. Furthermore, ApoJ’s cardioprotective effects are associated with the downregulation of the expression of tumor necrosis factor-α (TNF-α) and Bcl-2-associated X protein (BAX) through the inhibition of inflammatory signaling via the NF-κB pathway [153]. This inhibition is equally relevant for reducing the activation of the NLRP3 inflammasome and protein transcription mechanisms.
Thacker et al. [86] explored an alternative mechanism in which HDL might influence NLRP3 inflammasome activation. They conducted in vitro experiments using DAMPs mimetics. In this study, they treated human monocytes and macrophages (THP-1) with rHDL, consisting solely of ApoA-I and soybean phospholipids. The results revealed a noteworthy reduction in IL-1β levels by rHDL when these cells were exposed to cholesterol crystals, which act as danger signals capable of triggering a proinflammatory response. This suggests that HDL possesses the potential to exert anti-inflammatory effects by antagonizing NLRP3 and IL-1β transcription, as well as inhibiting caspase-1 activation. Both HDL actions on NLRP3 signaling and caspase-1 activation may prevent the disruption of lysosomal membrane integrity [86]. Typically, after coronary reperfusion, there is lysosomal degradation coupled with an accumulation of autophagosomes, stemming from disruptions in the autophagic process. These changes are intricately linked to the activation of the NLRP3 inflammasome and the subsequent demise of cardiomyocytes [154][155][156].
8. The Molecular Changes and Pathogenic Effects Produced by HDL during MI
As mentioned above, in the acute phase of MI, significant alterations occur in the activity of cells within the cardiovascular and immuno-inflammatory systems, leading to a profound transformation in the molecular composition of HDL [99] (Figure 4). HDL particles in patients with MI exhibit several dysfunctions when compared to individuals with stable coronary artery disease.
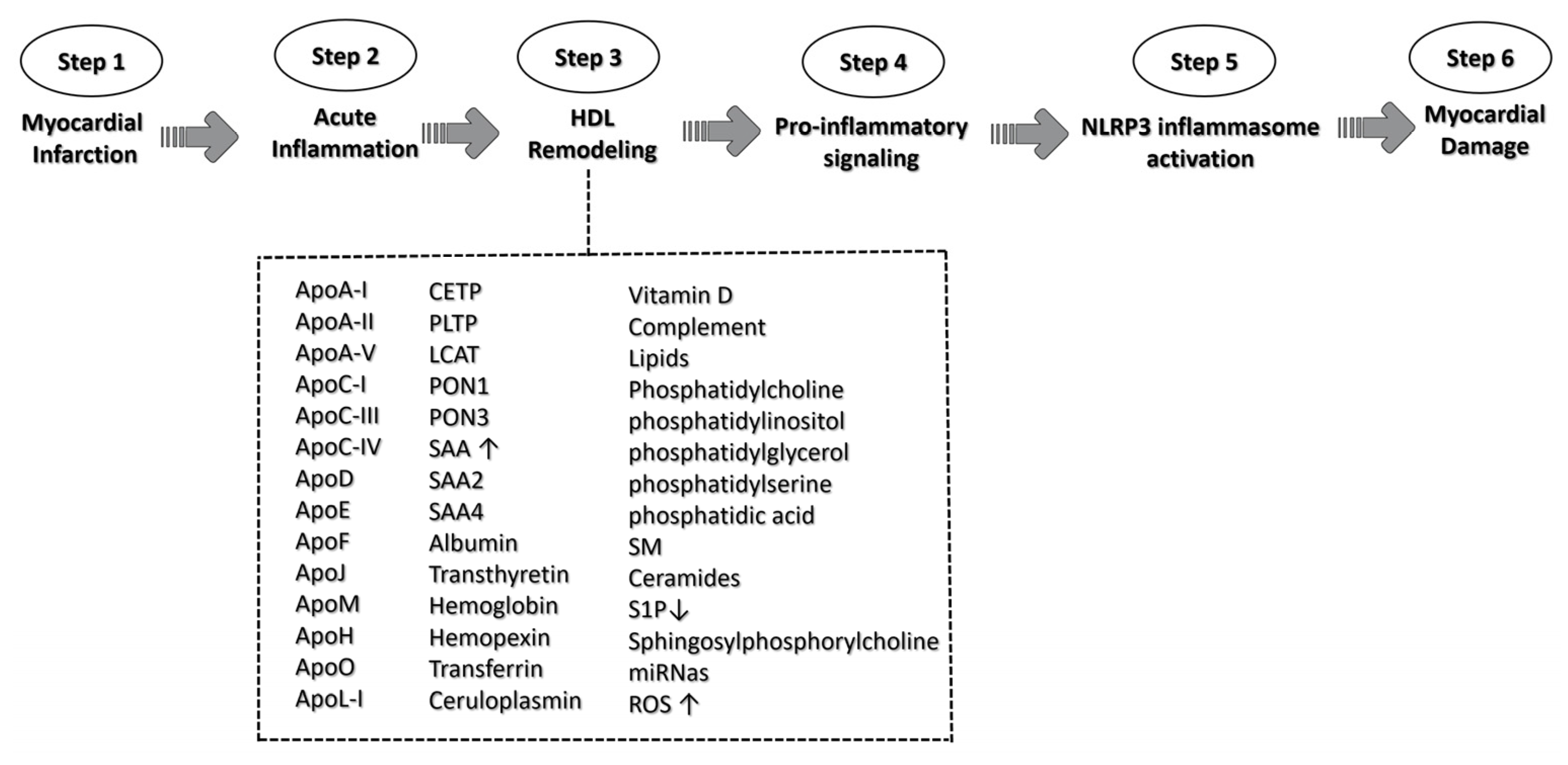
Figure 4. A schematic representation of HDL triggering NLRP3 activation signaling. The role of HDL in the context of myocardial infarction is illustrated along the six pivotal steps, culminating in NLRP3 activation signaling and the ultimate myocardial damage. HDL complement acronyms that could participate in the remodeling process: apolipoprotein A-I (ApoA-I); apolipoprotein A-II (ApoA-II); apolipoprotein A-V (ApoA-V); apolipoprotein C-I (ApoC-I); apolipoprotein C-III (ApoC-III); apolipoprotein C-IV (ApoC-IV); apolipoprotein D (ApoD); apolipoprotein E (ApoE); apolipoprotein F (ApoF); apolipoprotein J (ApoJ); apolipoprotein M (ApoM); apolipoprotein H (ApoH); apolipoprotein O (ApoO); apolipoprotein (ApoL-I); cholesteryl ester transfer protein (CETP); phospholipid transfer protein (PLTP); lecithin cholesterol acyltransferase (LCAT); paraoxonase-1 (PON1); paraoxonase-1 (PON3); serum amyloid A (SAA); serum amyloid A-2 (SAA2); serum amyloid A-4 (SAA4); Sphingomyelin (SM); Sphingosine 1-phosphate (S1P); microRNA (miRNas); and reactive oxygen species (ROS). ↑ means increase; ↓ means decrease.
This transformation encompasses a reduction in particle size associated with increased density, shifts in miRNA profiles, the oxidative modification of lipids and proteins due to oxidative stress, the incorporation of inflammatory proteins, and the depletion of protective proteins. Importantly, this metamorphosis in HDL commences even prior to the occurrence of MI, prompted by cardiovascular risk factors, and intensifies in the days following the onset of MI symptoms [157]. These changes in the lipid–protein–miRNA triad transform the phenotype of HDL particles, rendering them dysfunctional and potentially pathogenic [99][158][159].
9. Oxidation in HDL Components during MI and Inflammasome Activation
The alteration in HDL functionality is primarily attributed to its increased exposure to reactive oxygen and nitrogen species secreted by immune system cells, as well as its capacity to absorb oxidized molecules from other lipoproteins, notably LDL [160]. This intricate process can modify HDL functionality through quantitative and qualitative changes within its components, including various apolipoproteins (ApoA-I, ApoA-II, ApoA-IV, ApoD, ApoE, ApoF, ApoJ, ApoL1, and ApoM) and enzyme ligands (serum amyloid A [SAA], Paraoxonase-1 [PON1], Paraoxonase 3 [PON3], Lecithin cholesterol acyl transferase [LCAT], platelet-activating factor acetylhydrolase [PAF-AH], Glutathione peroxidase 3 [GPX3], Cholesteryl ester transfer protein [CETP], and phospholipid transfer protein [PLTP]) [161][162]. During this period of acute phase MI, when oxidative processes are heightened, there may also be shifts in the distribution of these components across different HDL subfractions. Notably, a study involving 69 patients with ST-segment elevation myocardial infarction (STEMI), in comparison to 67 healthy controls, revealed that the reduction in antioxidant capacity has a more significant impact on smaller HDL subtypes, particularly HDL3 [163]. This change was correlated with the decreased concentration of PON1 in this specific HDL subtype, a crucial enzyme in hydrolyzing oxidized LDL and mitigating the harmful effects of products resulting from phospholipid peroxidation [164]. Consequently, oxidative stress during the acute phase of MI is closely associated with the development of inflammation [165].
However, while research in atherosclerotic disease has indicated that the oxidation of methionine residues in human apoA-I can serve as a potent inflammasome activator [166], no clinical studies have definitively established a direct link between oxidized HDL components and the activation of the NLRP3 inflammasome signaling pathway during MI. Therefore, it is imperative to conduct further investigations to explore the specific pro-inflammatory mechanisms arising from oxidized HDL components.
10. HDL Lipid and Protein Composition Changes during MI and Inflammasome Activation
Observational studies have reported notable alterations in plasma lipid species during MI [167][168]. Specifically, in HDL, enzymes like CETP, which facilitate lipid transport among lipoproteins, exhibit heightened activity during the acute MI phase [169]. Consequently, the lipid composition of HDL undergoes significant changes, including a reduction in phospholipids, ester cholesterol, and S1P, along with an elevation in free cholesterol, triglycerides, saturated fatty acids, lysophosphatidylcholine, and lipid peroxidation products [170]. These shifts in HDL composition appear to be particularly relevant within the phospholipid category when comparing patients diagnosed with acute coronary syndrome compared to those with stable coronary disease [171]. Within the realm of phospholipids, a specific subclass known as plasmalogens emerges as particularly noteworthy [171][172]. Although the precise function of plasmalogen remains incompletely understood, its concentration, inversely related to inflammatory diseases, warrants further investigation to establish its clinical significance [172]. In pre-clinical in vivo models, dietary modifications aimed at increasing plasmalogen levels demonstrated a reduction in the inflammatory vascular marker vascular cell adhesion molecule 1 (VCAM1) [173]. Additionally, it is important to consider plasmalogen’s role as a carrier, as indicated by the significant attenuation of NLRP3 inflammasome pathways in vitro when immune microglial cells (BV-2) were pre-treated with eicosapentaenoic acid (EPA)-enriched ethanolamine plasmalogen [174].
During MI, there is a notable divergence among patients in terms of triglyceride levels, with some experiencing an increase while others experience a reduction [175]. Remarkably, the APOC-III protein levels within both LDL and HDL2 exhibit an elevation of approximately 45%, corresponding proportionately with the rise in triglycerides [176]. While the precise mechanism behind this shift in APOC-III remains elusive, its clinical ramifications are of considerable importance. Apo-CIII has been identified as a trigger for pro-inflammatory signaling, particularly via an alternate NLRP3 inflammasome pathway, as observed in human monocytes. Apo-CIII is predominantly found in very-low-density lipoproteins (VLDL) and is least abundant in HDL. However, the pro-inflammatory effects of Apo-CIII are solely elicited in the presence of VLDL alone. This is because the existence of other anti-inflammatory apolipoproteins, such as ApoA-I present in HDL, may potentially counterbalance its deleterious impact, as suggested by Zewinger et al. [177]. Furthermore, it is noteworthy that delipidated Apo-CIII has the capacity to induce the release of IL-1β from human monocytes without requiring priming with lipopolysaccharide. This induction occurs through the initiation of Toll-like receptor (TLR) 2 and TLR4 dimerization, Ca2+-dependent superoxide production, and the activation of caspase 8, presenting an alternative pathway for NLRP3 inflammasome activation [177].
The SAA, a prominent acute-phase inflammatory protein, is synthesized in abundance during the acute phase of MI and is primarily transported by HDL, accounting for approximately 95% of its transport [178]. The distinct cone-shaped structure of SAA, characterized by hydrophobic and hydrophilic regions on helices 1 and 3, promotes intricate molecular interactions on the lipid surface of HDL [179]. Initial investigations proposed that SAA diminishes the anti-inflammatory and antioxidative attributes of HDL, thereby compromising its functionality. However, it has since been revealed that the enrichment of HDL with SAA serves as a safeguard against lipoprotein oxidation. Furthermore, the mild oxidation of SAA-enriched HDL triggers the release of SAA, which in turn exhibits notable antioxidant properties (see this review for more details [176]). This is consistently supported by findings demonstrating that HDL derived from individuals with elevated SAA levels displays heightened antioxidant activity in comparison to control groups [180]. In summary, while an array of in vitro assays has extensively documented the impairment of HDL function during inflammation, the role of SAA in mediating HDL dysfunction in vivo lacks robust substantiation.
Delipidated human SAA can replace apoA-I within HDL, a phenomenon partially facilitated by CETP [181]. This process not only reduces circulating SAA levels but indirectly limits its bioavailability, thus mitigating its impact on detrimental pathways [182]. Furthermore, recent evidence has revealed that the incorporation of SAA into HDL prevents SAA from inducing the release of IL-1β, activating NLRP3 inflammasomes, and promoting ROS generation, potentially leading to a reduction in SAA’s proinflammatory influence [183]. Consequently, the capture of SAA can be viewed as a protective mechanism undertaken by HDL or a dampening of SAA’s pro-inflammatory activities in the circulation.
The acquisition of SAA by HDL implies the loss of ApoA-I, which can lead to a decline in the functionality of this particle. ApoA-I represents the major apolipoprotein in HDL and is essential for their formation and protective activities [184]. Research conducted in animal models has yielded compelling evidence regarding the positive impact of ApoA-I administration immediately following myocardial IRI. The introduction of ApoA-I during reperfusion sets in motion a cascade of highly beneficial processes within the myocardium. Firstly, it promotes enhanced glucose uptake by the myocardial tissue, effectively bolstering metabolic support [185]. Simultaneously, ApoA-I functions as a safeguard, shielding against post-ischemic mitochondrial damage, thereby preserving vital cellular energy production [186]. Furthermore, ApoA-I facilitates heightened perfusion in the injured myocardium by triggering the release of nitric oxide, a potent vasodilator that amplifies blood flow [187]. Intriguingly, in a mouse model of MI, ApoA-I has demonstrated a predilection for binding to pro-inflammatory monocyte subtypes. This binding, in turn, was correlated with an augmentation in anti-inflammatory subtypes, resulting in a marked enhancement in cardiac function [187]. Collectively, this body of evidence highlights the considerable potential of ApoA-I in mitigating cardiac inflammation and, consequently, its potential to attenuate the development of heart failure post-MI.
It is worth noting that not only a decrease in the availability of ApoA-I but also its displacement from HDL can yield adverse outcomes following MI. Remarkably, the ability of ApoA-I to inhibit inflammasome activation hinges on its association with HDL; lipid-free ApoA-I fails to suppress inflammasome activation. ApoA-I’s tertiary structure is known for its dynamic nature, undergoing substantial alterations contingent upon lipid content and HDL particle size. Different HDL subtypes may possess varying capacities to impede inflammasome activation due to the conformational characteristics of ApoA-I. This is particularly relevant given that HDL tends to reduce in size in patients during the acute phase of MI.
11. HDL microRNA Composition Change during MI
Approximately 85% of the human genome contains non-coding RNAs, with microRNAs (miRNAs) being a prominent subset consisting of roughly 22 nucleotides. MiRNAs play a pivotal role in post-transcriptional gene regulation, influencing critical processes like inflammatory responses, cholesterol homeostasis, oxidative stress, and hypertension—all of which are fundamental to cardiovascular disease [188]. Cells utilize three well-established mechanisms for miRNA secretion: (a) exosomes, (b) miRNA complexes containing Aug2, and (c) miRNA complexes associated with HDL. Notably, miRNA: HDL complexes are readily taken up by various cell types, including macrophages and cardiomyocytes [189].
During MI, changes in miRNA bound to HDL have been documented because of the systemic inflammatory response and as a direct effect of HDL. For instance, in a cohort comprising 93 individuals, HDL’s capacity to suppress pro-inflammatory miRNA expression was significantly compromised during MI [190]. HDL-mediated Stat3 activation plays a pivotal role in downregulating the expression of miR-34B and miR-337 [191]. These two specific miRNAs, miR-34B, and miR-337, exhibit heightened levels in myocardial tissues in animal models of IRI [192], as well as in the blood of patients with MI [193] or atherosclerotic coronary disease [194].
Pre-clinical and clinical investigations have provided robust validation for specific miRNAs that play pivotal roles in regulating the pro-inflammatory NLRP3 inflammasome pathway during myocardial injury, exacerbating pyroptosis and cardiac damage. These miRNAs include miR-1, miR-15, miR-29a, miR-29b, miR-30d, miR-125-5p, miR-149, miR-155, miR-383, and miR-424, as elaborated in references [195][196]. Conversely, there is an increased expression of other miRNAs associated with myocardial protection, acting as negative regulators of the NLRP3 inflammasome pathway during myocardial injury. These protective miRNAs encompass miR-9, miR-30c-5p, miR-100-5p, miR-133a-3p, miR-135b, miR-148a, miR-181b-5p, miR-223-3p, MicroRNA-330-5p, miR-320, miR-351, miR-495-3p, miR-590-3p, and miR-703, as documented in references [195][197][198][199]. Among these miRNAs, miR-21’s role remains somewhat enigmatic. While it exerts a protective effect against myocardial injury and pro-inflammatory modulation in the context of myocardial infarction, miR-21 is also implicated in the promotion of cardiac fibrosis post-MI, contributing to adverse cardiac remodeling [200][201][202]. Importantly, an intriguing hypothesis for future studies revolves around whether HDL particles have the capacity to modulate miRNA expression pathways or serve as carriers for specific miRNAs targeting cells during MI [203]. This research field holds significant promise in further elucidating the intricate mechanisms underlying cardiac injury and protection [203].
References
- Goffart, S.; von Kleist-Retzow, J.C.; Wiesner, R.J. Regulation of mitochondrial proliferation in the heart: Power-plant failure contributes to cardiac failure in hypertrophy. Cardiovasc. Res. 2004, 64, 198–207.
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258.
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317.
- Schelbert, E.B.; Cao, J.J.; Sigurdsson, S.; Aspelund, T.; Kellman, P.; Aletras, A.H.; Dyke, C.K.; Thorgeirsson, G.; Eiriksdottir, G.; Launer, L.J.; et al. Prevalence and prognosis of unrecognized myocardial infarction determined by cardiac magnetic resonance in older adults. JAMA 2012, 308, 890–896.
- El Aidi, H.; Adams, A.; Moons, K.G.M.; Den Ruijter, H.M.; Mali, W.P.T.M.; Doevendans, P.A.; Nagel, E.; Schalla, S.; Bots, M.L.; Leiner, T. Cardiac Magnetic Resonance Imaging Findings and the Risk of Cardiovascular Events in Patients with Recent Myocardial Infarction or Suspected or Known Coronary Artery Disease. J. Am. Coll. Cardiol. 2014, 63, 1031–1045.
- Wu, E.; Ortiz, J.T.; Tejedor, P.; Lee, D.C.; Bucciarelli-Ducci, C.; Kansal, P.; Carr, J.C.; Holly, T.A.; Lloyd-Jones, D.; Klocke, F.J.; et al. Infarct size by contrast enhanced cardiac magnetic resonance is a stronger predictor of outcomes than left ventricular ejection fraction or end-systolic volume index: Prospective cohort study. Heart 2008, 94, 730–736.
- Stone, G.W.; Selker, H.P.; Thiele, H.; Patel, M.R.; Udelson, J.E.; Ohman, E.M.; Maehara, A.; Eitel, I.; Granger, C.B.; Jenkins, P.L.; et al. Relationship Between Infarct Size and Outcomes Following Primary PCI: Patient-Level Analysis From 10 Randomized Trials. J. Am. Coll. Cardiol. 2016, 67, 1674–1683.
- Redfors, B.; Mohebi, R.; Giustino, G.; Chen, S.; Selker, H.P.; Thiele, H.; Patel, M.R.; Udelson, J.E.; Ohman, E.M.; Eitel, I.; et al. Time Delay, Infarct Size, and Microvascular Obstruction After Primary Percutaneous Coronary Intervention for ST-Segment-Elevation Myocardial Infarction. Circ. Cardiovasc. Interv. 2021, 14, e009879.
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135.
- Toldo, S.; Mauro, A.G.; Cutter, Z.; Abbate, A. Inflammasome, pyroptosis, and cytokines in myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1553–H1568.
- Neri, M.; Riezzo, I.; Pascale, N.; Pomara, C.; Turillazzi, E. Ischemia/Reperfusion Injury following Acute Myocardial Infarction: A Critical Issue for Clinicians and Forensic Pathologists. Mediat. Inflamm. 2017, 2017, 7018393.
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 588–606.
- Takahashi, M. Cell-Specific Roles of NLRP3 Inflammasome in Myocardial Infarction. J. Cardiovasc. Pharmacol. 2019, 74, 188–193.
- Shi, H.; Gao, Y.; Dong, Z.; Yang, J.; Gao, R.; Li, X.; Zhang, S.; Ma, L.; Sun, X.; Wang, Z.; et al. GSDMD-Mediated Cardiomyocyte Pyroptosis Promotes Myocardial I/R Injury. Circ. Res. 2021, 129, 383–396.
- Dinarello, C.A. The IL-1 family and inflammatory diseases. Clin. Exp. Rheumatol. 2002, 20 (Suppl. S27), S1–S13.
- Braddock, M.; Quinn, A. Targeting IL-1 in inflammatory disease: New opportunities for therapeutic intervention. Nat. Rev. Drug Discov. 2004, 3, 330–339.
- Mocanu, M.M.; Baxter, G.F.; Yellon, D.M. Caspase inhibition and limitation of myocardial infarct size: Protection against lethal reperfusion injury. Br. J. Pharmacol. 2000, 130, 197–200.
- Do Carmo, H.; Arjun, S.; Petrucci, O.; Yellon, D.M.; Davidson, S.M. The Caspase 1 Inhibitor VX-765 Protects the Isolated Rat Heart via the RISK Pathway. Cardiovasc. Drugs Ther. 2018, 32, 165–168.
- Zheng, Y.; Gardner, S.E.; Clarke, M.C. Cell death, damage-associated molecular patterns, and sterile inflammation in cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2781–2786.
- Zhaolin, Z.; Guohua, L.; Shiyuan, W.; Zuo, W. Role of pyroptosis in cardiovascular disease. Cell Prolif. 2019, 52, e12563.
- Durrington, P.N.; Bashir, B.; Soran, H. Paraoxonase 1 and atherosclerosis. Front. Cardiovasc. Med. 2023, 10, 1065967.
- Rock, K.L.; Latz, E.; Ontiveros, F.; Kono, H. The sterile inflammatory response. Annu. Rev. Immunol. 2010, 28, 321–342.
- Land, W.G. The Role of Damage-Associated Molecular Patterns (DAMPs) in Human Diseases: Part II: DAMPs as diagnostics, prognostics and therapeutics in clinical medicine. Sultan Qaboos Univ. Med. J. 2015, 15, e157–e170.
- Zhang, W.; Lavine, K.J.; Epelman, S.; Evans, S.A.; Weinheimer, C.J.; Barger, P.M.; Mann, D.L. Necrotic myocardial cells release damage-associated molecular patterns that provoke fibroblast activation in vitro and trigger myocardial inflammation and fibrosis in vivo. J. Am. Heart Assoc. 2015, 4, e001993.
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273.
- Toldo, S.; Abbate, A. The NLRP3 inflammasome in acute myocardial infarction. Nat. Rev. Cardiol. 2018, 15, 203–214.
- Bayer, A.L.; Alcaide, P. MyD88: At the heart of inflammatory signaling and cardiovascular disease. J. Mol. Cell. Cardiol. 2021, 161, 75–85.
- Jaen, R.I.; Val-Blasco, A.; Prieto, P.; Gil-Fernandez, M.; Smani, T.; Lopez-Sendon, J.L.; Delgado, C.; Bosca, L.; Fernandez-Velasco, M. Innate Immune Receptors, Key Actors in Cardiovascular Diseases. JACC Basic Transl. Sci. 2020, 5, 735–749.
- Maltez, V.I.; Miao, E.A. Reassessing the Evolutionary Importance of Inflammasomes. J. Immunol. 2016, 196, 956–962.
- Mezzaroma, E.; Toldo, S.; Farkas, D.; Seropian, I.M.; Van Tassell, B.W.; Salloum, F.N.; Kannan, H.R.; Menna, A.C.; Voelkel, N.F.; Abbate, A. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc. Natl. Acad. Sci. USA 2011, 108, 19725–19730.
- Merkle, S.; Frantz, S.; Schon, M.P.; Bauersachs, J.; Buitrago, M.; Frost, R.J.; Schmitteckert, E.M.; Lohse, M.J.; Engelhardt, S. A role for caspase-1 in heart failure. Circ. Res. 2007, 100, 645–653.
- Toldo, S.; Marchetti, C.; Mauro, A.G.; Chojnacki, J.; Mezzaroma, E.; Carbone, S.; Zhang, S.; Van Tassell, B.; Salloum, F.N.; Abbate, A. Inhibition of the NLRP3 inflammasome limits the inflammatory injury following myocardial ischemia-reperfusion in the mouse. Int. J. Cardiol. 2016, 209, 215–220.
- Bugger, H.; Pfeil, K. Mitochondrial ROS in myocardial ischemia reperfusion and remodeling. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165768.
- Liu, Y.; Lian, K.; Zhang, L.; Wang, R.; Yi, F.; Gao, C.; Xin, C.; Zhu, D.; Li, Y.; Yan, W.; et al. TXNIP mediates NLRP3 inflammasome activation in cardiac microvascular endothelial cells as a novel mechanism in myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 2014, 109, 415.
- Liu, Y.; Li, C.; Yin, H.; Zhang, X.; Li, Y. NLRP3 Inflammasome: A Potential Alternative Therapy Target for Atherosclerosis. Evid. Based Complement. Altern. Med. 2020, 2020, 1561342.
- Yoshioka, J.; Chutkow, W.A.; Lee, S.; Kim, J.B.; Yan, J.; Tian, R.; Lindsey, M.L.; Feener, E.P.; Seidman, C.E.; Seidman, J.G.; et al. Deletion of thioredoxin-interacting protein in mice impairs mitochondrial function but protects the myocardium from ischemia-reperfusion injury. J. Clin. Investig. 2012, 122, 267–279.
- Kinnunen, K.; Piippo, N.; Loukovaara, S.; Hytti, M.; Kaarniranta, K.; Kauppinen, A. Lysosomal destabilization activates the NLRP3 inflammasome in human umbilical vein endothelial cells (HUVECs). J. Cell Commun. Signal. 2017, 11, 275–279.
- Toldo, S.; Mezzaroma, E.; Mauro, A.G.; Salloum, F.; Van Tassell, B.W.; Abbate, A. The inflammasome in myocardial injury and cardiac remodeling. Antioxid. Redox Signal. 2015, 22, 1146–1161.
- Zhang, X.; Zeng, W.; Zhang, Y.; Yu, Q.; Zeng, M.; Gan, J.; Zhang, W.; Jiang, X.; Li, H. Focus on the role of mitochondria in NLRP3 inflammasome activation: A prospective target for the treatment of ischemic stroke (Review). Int. J. Mol. Med. 2022, 49, 74.
- Lemasters, J.J.; Theruvath, T.P.; Zhong, Z.; Nieminen, A.L. Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta 2009, 1787, 1395–1401.
- Kahlenberg, J.M.; Dubyak, G.R. Mechanisms of caspase-1 activation by P2X7 receptor-mediated K+ release. Am. J. Physiol. Cell Physiol. 2004, 286, C1100–C1108.
- Zhou, J.; Zhou, Z.; Liu, X.; Yin, H.Y.; Tang, Y.; Cao, X. P2X7 Receptor-Mediated Inflammation in Cardiovascular Disease. Front. Pharmacol. 2021, 12, 654425.
- Katsnelson, M.A.; Rucker, L.G.; Russo, H.M.; Dubyak, G.R. K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J. Immunol. 2015, 194, 3937–3952.
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Nunez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357.
- Li, Y.; Sun, X.; Liu, X.; Li, J.; Li, X.; Wang, G.; Liu, Y.; Lu, X.; Cui, L.; Shao, M.; et al. P2X7R-NEK7-NLRP3 Inflammasome Activation: A Novel Therapeutic Pathway of Qishen Granule in the Treatment of Acute Myocardial Ischemia. J. Inflamm. Res. 2022, 15, 5309–5326.
- Iyer, S.S.; Pulskens, W.P.; Sadler, J.J.; Butter, L.M.; Teske, G.J.; Ulland, T.K.; Eisenbarth, S.C.; Florquin, S.; Flavell, R.A.; Leemans, J.C.; et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc. Natl. Acad. Sci. USA 2009, 106, 20388–20393.
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 2011, 123, 594–604.
- Vijay, K. Toll-like receptors in immunity and inflammatory diseases: Past, present, and future. Int. Immunopharmacol. 2018, 59, 391–412.
- Sameer, A.S.; Nissar, S. Toll-Like Receptors (TLRs): Structure, Functions, Signaling, and Role of Their Polymorphisms in Colorectal Cancer Susceptibility. Biomed Res. Int. 2021, 2021, 1157023.
- Boyd, J.H.; Mathur, S.; Wang, Y.; Bateman, R.M.; Walley, K.R. Toll-like receptor stimulation in cardiomyoctes decreases contractility and initiates an NF-kappaB dependent inflammatory response. Cardiovasc. Res. 2006, 72, 384–393.
- Yu, L.; Feng, Z. The Role of Toll-Like Receptor Signaling in the Progression of Heart Failure. Mediat. Inflamm. 2018, 2018, 9874109.
- Dai, Y.; Song, J.; Li, W.; Yang, T.; Yue, X.; Lin, X.; Yang, X.; Luo, W.; Guo, J.; Wang, X.; et al. RhoE Fine-Tunes Inflammatory Response in Myocardial Infarction. Circulation 2019, 139, 1185–1198.
- Sandanger, O.; Ranheim, T.; Vinge, L.E.; Bliksoen, M.; Alfsnes, K.; Finsen, A.V.; Dahl, C.P.; Askevold, E.T.; Florholmen, G.; Christensen, G.; et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 2013, 99, 164–174.
- Zhang, W.; Xu, X.; Kao, R.; Mele, T.; Kvietys, P.; Martin, C.M.; Rui, T. Cardiac fibroblasts contribute to myocardial dysfunction in mice with sepsis: The role of NLRP3 inflammasome activation. PLoS ONE 2014, 9, e107639.
- Xi, H.; Zhang, Y.; Xu, Y.; Yang, W.Y.; Jiang, X.; Sha, X.; Cheng, X.; Wang, J.; Qin, X.; Yu, J.; et al. Caspase-1 Inflammasome Activation Mediates Homocysteine-Induced Pyrop-Apoptosis in Endothelial Cells. Circ. Res. 2016, 118, 1525–1539.
- Van Gorp, H.; Lamkanfi, M. The emerging roles of inflammasome-dependent cytokines in cancer development. EMBO Rep. 2019, 20, e47575.
- Rauf, A.; Shah, M.; Yellon, D.M.; Davidson, S.M. Role of Caspase 1 in Ischemia/Reperfusion Injury of the Myocardium. J. Cardiovasc. Pharmacol. 2019, 74, 194–200.
- Molla, M.D.; Akalu, Y.; Geto, Z.; Dagnew, B.; Ayelign, B.; Shibabaw, T. Role of Caspase-1 in the Pathogenesis of Inflammatory-Associated Chronic Noncommunicable Diseases. J. Inflamm. Res. 2020, 13, 749–764.
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328.
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128.
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516.
- Kristen, A.V.; Ackermann, K.; Buss, S.; Lehmann, L.; Schnabel, P.A.; Haunstetter, A.; Katus, H.A.; Hardt, S.E. Inhibition of apoptosis by the intrinsic but not the extrinsic apoptotic pathway in myocardial ischemia-reperfusion. Cardiovasc. Pathol. 2013, 22, 280–286.
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114.
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254.
- Xu, Y.J.; Zheng, L.; Hu, Y.W.; Wang, Q. Pyroptosis and its relationship to atherosclerosis. Clin. Chim. Acta 2018, 476, 28–37.
- Lamkanfi, M.; Dixit, V.M. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe 2010, 8, 44–54.
- Land, W.G. The Role of Damage-Associated Molecular Patterns in Human Diseases: Part I—Promoting inflammation and immunity. Sultan Qaboos Univ. Med. J. 2015, 15, e9–e21.
- Liu, X.; Lieberman, J. A Mechanistic Understanding of Pyroptosis: The Fiery Death Triggered by Invasive Infection. Adv. Immunol. 2017, 135, 81–117.
- Yang, X.M.; Downey, J.M.; Cohen, M.V.; Housley, N.A.; Alvarez, D.F.; Audia, J.P. The Highly Selective Caspase-1 Inhibitor VX-765 Provides Additive Protection Against Myocardial Infarction in Rat Hearts When Combined with a Platelet Inhibitor. J. Cardiovasc. Pharmacol. Ther. 2017, 22, 574–578.
- Mezzaroma, E.; Abbate, A.; Toldo, S. NLRP3 Inflammasome Inhibitors in Cardiovascular Diseases. Molecules 2021, 26, 976.
- Vigano, E.; Diamond, C.E.; Spreafico, R.; Balachander, A.; Sobota, R.M.; Mortellaro, A. Human caspase-4 and caspase-5 regulate the one-step non-canonical inflammasome activation in monocytes. Nat. Commun. 2015, 6, 8761.
- Lagrange, B.; Benaoudia, S.; Wallet, P.; Magnotti, F.; Provost, A.; Michal, F.; Martin, A.; Di Lorenzo, F.; Py, B.F.; Molinaro, A.; et al. Human caspase-4 detects tetra-acylated LPS and cytosolic Francisella and functions differently from murine caspase-11. Nat. Commun. 2018, 9, 242.
- Yanpiset, P.; Maneechote, C.; Sriwichaiin, S.; Siri-Angkul, N.; Chattipakorn, S.C.; Chattipakorn, N. Gasdermin D-mediated pyroptosis in myocardial ischemia and reperfusion injury: Cumulative evidence for future cardioprotective strategies. Acta Pharm. Sin. B 2022, 13, 29–53.
- Jun, H.K.; Jung, Y.J.; Ji, S.; An, S.J.; Choi, B.K. Caspase-4 activation by a bacterial surface protein is mediated by cathepsin G in human gingival fibroblasts. Cell Death Differ. 2018, 25, 380–391.
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signaling. Nature 2015, 526, 666–671.
- Yang, D.; He, Y.; Munoz-Planillo, R.; Liu, Q.; Nunez, G. Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity 2015, 43, 923–932.
- Sun, W.; Lu, H.; Dong, S.; Li, R.; Chu, Y.; Wang, N.; Zhao, Y.; Zhang, Y.; Wang, L.; Sun, L.; et al. Beclin1 controls caspase-4 inflammsome activation and pyroptosis in mouse myocardial reperfusion-induced microvascular injury. Cell Commun. Signal. 2021, 19, 107.
- Hitomi, J.; Katayama, T.; Eguchi, Y.; Kudo, T.; Taniguchi, M.; Koyama, Y.; Manabe, T.; Yamagishi, S.; Bando, Y.; Imaizumi, K.; et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J. Cell Biol. 2004, 165, 347–356.
- Kim, S.J.; Zhang, Z.; Hitomi, E.; Lee, Y.C.; Mukherjee, A.B. Endoplasmic reticulum stress-induced caspase-4 activation mediates apoptosis and neurodegeneration in INCL. Hum. Mol. Genet. 2006, 15, 1826–1834.
- Mo, G.; Liu, X.; Zhong, Y.; Mo, J.; Li, Z.; Li, D.; Zhang, L.; Liu, Y. IP3R1 regulates Ca2+ transport and pyroptosis through the NLRP3/Caspase-1 pathway in myocardial ischemia/reperfusion injury. Cell Death Discov. 2021, 7, 31.
- Shen, S.; Wang, Z.; Sun, H.; Ma, L. Role of NLRP3 Inflammasome in Myocardial Ischemia-Reperfusion Injury and Ventricular Remodeling. Med. Sci. Monit. 2022, 28, e934255.
- Tall, A.R. HDL in Morbidity and Mortality: A 40+ Year Perspective. Clin. Chem. 2021, 67, 19–23.
- Sposito, A.C.; Carmo, H.R.; Barreto, J.; Sun, L.; Carvalho, L.S.F.; Feinstein, S.B.; Zanotti, I.; Kontush, A.; Remaley, A. HDL-Targeted Therapies During Myocardial Infarction. Cardiovasc. Drugs Ther. 2019, 33, 371–381.
- Michell, D.L.; Vickers, K.C. Lipoprotein carriers of microRNAs. Biochim. Biophys. Acta 2016, 1861, 2069–2074.
- Richart, A.L.; Calkin, A.C.; Kingwell, B.A. Apolipoprotein A-I for Cardiac Recovery Post-Myocardial Infarction. JACC Basic Transl. Sci. 2021, 6, 768–771.
- Thacker, S.G.; Zarzour, A.; Chen, Y.; Alcicek, M.S.; Freeman, L.A.; Sviridov, D.O.; Demosky, S.J., Jr.; Remaley, A.T. High-density lipoprotein reduces inflammation from cholesterol crystals by inhibiting inflammasome activation. Immunology 2016, 149, 306–319.
- Colliva, A.; Braga, L.; Giacca, M.; Zacchigna, S. Endothelial cell-cardiomyocyte crosstalk in heart development and disease. J. Physiol. 2019, 598, 2923–2939.
- Durham, K.K.; Chathely, K.M.; Trigatti, B.L. High-density lipoprotein protects cardiomyocytes against necrosis induced by oxygen and glucose deprivation through SR-B1, PI3K, and AKT1 and 2. Biochem. J. 2018, 475, 1253–1265.
- Robert, J.; Osto, E.; von Eckardstein, A. The Endothelium Is Both a Target and a Barrier of HDL’s Protective Functions. Cells 2021, 10, 1041.
- Nofer, J.R. Signal transduction by HDL: Agonists, receptors, and signaling cascades. Handb. Exp. Pharmacol. 2015, 224, 229–256.
- Yellon, D.M.; Beikoghli Kalkhoran, S.; Davidson, S.M. The RISK pathway leading to mitochondria and cardioprotection: How everything started. Basic Res. Cardiol. 2023, 118, 22.
- Rossello, X.; Yellon, D.M. The RISK pathway and beyond. Basic Res. Cardiol. 2017, 113, 2.
- White, C.R.; Giordano, S.; Anantharamaiah, G.M. High-density lipoprotein, mitochondrial dysfunction and cell survival mechanisms. Chem. Phys. Lipids 2016, 199, 161–169.
- Heusch, G. Treatment of Myocardial Ischemia/Reperfusion Injury by Ischemic and Pharmacological Postconditioning. Compr. Physiol. 2015, 5, 1123–1145.
- Roberts, D.J.; Tan-Sah, V.P.; Smith, J.M.; Miyamoto, S. Akt phosphorylates HK-II at Thr-473 and increases mitochondrial HK-II association to protect cardiomyocytes. J. Biol. Chem. 2013, 288, 23798–23806.
- Gurel, E.; Ustunova, S.; Kapucu, A.; Yilmazer, N.; Eerbeek, O.; Nederlof, R.; Hollmann, M.W.; Demirci-Tansel, C.; Zuurbier, C.J. Hexokinase cellular trafficking in ischemia-reperfusion and ischemic preconditioning is altered in type I diabetic heart. Mol. Biol. Rep. 2013, 40, 4153–4160.
- McCommis, K.S.; Douglas, D.L.; Krenz, M.; Baines, C.P. Cardiac-specific hexokinase 2 overexpression attenuates hypertrophy by increasing pentose phosphate pathway flux. J. Am. Heart Assoc. 2013, 2, e000355.
- Beutler, E. Disorders due to enzyme defects in the red blood cell. Adv. Metab. Disord. 1972, 60, 131–160.
- Sposito, A.C.; de Lima-Junior, J.C.; Moura, F.A.; Barreto, J.; Bonilha, I.; Santana, M.; Virginio, V.W.; Sun, L.; Carvalho, L.S.F.; Soares, A.A.S.; et al. Reciprocal Multifaceted Interaction Between HDL (High-Density Lipoprotein) and Myocardial Infarction. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1550–1564.
- Mastrocola, R.; Penna, C.; Tullio, F.; Femmino, S.; Nigro, D.; Chiazza, F.; Serpe, L.; Collotta, D.; Alloatti, G.; Cocco, M.; et al. Pharmacological Inhibition of NLRP3 Inflammasome Attenuates Myocardial Ischemia/Reperfusion Injury by Activation of RISK and Mitochondrial Pathways. Oxid. Med. Cell. Longev. 2016, 2016, 5271251.
- Sandanger, O.; Gao, E.; Ranheim, T.; Bliksoen, M.; Kaasboll, O.J.; Alfsnes, K.; Nymo, S.H.; Rashidi, A.; Ohm, I.K.; Attramadal, H.; et al. NLRP3 inflammasome activation during myocardial ischemia reperfusion is cardioprotective. Biochem. Biophys. Res. Commun. 2016, 469, 1012–1020.
- Liang, J.; Zhao, H.; Yao, L.; Tang, H.; Dong, H.; Wu, Y.; Liu, L.; Zou, F.; Cai, S. Phosphatidylinositol 3-kinases pathway mediates lung caspase-1 activation and high mobility group box 1 production in a toluene-diisocyanate induced murine asthma model. Toxicol. Lett. 2015, 236, 25–33.
- Nakahira, K.; Hisata, S.; Choi, A.M. The Roles of Mitochondrial Damage-Associated Molecular Patterns in Diseases. Antioxid. Redox Signal. 2015, 23, 1329–1350.
- Garg, M.; Johri, S.; Chakraborty, K. Immunomodulatory role of mitochondrial DAMPs: A missing link in pathology? FEBS J. 2022, 290, 4395–4418.
- Zhao, W.; Shi, C.S.; Harrison, K.; Hwang, I.Y.; Nabar, N.R.; Wang, M.; Kehrl, J.H. AKT Regulates NLRP3 Inflammasome Activation by Phosphorylating NLRP3 Serine 5. J. Immunol. 2020, 205, 2255–2264.
- Chen, L.; Qin, Y.; Liu, B.; Gao, M.; Li, A.; Li, X.; Gong, G. PGC-1α-Mediated Mitochondrial Quality Control: Molecular Mechanisms and Implications for Heart Failure. Front. Cell Dev. Biol. 2022, 10, 871357.
- Nam, B.Y.; Jhee, J.H.; Park, J.; Kim, S.; Kim, G.; Park, J.T.; Yoo, T.H.; Kang, S.W.; Yu, J.W.; Han, S.H. PGC-1α inhibits the NLRP3 inflammasome via preserving mitochondrial viability to protect kidney fibrosis. Cell Death Dis. 2022, 13, 31.
- Kang, Y.; Zhang, H.; Zhao, Y.; Wang, Y.; Wang, W.; He, Y.; Zhang, W.; Zhang, W.; Zhu, X.; Zhou, Y.; et al. Telomere Dysfunction Disturbs Macrophage Mitochondrial Metabolism and the NLRP3 Inflammasome through the PGC-1α/TNFAIP3 Axis. Cell Rep. 2018, 22, 3493–3506.
- Liu, X.S.; Zeng, J.; Yang, Y.X.; Qi, C.L.; Xiong, T.; Wu, G.Z.; Zeng, C.Y.; Wang, D.X. DRD4 Mitigates Myocardial Ischemia/Reperfusion Injury in Association with PI3K/AKT Mediated Glucose Metabolism. Front. Pharmacol. 2020, 11, 619426.
- Yuan, Y.; Liu, X.; Miao, H.; Huang, B.; Liu, Z.; Chen, J.; Quan, X.; Zhu, L.; Dong, H.; Zhang, Z. PEDF increases GLUT4-mediated glucose uptake in rat ischemic myocardium via PI3K/AKT pathway in a PEDFR-dependent manner. Int. J. Cardiol. 2019, 283, 136–143.
- Frias, M.A.; Lecour, S.; James, R.W.; Pedretti, S. High density lipoprotein/sphingosine-1-phosphate-induced cardioprotection: Role of STAT3 as part of the SAFE pathway. JAKSTAT 2012, 1, 92–100.
- Somers, S.J.; Frias, M.; Lacerda, L.; Opie, L.H.; Lecour, S. Interplay between SAFE and RISK pathways in sphingosine-1-phosphate-induced cardioprotection. Cardiovasc. Drugs Ther. 2012, 26, 227–237.
- Kalakech, H.; Hibert, P.; Prunier-Mirebeau, D.; Tamareille, S.; Letournel, F.; Macchi, L.; Pinet, F.; Furber, A.; Prunier, F. RISK and SAFE signaling pathway involvement in apolipoprotein A-I-induced cardioprotection. PLoS ONE 2014, 9, e107950.
- Feuerborn, R.; Becker, S.; Poti, F.; Nagel, P.; Brodde, M.; Schmidt, H.; Christoffersen, C.; Ceglarek, U.; Burkhardt, R.; Nofer, J.R. High density lipoprotein (HDL)-associated sphingosine 1-phosphate (S1P) inhibits macrophage apoptosis by stimulating STAT3 activity and survivin expression. Atherosclerosis 2017, 257, 29–37.
- Kang, J.-H.; Lee, S.-B.; Seok, J.; Kim, D.-H.; Ma, G.; Park, J.; Jeong, A.J.; Ye, S.-K.; Kang, T.-B. Novel Activity of ODZ10117, a STAT3 Inhibitor, for Regulation of NLRP3 Inflammasome Activation. Int. J. Mol. Sci. 2023, 24, 6079.
- Zhu, L.; Wang, Z.; Sun, X.; Yu, J.; Li, T.; Zhao, H.; Ji, Y.; Peng, B.; Du, M. STAT3/Mitophagy Axis Coordinates Macrophage NLRP3 Inflammasome Activation and Inflammatory Bone Loss. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2023, 38, 335–353.
- Bai, H.; Zhang, Q.F.; Duan, J.J.; Yu, D.J.; Liu, L.J. Downregulation of signal transduction and STAT3 expression exacerbates oxidative stress mediated by NLRP3 inflammasome. Neural Regen. Res. 2018, 13, 2147–2155.
- Alvarez, S.E.; Harikumar, K.B.; Hait, N.C.; Allegood, J.; Strub, G.M.; Kim, E.Y.; Maceyka, M.; Jiang, H.; Luo, C.; Kordula, T.; et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 2010, 465, 1084–1088.
- Takabe, K.; Paugh, S.W.; Milstien, S.; Spiegel, S. “Inside-out” signaling of sphingosine-1-phosphate: Therapeutic targets. Pharmacol. Rev. 2008, 60, 181–195.
- Shen, Y.H.; Abe, J.I. Enigma of Inflammasome Activation by Kinases. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1501–1503.
- Abderrahmani, A.; Niederhauser, G.; Favre, D.; Abdelli, S.; Ferdaoussi, M.; Yang, J.Y.; Regazzi, R.; Widmann, C.; Waeber, G. Human high-density lipoprotein particles prevent activation of the JNK pathway induced by human oxidised low-density lipoprotein particles in pancreatic beta cells. Diabetologia 2007, 50, 1304–1314.
- Muller, C.; Salvayre, R.; Nègre-Salvayre, A.; Vindis, C. HDLs inhibit endoplasmic reticulum stress and autophagic response induced by oxidized LDLs. Cell Death Differ. 2011, 18, 817–828.
- Sandall, C.F.; MacDonald, J.A. Effects of phosphorylation on the NLRP3 inflammasome. Arch. Biochem. Biophys. 2019, 670, 43–57.
- Mulay, V.; Wood, P.; Rentero, C.; Enrich, C.; Grewal, T. Signal transduction pathways provide opportunities to enhance HDL and apoAI-dependent reverse cholesterol transport. Curr. Pharm. Biotechnol. 2012, 13, 352–364.
- Pulli, I.; Asghar, M.Y.; Kemppainen, K.; Tornquist, K. Sphingolipid-mediated calcium signaling and its pathological effects. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1668–1677.
- Ke, M.; Tang, Q.; Pan, Z.; Yin, Y.; Zhang, L.; Wen, K. Sphingosine-1-phosphate attenuates hypoxia/reoxygenation-induced cardiomyocyte injury via a mitochondrial pathway. Biochem. Biophys. Res. Commun. 2019, 510, 142–148.
- Kalayci, A.; Gibson, C.M.; Ridker, P.M.; Wright, S.D.; Kingwell, B.A.; Korjian, S.; Chi, G.; Lee, J.J.; Tricoci, P.; Kazmi, S.H.; et al. ApoA-I Infusion Therapies Following Acute Coronary Syndrome: Past, Present, and Future. Curr. Atheroscler. Rep. 2022, 24, 585–597.
- Orekhov, A.N.; Misharin, A.; Tertov, V.V.; Khashimov Kh, A.; Pokrovsky, S.N.; Repin, V.S.; Smirnov, V.N. Artificial HDL as an anti-atherosclerotic drug. Lancet 1984, 324, 1149–1150.
- Krause, B.R.; Remaley, A.T. Reconstituted HDL for the acute treatment of acute coronary syndrome. Curr. Opin. Lipidol. 2013, 24, 480–486.
- Hubsch, A.P.; Casas, A.T.; Doran, J.E. Protective effects of reconstituted high-density lipoprotein in rabbit gram-negative bacteremia models. J. Lab. Clin. Med. 1995, 126, 548–558.
- Casas, A.T.; Hubsch, A.P.; Rogers, B.C.; Doran, J.E. Reconstituted high-density lipoprotein reduces LPS-stimulated TNF alpha. J. Surg. Res. 1995, 59, 544–552.
- Pajkrt, D.; Doran, J.E.; Koster, F.; Lerch, P.G.; Arnet, B.; van der Poll, T.; ten Cate, J.W.; van Deventer, S.J. Antiinflammatory effects of reconstituted high-density lipoprotein during human endotoxemia. J. Exp. Med. 1996, 184, 1601–1608.
- Didichenko, S.A.; Navdaev, A.V.; Cukier, A.M.; Gille, A.; Schuetz, P.; Spycher, M.O.; Therond, P.; Chapman, M.J.; Kontush, A.; Wright, S.D. Enhanced HDL Functionality in Small HDL Species Produced Upon Remodeling of HDL by Reconstituted HDL, CSL112: Effects on Cholesterol Efflux, Anti-Inflammatory and Antioxidative Activity. Circ. Res. 2016, 119, 751–763.
- Morin, E.E.; Guo, L.; Schwendeman, A.; Li, X.A. HDL in sepsis—Risk factor and therapeutic approach. Front. Pharmacol. 2015, 6, 244.
- Tardif, J.C.; Gregoire, J.; L’Allier, P.L.; Ibrahim, R.; Lesperance, J.; Heinonen, T.M.; Kouz, S.; Berry, C.; Basser, R.; Lavoie, M.A.; et al. Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: A randomized controlled trial. Jama 2007, 297, 1675–1682.
- Christoffersen, C.; Obinata, H.; Kumaraswamy, S.B.; Galvani, S.; Ahnstrom, J.; Sevvana, M.; Egerer-Sieber, C.; Muller, Y.A.; Hla, T.; Nielsen, L.B.; et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc. Natl. Acad. Sci. USA 2011, 108, 9613–9618.
- Newton, J.; Lima, S.; Maceyka, M.; Spiegel, S. Revisiting the sphingolipid rheostat: Evolving concepts in cancer therapy. Exp. Cell Res. 2015, 333, 195–200.
- Bandhuvula, P.; Honbo, N.; Wang, G.Y.; Jin, Z.Q.; Fyrst, H.; Zhang, M.; Borowsky, A.D.; Dillard, L.; Karliner, J.S.; Saba, J.D. S1P lyase: A novel therapeutic target for ischemia-reperfusion injury of the heart. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1753–H1761.
- Cannavo, A.; Liccardo, D.; Komici, K.; Corbi, G.; de Lucia, C.; Femminella, G.D.; Elia, A.; Bencivenga, L.; Ferrara, N.; Koch, W.J.; et al. Sphingosine Kinases and Sphingosine 1-Phosphate Receptors: Signaling and Actions in the Cardiovascular System. Front. Pharmacol. 2017, 8, 556.
- Marsolais, D.; Rosen, H. Chemical modulators of sphingosine-1-phosphate receptors as barrier-oriented therapeutic molecules. Nat. Rev. Drug Discov. 2009, 8, 297–307.
- Nishi, T.; Kobayashi, N.; Hisano, Y.; Kawahara, A.; Yamaguchi, A. Molecular and physiological functions of sphingosine 1-phosphate transporters. Biochim. Et Biophys. Acta 2014, 1841, 759–765.
- Zhang, F.; Xia, Y.; Yan, W.; Zhang, H.; Zhou, F.; Zhao, S.; Wang, W.; Zhu, D.; Xin, C.; Lee, Y.; et al. Sphingosine 1-phosphate signaling contributes to cardiac inflammation, dysfunction, and remodeling following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H250–H261.
- Meshcheryakova, A.; Svoboda, M.; Tahir, A.; Köfeler, H.C.; Triebl, A.; Mungenast, F.; Heinze, G.; Gerner, C.; Zimmermann, P.; Jaritz, M.; et al. Exploring the role of sphingolipid machinery during the epithelial to mesenchymal transition program using an integrative approach. Oncotarget 2016, 7, 22295–22323.
- Feingold, K.R.; Shigenaga, J.K.; Chui, L.G.; Moser, A.; Khovidhunkit, W.; Grunfeld, C. Infection and inflammation decrease apolipoprotein M expression. Atherosclerosis 2008, 199, 19–26.
- Brulhart-Meynet, M.C.; Braunersreuther, V.; Brinck, J.; Montecucco, F.; Prost, J.C.; Thomas, A.; Galan, K.; Pelli, G.; Pedretti, S.; Vuilleumier, N.; et al. Improving Reconstituted HDL Composition for Efficient Post-Ischemic Reduction of Ischemia Reperfusion Injury. PLoS ONE 2015, 10, e0119664.
- Morel, S.; Christoffersen, C.; Axelsen, L.N.; Montecucco, F.; Rochemont, V.; Frias, M.A.; Mach, F.; James, R.W.; Naus, C.C.; Chanson, M.; et al. Sphingosine-1-phosphate reduces ischaemia-reperfusion injury by phosphorylating the gap junction protein Connexin43. Cardiovasc. Res. 2016, 109, 385–396.
- O’Sullivan, C.; Dev, K.K. The structure and function of the S1P1 receptor. Trends Pharmacol. Sci. 2013, 34, 401–412.
- Pyne, N.J.; Pyne, S. Sphingosine 1-Phosphate Receptor 1 Signaling in Mammalian Cells. Molecules 2017, 22, 344.
- Weigert, A.; Cremer, S.; Schmidt, M.V.; von Knethen, A.; Angioni, C.; Geisslinger, G.; Brune, B. Cleavage of sphingosine kinase 2 by caspase-1 provokes its release from apoptotic cells. Blood 2010, 115, 3531–3540.
- White, C.R.; Datta, G.; Giordano, S. High-Density Lipoprotein Regulation of Mitochondrial Function. Adv. Exp. Med. Biol. 2017, 982, 407–429.
- Traxler, D.; Spannbauer, A.; Einzinger, P.; Mester-Tonczar, J.; Lukovic, D.; Winkler, J.; Zlabinger, K.; Gugerell, A.; Mandic, L.; Gyöngyösi, M.; et al. Early Elevation of Systemic Plasma Clusterin after Reperfused Acute Myocardial Infarction in a Preclinical Porcine Model of Ischemic Heart Disease. Int. J. Mol. Sci. 2020, 21, 4591.
- Jun, H.O.; Kim, D.H.; Lee, S.W.; Lee, H.S.; Seo, J.H.; Kim, J.H.; Kim, J.H.; Yu, Y.S.; Min, B.H.; Kim, K.W. Clusterin protects H9c2 cardiomyocytes from oxidative stress-induced apoptosis via Akt/GSK-3β signaling pathway. Exp. Mol. Med. 2011, 43, 53–61.
- Rodriguez-Rivera, C.; Garcia, M.M.; Molina-Alvarez, M.; Gonzalez-Martin, C.; Goicoechea, C. Clusterin: Always protecting. Synthesis, function and potential issues. Biomed. Pharmacother. 2021, 134, 111174.
- Biasizzo, M.; Kopitar-Jerala, N. Interplay Between NLRP3 Inflammasome and Autophagy. Front. Immunol. 2020, 11, 591803.
- Sun, Q.; Fan, J.; Billiar, T.R.; Scott, M.J. Inflammasome and autophagy regulation—A two-way street. Mol. Med. 2017, 23, 188–195.
- Ikeda, S.; Zablocki, D.; Sadoshima, J. The role of autophagy in death of cardiomyocytes. J. Mol. Cell. Cardiol. 2022, 165, 1–8.
- Carmo, H.R.P.; Yoshinaga, M.Y.; Castillo, A.R.; Britto Chaves-Filho, A.; Bonilha, I.; Barreto, J.; Muraro, S.P.; de Souza, G.F.; Davanzo, G.G.; Perroud, M.W., Jr.; et al. Phenotypic changes in low-density lipoprotein particles as markers of adverse clinical outcomes in COVID-19. Mol. Genet. Metab. 2023, 138, 107552.
- Smith, J.D. Dysfunctional HDL as a diagnostic and therapeutic target. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 151–155.
- Schoch, L.; Badimon, L.; Vilahur, G. Unraveling the Complexity of HDL Remodeling: On the Hunt to Restore HDL Quality. Biomedicines 2021, 9, 805.
- Giordano, F.J. Oxygen, oxidative stress, hypoxia, and heart failure. J. Clin. Investig. 2005, 115, 500–508.
- Brites, F.; Martin, M.; Guillas, I.; Kontush, A. Antioxidative activity of high-density lipoprotein (HDL): Mechanistic insights into potential clinical benefit. BBA Clin. 2017, 8, 66–77.
- Bindu, G.H.; Rao, V.S.; Kakkar, V.V. Friend Turns Foe: Transformation of Anti-Inflammatory HDL to Proinflammatory HDL during Acute-Phase Response. Cholesterol 2011, 2011, 274629.
- Annema, W.; Willemsen, H.M.; de Boer, J.F.; Dikkers, A.; van der Giet, M.; Nieuwland, W.; Muller Kobold, A.C.; van Pelt, L.J.; Slart, R.H.; van der Horst, I.C.; et al. HDL function is impaired in acute myocardial infarction independent of plasma HDL cholesterol levels. J. Clin. Lipidol. 2016, 10, 1318–1328.
- Vyletelová, V.; Nováková, M.; Pašková, Ľ. Alterations of HDL’s to piHDL’s Proteome in Patients with Chronic Inflammatory Diseases, and HDL-Targeted Therapies. Pharmaceuticals 2022, 15, 1278.
- Jaimes, E.A.; Sweeney, C.; Raij, L. Effects of the reactive oxygen species hydrogen peroxide and hypochlorite on endothelial nitric oxide production. Hypertension 2001, 38, 877–883.
- Witkowski, A.; Carta, S.; Lu, R.; Yokoyama, S.; Rubartelli, A.; Cavigiolio, G. Oxidation of methionine residues in human apolipoprotein A-I generates a potent pro-inflammatory molecule. J. Biol. Chem. 2019, 294, 3634–3646.
- Sutter, I.; Klingenberg, R.; Othman, A.; Rohrer, L.; Landmesser, U.; Heg, D.; Rodondi, N.; Mach, F.; Windecker, S.; Matter, C.M.; et al. Decreased phosphatidylcholine plasmalogens—A putative novel lipid signature in patients with stable coronary artery disease and acute myocardial infarction. Atherosclerosis 2016, 246, 130–140.
- Moxon, J.V.; Jones, R.E.; Wong, G.; Weir, J.M.; Mellett, N.A.; Kingwell, B.A.; Meikle, P.J.; Golledge, J. Baseline serum phosphatidylcholine plasmalogen concentrations are inversely associated with incident myocardial infarction in patients with mixed peripheral artery disease presentations. Atherosclerosis 2017, 263, 301–308.
- Carvalho, L.S.; Virginio, V.W.; Panzoldo, N.B.; Figueiredo, V.N.; Santos, S.N.; Modolo, R.G.; Andrade, J.M.; Quinaglia, E.S.J.C.; Nadruz-Junior, W.; de Faria, E.C.; et al. Elevated CETP activity during acute phase of myocardial infarction is independently associated with endothelial dysfunction and adverse clinical outcome. Atherosclerosis 2014, 237, 777–783.
- Ndrepepa, G. High-density lipoprotein: A double-edged sword in cardiovascular physiology and pathophysiology. J. Lab. Precis. Med. 2021, 6, 28.
- Meikle, P.J.; Formosa, M.F.; Mellett, N.A.; Jayawardana, K.S.; Giles, C.; Bertovic, D.A.; Jennings, G.L.; Childs, W.; Reddy, M.; Carey, A.L.; et al. HDL Phospholipids, but Not Cholesterol Distinguish Acute Coronary Syndrome from Stable Coronary Artery Disease. J. Am. Heart Assoc. 2019, 8, e011792.
- Bozelli, J.C., Jr.; Azher, S.; Epand, R.M. Plasmalogens and Chronic Inflammatory Diseases. Front. Physiol. 2021, 12, 730829.
- Rasmiena, A.A.; Barlow, C.K.; Stefanovic, N.; Huynh, K.; Tan, R.; Sharma, A.; Tull, D.; de Haan, J.B.; Meikle, P.J. Plasmalogen modulation attenuates atherosclerosis in ApoE- and ApoE/GPx1-deficient mice. Atherosclerosis 2015, 243, 598–608.
- Yang, T.X.; Zhu, Y.F.; Wang, C.C.; Yang, J.Y.; Xue, C.H.; Huang, Q.R.; Wang, Y.M.; Zhang, T.T. EPA-enriched plasmalogen attenuates the cytotoxic effects of LPS-stimulated microglia on the SH-SY5Y neuronal cell line. Brain Res. Bull. 2022, 186, 143–152.
- Correia, L.C.; Magalhães, L.P.; Braga, J.C.; Rocha, M.S.; Lima, J.C.; Passos, L.C.; D’Oliveira, A.; Péricles Esteves, J.; Spósito, A.C. Decrease of plasma triglycerides during the acute phase of unstable angina or non-ST elevation myocardial infarction is a marker of recurrent ischemia. Atherosclerosis 2004, 177, 71–76.
- Cho, K.H.; Shin, D.G.; Baek, S.H.; Kim, J.R. Myocardial infarction patients show altered lipoprotein properties and functions when compared with stable angina pectoris patients. Exp. Mol. Med. 2009, 41, 67–76.
- Zewinger, S.; Reiser, J.; Jankowski, V.; Alansary, D.; Hahm, E.; Triem, S.; Klug, M.; Schunk, S.J.; Schmit, D.; Kramann, R.; et al. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat. Immunol. 2020, 21, 30–41.
- Webb, N.R. High-Density Lipoproteins and Serum Amyloid A (SAA). Curr. Atheroscler. Rep. 2021, 23, 7.
- Lu, J.; Yu, Y.; Zhu, I.; Cheng, Y.; Sun, P.D. Structural mechanism of serum amyloid A-mediated inflammatory amyloidosis. Proc. Natl. Acad. Sci. USA 2014, 111, 5189–5194.
- Tsunoda, F.; Lamon-Fava, S.; Horvath, K.V.; Schaefer, E.J.; Asztalos, B.F. Comparing fluorescence-based cell-free assays for the assessment of antioxidative capacity of high-density lipoproteins. Lipids Health Dis. 2016, 15, 163.
- Wilson, P.G.; Thompson, J.C.; Shridas, P.; McNamara, P.J.; de Beer, M.C.; de Beer, F.C.; Webb, N.R.; Tannock, L.R. Serum Amyloid A Is an Exchangeable Apolipoprotein. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1890–1900.
- Benditt, E.P.; Eriksen, N. Amyloid protein SAA is associated with high density lipoprotein from human serum. Proc. Natl. Acad. Sci. USA 1977, 74, 4025–4028.
- Shridas, P.; De Beer, M.C.; Webb, N.R. High-density lipoprotein inhibits serum amyloid A-mediated reactive oxygen species generation and NLRP3 inflammasome activation. J. Biol. Chem. 2018, 293, 13257–13269.
- Phillips, M.C. New insights into the determination of HDL structure by apolipoproteins: Thematic review series: High density lipoprotein structure, function, and metabolism. J. Lipid Res. 2013, 54, 2034–2048.
- Heywood, S.E.; Richart, A.L.; Henstridge, D.C.; Alt, K.; Kiriazis, H.; Zammit, C.; Carey, A.L.; Kammoun, H.L.; Delbridge, L.M.; Reddy, M.; et al. High-density lipoprotein delivered after myocardial infarction increases cardiac glucose uptake and function in mice. Sci. Transl. Med. 2017, 9, 6084.
- Dadabayev, A.R.; Yin, G.; Latchoumycandane, C.; McIntyre, T.M.; Lesnefsky, E.J.; Penn, M.S. Apolipoprotein A1 regulates coenzyme Q10 absorption, mitochondrial function, and infarct size in a mouse model of myocardial infarction. J. Nutr. 2014, 144, 1030–1036.
- Richart, A.L.; Reddy, M.; Khalaji, M.; Natoli, A.L.; Heywood, S.E.; Siebel, A.L.; Lancaster, G.L.; Murphy, A.J.; Carey, A.L.; Drew, B.G.; et al. Apo AI Nanoparticles Delivered Post Myocardial Infarction Moderate Inflammation. Circ. Res. 2020, 127, 1422–1436.
- Michell, D.L.; Vickers, K.C. HDL and microRNA therapeutics in cardiovascular disease. Pharmacol. Ther. 2016, 168, 43–52.
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell Biol. 2011, 13, 423–433.
- Dullaart, R.P.; Annema, W.; Tio, R.A.; Tietge, U.J. The HDL anti-inflammatory function is impaired in myocardial infarction and may predict new cardiac events independent of HDL cholesterol. Clin. Chim. Acta 2014, 433, 34–38.
- Pedretti, S.; Brulhart-Meynet, M.C.; Montecucco, F.; Lecour, S.; James, R.W.; Frias, M.A. HDL protects against myocardial ischemia reperfusion injury via miR-34b and miR-337 expression which requires STAT3. PLoS ONE 2019, 14, e0218432.
- Liu, J.; Xing, J.; Li, Y.; Liu, J.; Tian, M.; Xu, Z.-S. Protective effects of miR-34b and miR-337 against myocardial ischemia/reperfusion injury in rats by liposomal nano-delivery system. Mater. Express 2021, 11, 1147–1153.
- Bernardo, B.C.; Gao, X.M.; Winbanks, C.E.; Boey, E.J.; Tham, Y.K.; Kiriazis, H.; Gregorevic, P.; Obad, S.; Kauppinen, S.; Du, X.J.; et al. Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc. Natl. Acad. Sci. USA 2012, 109, 17615–17620.
- D’Alessandra, Y.; Carena, M.C.; Spazzafumo, L.; Martinelli, F.; Bassetti, B.; Devanna, P.; Rubino, M.; Marenzi, G.; Colombo, G.I.; Achilli, F.; et al. Diagnostic potential of plasmatic MicroRNA signatures in stable and unstable angina. PLoS ONE 2013, 8, e80345.
- Gao, J.; Chen, X.; Wei, P.; Wang, Y.; Li, P.; Shao, K. Regulation of pyroptosis in cardiovascular pathologies: Role of noncoding RNAs. Mol. Ther. Nucleic Acids 2021, 25, 220–236.
- Tong, R.; Jia, T.; Shi, R.; Yan, F. Inhibition of microRNA-15 protects H9c2 cells against CVB3-induced myocardial injury by targeting NLRX1 to regulate the NLRP3 inflammasome. Cell. Mol. Biol. Lett. 2020, 25, 6.
- Li, X.; Long, J.; Zong, L.; Zhang, C.; Yang, Z.; Guo, S. ZNF561-AS1 Regulates Cell Proliferation and Apoptosis in Myocardial Infarction Through miR-223-3p/NLRP3 Axis. Cell Transplant. 2022, 31, 9636897221077928.
- Zhou, T.; Xiang, D.K.; Li, S.N.; Yang, L.H.; Gao, L.F.; Feng, C. MicroRNA-495 Ameliorates Cardiac Microvascular Endothelial Cell Injury and Inflammatory Reaction by Suppressing the NLRP3 Inflammasome Signaling Pathway. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 49, 798–815.
- Zuo, W.; Tian, R.; Chen, Q.; Wang, L.; Gu, Q.; Zhao, H.; Huang, C.; Liu, Y.; Li, J.; Yang, X.; et al. miR-330-5p inhibits NLRP3 inflammasome-mediated myocardial ischaemia-reperfusion injury by targeting TIM3. Cardiovasc. Drugs Ther. 2021, 35, 691–705.
- Yang, L.; Wang, B.; Zhou, Q.; Wang, Y.; Liu, X.; Liu, Z.; Zhan, Z. MicroRNA-21 prevents excessive inflammation and cardiac dysfunction after myocardial infarction through targeting KBTBD7. Cell Death Dis. 2018, 9, 769.
- Yuan, J.; Chen, H.; Ge, D.; Xu, Y.; Xu, H.; Yang, Y.; Gu, M.; Zhou, Y.; Zhu, J.; Ge, T.; et al. Mir-21 Promotes Cardiac Fibrosis After Myocardial Infarction Via Targeting Smad7. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 42, 2207–2219.
- Sothivelr, V.; Hasan, M.Y.; Mohd Saffian, S.; Zainalabidin, S.; Ugusman, A.; Mahadi, M.K. Revisiting miRNA-21 as a Therapeutic Strategy for Myocardial Infarction: A Systematic Review. J. Cardiovasc. Pharmacol. 2022, 80, 393–406.
- Rayner, K.J.; Moore, K.J. MicroRNA control of high-density lipoprotein metabolism and function. Circ. Res. 2014, 114, 183–192.
More
Information
Subjects:
Cardiac & Cardiovascular Systems
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
609
Revisions:
2 times
(View History)
Update Date:
24 Jan 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No