 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jee-Woong Park | -- | 4491 | 2024-01-23 09:34:54 | | | |
| 2 | Jessie Wu | -1 word(s) | 4490 | 2024-01-24 02:19:15 | | |
Video Upload Options
The field of drug delivery has witnessed remarkable progress, driven by the quest for more effective and precise therapeutic interventions. Among the myriad strategies employed, the integration of aptamers as targeting moieties and stimuli-responsive systems has emerged as a promising avenue, particularly in the context of anticancer therapy. The conventional chemotherapy paradigm often suffers from systemic toxicity, as potent cytotoxic agents are indiscriminately delivered throughout the body, causing adverse effects on healthy tissues. To address this limitation, the integration of smart targeting mechanisms has gained prominence. Within this paradigm, aptamers, short nucleic acid sequences with a unique ability to bind specifically to target molecules, have emerged as valuable targeting ligands. Aptamers share similarities with antibodies as they exhibit a high affinity for specific targets, making them a focus of research in disease-targeted therapy owing to their remarkable selectivity. Regarded as promising therapeutic agents, aptamers possess attributes such as non-immunogenicity, high specificity, and stability.
1. Aptamers
2. Aptamer-Modified Nanomaterials
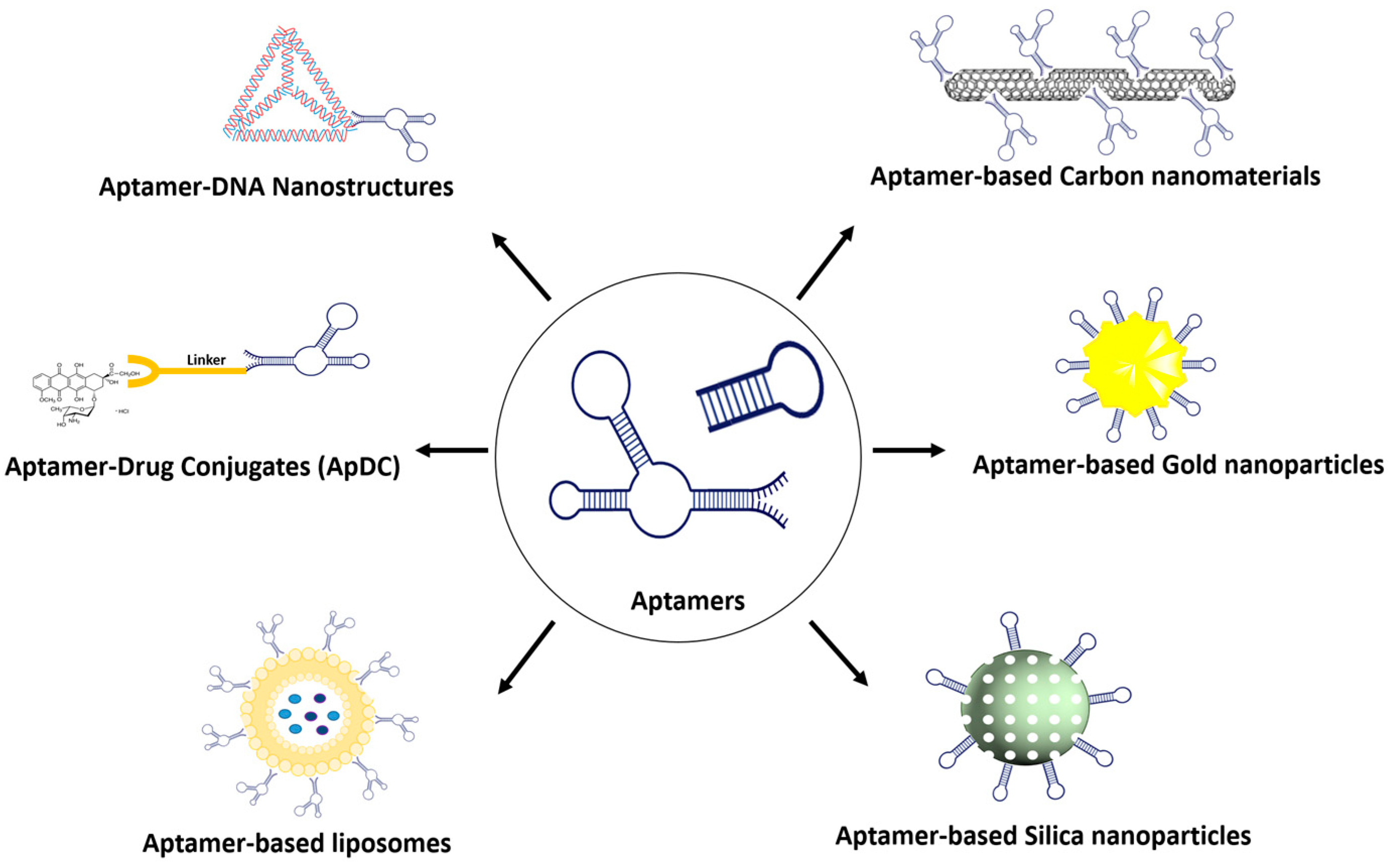
2.1. Aptamer–Gold Conjugation
2.2. Aptamer–Silica Conjugation
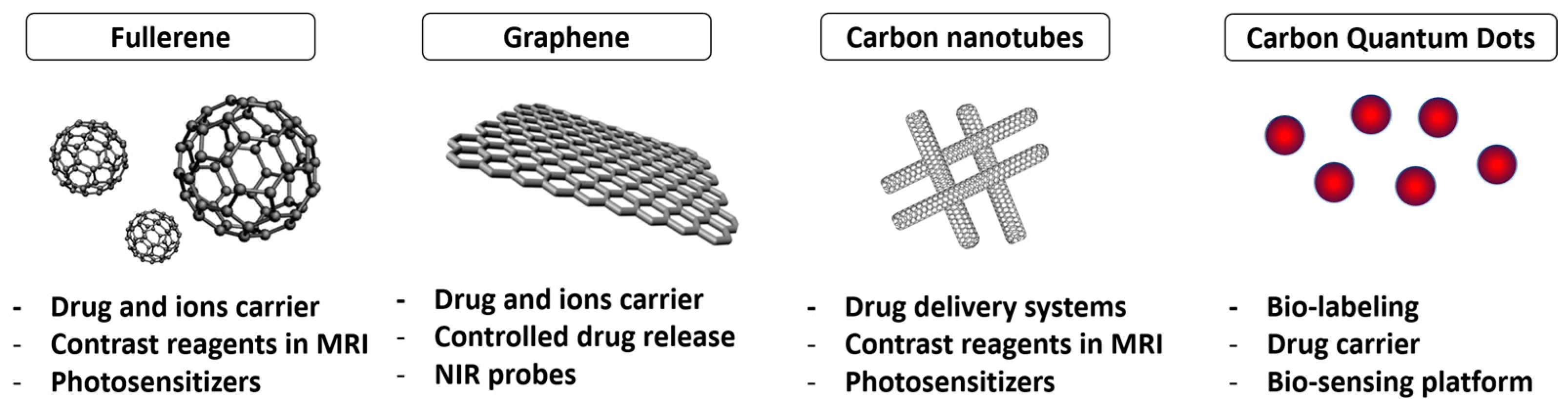
2.3. Aptamer–Carbon Conjugation
3. Aptamer–Drug Conjugate
4. Aptamer Conjugation with Organic Material
4.1. Aptamer–Liposome Conjugation
4.2. Aptamer–Micelle Conjugation
4.3. Aptamer–siRNA Chimeras
References
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822.
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510.
- Alshaer, W.; Hillaireau, H.; Fattal, E. Aptamer-guided nanomedicines for anticancer drug delivery. Adv. Drug Deliv. Rev. 2018, 134, 122–137.
- Choi, J.W.; Seo, M.; Kim, K.; Kim, A.R.; Lee, H.; Kim, H.S.; Park, C.G.; Cho, S.W.; Kang, J.H.; Joo, J.; et al. Aptamer Nanoconstructs Crossing Human Blood-Brain Barrier Discovered via Microphysiological System-Based SELEX Technology. ACS Nano 2023, 17, 8153–8166.
- Nguyen, T.T.Q.; Kim, E.R.; Gu, M.B. A new cognate aptamer pair-based sandwich-type electrochemical biosensor for sensitive detection of Staphylococcus aureus. Biosens. Bioelectron. 2022, 198, 113835.
- Wan, L.Y.; Yuan, W.F.; Ai, W.B.; Ai, Y.W.; Wang, J.J.; Chu, L.Y.; Zhang, Y.Q.; Wu, J.F. An exploration of aptamer internalization mechanisms and their applications in drug delivery. Expert Opin. Drug Deliv. 2019, 16, 207–218.
- Röthlisberger, P.; Hollenstein, M. Aptamer chemistry. Adv. Drug Deliv. Rev. 2018, 134, 3–21.
- Fu, Z.; Xiang, J. Aptamers, the nucleic acid antibodies, in cancer therapy. Int. J. Mol. Sci. 2020, 21, 2793.
- Zeng, Z.; Qi, J.; Wan, Q.; Zu, Y. Aptamers with Self-Loading Drug Payload and pH-Controlled Drug Release for Targeted Chemotherapy. Pharmaceutics 2021, 13, 1221.
- Zhang, Y.; Zhang, H.; Chan, D.W.H.; Ma, Y.; Lu, A.; Yu, S.; Zhang, B.; Zhang, G. Strategies for developing long-lasting therapeutic nucleic acid aptamer targeting circulating protein: The present and the future. Front. Cell Dev. Biol. 2022, 10, 1048148.
- Vazquez-Gonzalez, M.; Willner, I. Aptamer-Functionalized Micro- And Nanocarriers for Controlled Release. ACS Appl. Mater. Interfaces 2021, 13, 9520–9541.
- Li, S.L.; Jiang, P.; Jiang, F.L.; Liu, Y. Recent Advances in Nanomaterial-Based Nanoplatforms for Chemodynamic Cancer Therapy. Adv. Funct. Mater. 2021, 31, 2100243.
- Mahmoudpour, M.; Ding, S.; Lyu, Z.; Ebrahimi, G.; Du, D.; Ezzati Nazhad Dolatabadi, J.; Torbati, M.; Lin, Y. Aptamer functionalized nanomaterials for biomedical applications: Recent advances and new horizons. Nano Today 2021, 39, 101177.
- Shiao, Y.S.; Chiu, H.H.; Wu, P.H.; Huang, Y.F. Aptamer-functionalized gold nanoparticles as photoresponsive nanoplatform for Co-drug delivery. ACS Appl. Mater. Interfaces 2014, 6, 21832–21841.
- Yang, X.; Yang, M.; Pang, B.; Vara, M.; Xia, Y. Gold Nanomaterials at Work in Biomedicine. Chem. Rev. 2015, 115, 10410–10488.
- Lv, Q.-Y.; Cui, H.-F.; Song, X. Aptamer-based technology for gastric cancer theranostics. Anal. Methods 2023, 15, 2142–2153.
- Elghanian, R.; Storhoff, J.J.; Mucic, R.C.; Letsinger, R.L.; Mirkin, C.A. Selective Colorimetric Detection of Polynucleotides Based on the Distance-Dependent Optical Properties of Gold Nanoparticles. Science 1997, 277, 1078–1081.
- Hu, R.; Wen, W.; Wang, Q.; Xiong, H.; Zhang, X.; Gu, H.; Wang, S. Novel electrochemical aptamer biosensor based on an enzyme–gold nanoparticle dual label for the ultrasensitive detection of epithelial tumour marker MUC1. Biosens. Bioelectron. 2014, 53, 384–389.
- Yang, L.; Tseng, Y.-T.; Suo, G.; Chen, L.; Yu, J.; Chiu, W.-J.; Huang, C.-C.; Lin, C.-H. Photothermal Therapeutic Response of Cancer Cells to Aptamer–Gold Nanoparticle-Hybridized Graphene Oxide under NIR Illumination. ACS Appl. Mater. Interfaces 2015, 7, 5097–5106.
- Dou, B.; Xu, L.; Jiang, B.; Yuan, R.; Xiang, Y. Aptamer-Functionalized and Gold Nanoparticle Array-Decorated Magnetic Graphene Nanosheets Enable Multiplexed and Sensitive Electrochemical Detection of Rare Circulating Tumor Cells in Whole Blood. Anal. Chem. 2019, 91, 10792–10799.
- Khorshid, M.; Varshosaz, J.; Rostami, M.; Haghiralsadat, F.; Akbari, V.; Khorshid, P. Anti HER-2 aptamer functionalized gold nanoparticles of dasatinib for targeted chemo-radiotherapy in breast cancer cells. Biomater. Adv. 2023, 154, 213591.
- Navyatha, B.; Nara, S. Aptamer- and antibody-conjugated gold nanobipyramids—A study on cytotoxicity towards breast cancer cell lines. J. Nanoparticle Res. 2023, 25, 165.
- Manzano, M.; Vallet-Regí, M. Mesoporous Silica Nanoparticles for Drug Delivery. Adv. Funct. Mater. 2020, 30, 1902634.
- Fu, Z.; Xiang, J. Aptamer-Functionalized Nanoparticles in Targeted Delivery and Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 9123.
- Vandghanooni, S.; Barar, J.; Eskandani, M.; Omidi, Y. Aptamer-conjugated mesoporous silica nanoparticles for simultaneous imaging and therapy of cancer. TrAC Trends Anal. Chem. 2020, 123, 115759.
- Heydari, S.R.; Ghahremani, M.H.; Atyabi, F.; Bafkary, R.; Jaafari, M.R.; Dinarvand, R. Aptamer-modified chitosan-capped mesoporous silica nanoparticles for co-delivery of cytarabine and daunorubicin in leukemia. Int. J. Pharm. 2023, 646, 123495.
- Kianpour, M.; Huang, C.-W.; Vejvisithsakul, P.P.; Wang, J.-Y.; Li, C.-F.; Shiao, M.-S.; Pan, C.-T.; Shiue, Y.-L. Aptamer/doxorubicin-conjugated nanoparticles target membranous CEMIP2 in colorectal cancer. Int. J. Biol. Macromol. 2023, 245, 125510.
- Xie, X.; Li, F.; Zhang, H.; Lu, Y.; Lian, S.; Lin, H.; Gao, Y.; Jia, L. EpCAM aptamer-functionalized mesoporous silica nanoparticles for efficient colon cancer cell-targeted drug delivery. Eur. J. Pharm. Sci. 2016, 83, 28–35.
- Cui, X.; Xu, S.; Wang, X.; Chen, C. The nano-bio interaction and biomedical applications of carbon nanomaterials. Carbon 2018, 138, 436–450.
- Kościk, I.; Jankowski, D.; Jagusiak, A. Carbon Nanomaterials for Theranostic Use. C 2021, 8, 3.
- Yaghoubi, F.; Naghib, S.M.; Motlagh, N.S.H.; Haghiralsadat, F.; Jaliani, H.Z.; Tofighi, D.; Moradi, A. Multiresponsive carboxylated graphene oxide-grafted aptamer as a multifunctional nanocarrier for targeted delivery of chemotherapeutics and bioactive compounds in cancer therapy. Nanotechnol. Rev. 2021, 10, 1838–1852.
- Viraka Nellore, B.P.; Pramanik, A.; Chavva, S.R.; Sinha, S.S.; Robinson, C.; Fan, Z.; Kanchanapally, R.; Grennell, J.; Weaver, I.; Hamme, A.T.; et al. Aptamer-conjugated theranostic hybrid graphene oxide with highly selective biosensing and combined therapy capability. Faraday Discuss. 2014, 175, 257–271.
- Zhao, C.; Song, X.; Jin, W.; Wu, F.; Zhang, Q.; Zhang, M.; Zhou, N.; Shen, J. Image-guided cancer therapy using aptamer-functionalized cross-linked magnetic-responsive Fe3O4@carbon nanoparticles. Anal. Chim. Acta 2019, 1056, 108–116.
- Zavareh, H.S.; Pourmadadi, M.; Moradi, A.; Yazdian, F.; Omidi, M. Chitosan/carbon quantum dot/aptamer complex as a potential anticancer drug delivery system towards the release of 5-fluorouracil. Int. J. Biol. Macromol. 2020, 165, 1422–1430.
- Biju, V. Chemical modifications and bioconjugate reactions of nanomaterials for sensing, imaging, drug delivery and therapy. Chem. Soc. Rev. 2014, 43, 744–764.
- Liu, Q.; Xu, L.; Zhang, X.; Li, N.; Zheng, J.; Guan, M.; Fang, X.; Wang, C.; Shu, C. Enhanced Photodynamic Efficiency of an Aptamer-Guided Fullerene Photosensitizer toward Tumor Cells. Chem. Asian J. 2013, 8, 2370–2376.
- Jha, R.; Singh, A.; Sharma, P.K.; Fuloria, N.K. Smart carbon nanotubes for drug delivery system: A comprehensive study. J. Drug Deliv. Sci. Technol. 2020, 58, 101811.
- Chen, W.; Yang, S.; Wei, X.; Yang, Z.; Liu, D.; Pu, X.; He, S.; Zhang, Y. Construction of Aptamer-siRNA Chimera/PEI/5-FU/Carbon Nanotube/Collagen Membranes for the Treatment of Peritoneal Dissemination of Drug-Resistant Gastric Cancer. Adv. Healthc. Mater. 2020, 9, 2001153.
- Liu, J.; Cui, L.; Losic, D. Graphene and graphene oxide as new nanocarriers for drug delivery applications. Acta Biomater. 2013, 9, 9243–9257.
- Shahidi, M.; Haghiralsadat, B.F.; Abazari, O.; Hemati, M.; Dayati, P.; Jaliani, H.Z.; Motlagh, N.S.H.; Naghib, S.M.; Moradi, A. HB5 aptamer-tagged graphene oxide for co-delivery of doxorubicin and silibinin, and highly effective combination therapy in breast cancer. Cancer Nanotechnol. 2023, 14, 59.
- Liu, Z.; Robinson, J.T.; Sun, X.; Dai, H. PEGylated Nanographene Oxide for Delivery of Water-Insoluble Cancer Drugs. J. Am. Chem. Soc. 2008, 130, 10876–10877.
- Lu, J.; Zhang, A.; Zhang, F.; Linhardt, R.J.; Zhu, Z.; Yang, Y.; Zhang, T.; Lin, Z.; Zhang, S.; Zhao, H.; et al. Ganoderenic acid D-loaded functionalized graphene oxide-based carrier for active targeting therapy of cervical carcinoma. Biomed. Pharmacother. 2023, 164, 114947.
- Baneshi, M.; Dadfarnia, S.; Haji Shabani, A.M.; Sabbagh, S.K.; Bardania, H. AS1411 aptamer-functionalized graphene oxide-based nano-carrier for active-target and pH-sensitive delivery of curcumin. J. Iran. Chem. Soc. 2022, 19, 2367–2376.
- He, J.; Duan, Q.; Ran, C.; Fu, T.; Liu, Y.; Tan, W. Recent progress of aptamer–drug conjugates in cancer therapy. Acta Pharm. Sin. B 2023, 13, 1358–1370.
- Xuan, W.; Peng, Y.; Deng, Z.; Peng, T.; Kuai, H.; Li, Y.; He, J.; Jin, C.; Liu, Y.; Wang, R.; et al. A basic insight into aptamer-drug conjugates (ApDCs). Biomaterials 2018, 182, 216–226.
- Powell Gray, B.; Kelly, L.; Ahrens, D.P.; Barry, A.P.; Kratschmer, C.; Levy, M.; Sullenger, B.A. Tunable cytotoxic aptamer–drug conjugates for the treatment of prostate cancer. Proc. Natl. Acad. Sci. USA 2018, 115, 4761–4766.
- Sicco, E.; Cerecetto, H.; Calzada, V.; Moreno, M. Targeted-Lymphoma Drug Delivery System Based on the Sgc8-c Aptamer. Cancers 2023, 15, 922.
- Zhang, N.; Wang, J.; Bing, T.; Liu, X.; Shangguan, D. Transferrin receptor-mediated internalization and intracellular fate of conjugates of a DNA aptamer. Mol. Ther. Nucleic Acids 2022, 27, 1249–1259.
- Henri, J.L.; Nakhjavani, M.; McCoombe, S.; Shigdar, S. Cytotoxic effects of aptamer-doxorubicin conjugates in an ovarian cancer cell line. Biochimie 2023, 204, 108–117.
- Ma, W.; Yang, Y.; Liu, Z.; Zhao, R.; Wan, Q.; Chen, X.; Tang, B.; Zhou, Y.; Lin, Y. Self-Assembled Multivalent Aptamer Drug Conjugates: Enhanced Targeting and Cytotoxicity for HER2-Positive Gastric Cancer. ACS Appl. Mater. Interfaces 2023, 15, 43359–43373.
- Jo, J.; Bae, S.; Jeon, J.; Youn, H.; Lee, G.; Ban, C. Bifunctional G-Quadruplex Aptamer Targeting Nucleolin and Topoisomerase 1: Antiproliferative Activity and Synergistic Effect of Conjugated Drugs. Bioconjug. Chem. 2023, 34, 238–247.
- Liu, B.; Wang, J.; Peng, Y.; Zeng, H.; Zhang, Q.; Deng, M.; Xiang, W.; Liu, J.; Hu, X.; Liu, X.; et al. CD71/CD44 dual-aptamer-gemcitabine conjugate for tumor co-targeting treatment of bladder cancer. Chem. Eng. J. 2023, 464, 142597.
- Wu, Y.; Lin, B.; Lu, Y.; Li, L.; Deng, K.; Zhang, S.; Zhang, H.; Yang, C.; Zhu, Z. Aptamer-LYTACs for Targeted Degradation of Extracellular and Membrane Proteins. Angew. Chemie Int. Ed. 2023, 62, e202218106.
- He, S.; Gao, F.; Ma, J.; Ma, H.; Dong, G.; Sheng, C. Aptamer-PROTAC Conjugates (APCs) for Tumor-Specific Targeting in Breast Cancer. Angew. Chemie Int. Ed. 2021, 60, 23299–23305.
- Woo, J.; Chiu, G.N.C.; Karlsson, G.; Wasan, E.; Ickenstein, L.; Edwards, K.; Bally, M.B. Use of a passive equilibration methodology to encapsulate cisplatin into preformed thermosensitive liposomes. Int. J. Pharm. 2008, 349, 38–46.
- Jain Singhai, N.; Maheshwari, R.; Khatri, K. New insights in aptamer-targeted nanoliposomes for the treatment of breast cancer. J. Drug Deliv. Sci. Technol. 2023, 87, 104880.
- Moosavian, S.A.; Kesharwani, P.; Singh, V.; Sahebkar, A. 6-Aptamer-functionalized liposomes for targeted cancer therapy. In Woodhead Publishing Series in Biomaterials; Woodhead Publishing: Cambridge, UK, 2023; pp. 141–172. ISBN 978-0-323-85881-6.
- Iman, M.; Moosavian, S.A.; Zamani, P.; Jaafari, M.R. Preparation of AS1411 aptamer-modified PEGylated liposomal doxorubicin and evaluation of its anti-cancer effects in vitro and in vivo. J. Drug Deliv. Sci. Technol. 2023, 81, 104255.
- Han, S.; Xue, L.; Wei, Y.; Yong, T.; Jia, W.; Qi, Y.; Luo, Y.; Liang, J.; Wen, J.; Bie, N.; et al. Bone Lesion-Derived Extracellular Vesicles Fuel Prometastatic Cascades in Hepatocellular Carcinoma by Transferring ALKBH5-Targeting miR-3190-5p. Adv. Sci. 2023, 10, 2207080.
- Hu, Z.; He, J.; Gong, W.; Zhou, N.; Zhou, S.; Lai, Z.; Zheng, R.; Wang, Y.; Yang, X.; Yang, W.; et al. TLS11a Aptamer/CD3 Antibody Anti-Tumor System for Liver Cancer. J. Biomed. Nanotechnol. 2018, 14, 1645–1653.
- Khodarahmi, M.; Abbasi, H.; Kouchak, M.; Mahdavinia, M.; Handali, S.; Rahbar, N. Nanoencapsulation of aptamer-functionalized 5-Fluorouracil liposomes using alginate/chitosan complex as a novel targeting strategy for colon-specific drug delivery. J. Drug Deliv. Sci. Technol. 2022, 71, 103299.
- Manoochehri, H.; Jalali, A.; Tanzadehpanah, H.; Taherkhani, A.; Najafi, R. Aptamer-conjugated nanoliposomes containing COL1A1 siRNA sensitize CRC cells to conventional chemotherapeutic drugs. Colloids Surf. B Biointerfaces 2022, 218, 112714.
- Alven, S.; Aderibigbe, B.A. The Therapeutic Efficacy of Dendrimer and Micelle Formulations for Breast Cancer Treatment. Pharmaceutics 2020, 12, 1212.
- Biancacci, I.; De Lorenzi, F.; Theek, B.; Bai, X.; May, J.-N.; Consolino, L.; Baues, M.; Moeckel, D.; Gremse, F.; von Stillfried, S.; et al. Monitoring EPR Effect Dynamics during Nanotaxane Treatment with Theranostic Polymeric Micelles. Adv. Sci. 2022, 9, e2103745.
- Cho, H.; Lai, T.C.; Tomoda, K.; Kwon, G.S. Polymeric micelles for multi-drug delivery in cancer. AAPS PharmSciTech 2015, 16, 10–20.
- Vorobiev, V.; Adriouach, S.; Crowe, L.A.; Lenglet, S.; Thomas, A.; Chauvin, A.-S.; Allémann, E. Vascular-targeted micelles as a specific MRI contrast agent for molecular imaging of fibrin clots and cancer cells. Eur. J. Pharm. Biopharm. 2021, 158, 347–358.
- Prencipe, F.; Diaferia, C.; Rossi, F.; Ronga, L.; Tesauro, D. Forward Precision Medicine: Micelles for Active Targeting Driven by Peptides. Molecules 2021, 26, 4049.
- Salahpour-Anarjan, F.; Zare, F.; Hosseini, F.; Ahranjani, S.D.; Alipour, M.; Gozali, E. 7-Aptamer-functionalized micelles for targeted cancer therapy. In Woodhead Publishing Series in Biomaterials; Woodhead Publishing: Cambridge, UK, 2023; pp. 173–189. ISBN 978-0-323-85881-6.
- Tian, L.; Pei, R.; Zhong, L.; Ji, Y.; Zhou, D.; Zhou, S. Enhanced targeting of 3D pancreatic cancer spheroids by aptamer-conjugated polymeric micelles with deep tumor penetration. Eur. J. Pharmacol. 2021, 894, 173814.
- Chauhan, M.; Singh, R.P.; Yadav, B.; Shekhar, S.; Kumar, A.; Mehata, A.K.; Nayak, A.K.; Dutt, R.; Garg, V.; Kailashiya, V.; et al. Development and characterization of micelles for nucleolin-targeted co-delivery of docetaxel and upconversion nanoparticles for theranostic applications in brain cancer therapy. J. Drug Deliv. Sci. Technol. 2023, 87, 104808.
- Cao, Z.; Tong, R.; Mishra, A.; Xu, W.; Wong, G.C.L.; Cheng, J.; Lu, Y. Reversible Cell-Specific Drug Delivery with Aptamer-Functionalized. Angew. Chem. Int. Ed. 2009, 48, 6494–6498.
- McNamara, J.O.; Andrechek, E.R.; Wang, Y.; Viles, K.D.; Rempel, R.E.; Gilboa, E.; Sullenger, B.A.; Giangrande, P.H. Cell type–specific delivery of siRNAs with aptamer-siRNA chimeras. Nat. Biotechnol. 2006, 24, 1005–1015.
- Wei, J.; Song, R.; Sabbagh, A.; Marisetty, A.; Shukla, N.; Fang, D.; Najem, H.; Ott, M.; Long, J.; Zhai, L.; et al. Cell-directed aptamer therapeutic targeting for cancers including those within the central nervous system. Oncoimmunology 2022, 11, 2062827.
- Zhang, L.; Mu, C.; Zhang, T.; Wang, Y.; Wang, Y.; Fan, L.; Liu, C.; Chen, H.; Shen, J.; Wei, K.; et al. Systemic Delivery of Aptamer-Conjugated XBP1 siRNA Nanoparticles for Efficient Suppression of HER2+ Breast Cancer. ACS Appl. Mater. Interfaces 2020, 12, 32360–32371.

