 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Petr Pavloviсh Avdonin | -- | 4633 | 2024-01-19 17:36:21 | | | |
| 2 | Sirius Huang | Meta information modification | 4633 | 2024-01-22 02:24:48 | | |
Video Upload Options
Hemolytic uremic syndrome (HUS) is an acute disease and the most common cause of childhood acute renal failure. HUS is characterized by a triad of symptoms: microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury. In most of the cases, HUS occurs as a result of infection caused by Shiga toxin-producing microbes: hemorrhagic Escherichia coli and Shigella dysenteriae type 1. They account for up to 90% of all cases of HUS. The remaining 10% of cases grouped under the general term atypical HUS represent a heterogeneous group of diseases with similar clinical signs. Emerging evidence suggests that in addition to E. coli and S. dysenteriae type 1, a variety of bacterial and viral infections can cause the development of HUS. In particular, infectious diseases act as the main cause of aHUS recurrence. The pathogenesis of most cases of atypical HUS is based on congenital or acquired defects of complement system.
1. Introduction
- HUS caused by hemorrhagic Shiga toxin-producing E. coli (STEC-HUS);
- Secondary HUS (due to cancer, organ and tissue transplantation, medications, autoimmune disorders, malignant hypertension, and HIV infection);
- HUS associated with infections caused by the H1N1 influenza virus and S. pneumoniae;
- HUS associated with cobalamin C defect;
- HUS associated with mutations in the DGKE gene;
- HUS caused by dysregulation of the alternative complement pathway (mutations in complement genes and antibodies to factor H);
- HUS of unknown etiology.
2. Complement System
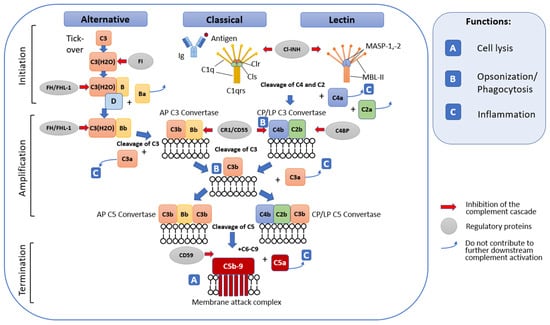
2.1. General Terminal Stage of Complement Activation
2.2. Complement Regulatory Mechanisms
3. Interactions of the Complement System with the Blood Coagulation System
3.1. Blood Coagulation System
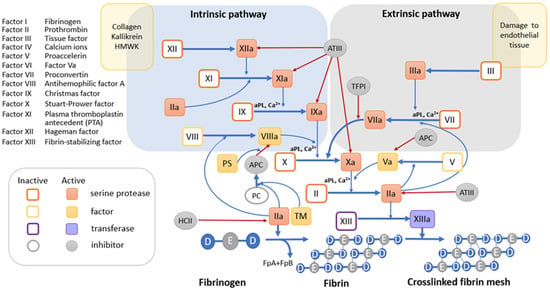
3.1.1. Tissue Factor Pathway
3.1.2. The Internal Contact Activation Pathway
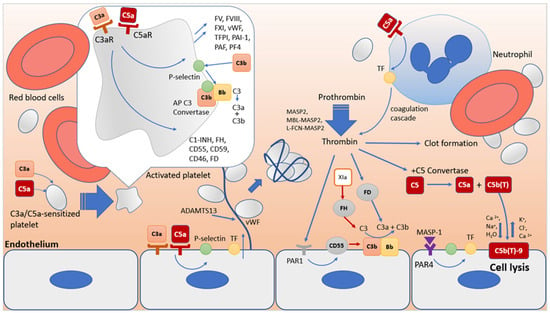
3.2. Synergism in the Functioning of the Complement System and the Blood Coagulation System as a Key Factor in Thrombus Formation in HUS
References
- Aigner, C.; Schmidt, A.; Gaggl, M.; Sunder-Plassmann, G. An updated classification of thrombotic microangiopathies and treatment of complement gene variant-mediated thrombotic microangiopathy. Clin. Kidney J. 2019, 12, 333–337.
- Brocklebank, V.; Wood, K.M.; Kavanagh, D. Thrombotic microangiopathy and the kidney. Clin. J. Am. Soc. Nephrol. CJASN 2018, 13, 300–317.
- Fakhouri, F.; Fila, M.; Hummel, A.; Ribes, D.; Sellier-Leclerc, A.L.; Ville, S.; Pouteil-Noble, C.; Coindre, J.P.; Le Quintrec, M.; Rondeau, E.; et al. Eculizumab discontinuation in children and adults with atypical hemolytic-uremic syndrome: A prospective multicenter study. Blood 2021, 137, 2438–2449.
- Loirat, C.; Fakhouri, F.; Ariceta, G.; Besbas, N.; Bitzan, M.; Bjerre, A.; Coppo, R.; Emma, F.; Johnson, S.; Karpman, D.; et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr. Nephrol. 2016, 31, 15–39.
- Warwicker, P.; Goodship, T.H.; Donne, R.L.; Pirson, Y.; Nicholls, A.; Ward, R.M.; Turnpenny, P.; Goodship, J.A. Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int. 1998, 53, 836–844.
- Zipfel, P.F.; Skerka, C. Complement regulators and inhibitory proteins. Nat. Rev. Immunol. 2009, 9, 729–740.
- Cserhalmi, M.; Papp, A.; Brandus, B.; Uzonyi, B.; Jozsi, M. Regulation of regulators: Role of the complement factor h-related proteins. Semin. Immunol. 2019, 45, 101341.
- Wurzner, R.; Joysey, V.C.; Lachmann, P.J. Complement component c7. Assessment of in vivo synthesis after liver transplantation reveals that hepatocytes do not synthesize the majority of human c7. J. Immunol. 1994, 152, 4624–4629.
- Carroll, M.C. The role of complement and complement receptors in induction and regulation of immunity. Annu. Rev. Immunol. 1998, 16, 545–568.
- Arbore, G.; Kemper, C.; Kolev, M. Intracellular complement—The complosome—In immune cell regulation. Mol. Immunol. 2017, 89, 2–9.
- Bhakdi, S.; Tranum-Jensen, J. C5b-9 assembly: Average binding of one c9 molecule to c5b-8 without poly-c9 formation generates a stable transmembrane pore. J. Immunol. 1986, 136, 2999–3005.
- Podack, E.R.; Tschoop, J.; Muller-Eberhard, H.J. Molecular organization of c9 within the membrane attack complex of complement. Induction of circular c9 polymerization by the c5b-8 assembly. J. Exp. Med. 1982, 156, 268–282.
- Ward, P.A.; Newman, L.J. A neutrophil chemotactic factor from human c′5. J. Immunol. 1969, 102, 93–99.
- Ogden, C.A.; Elkon, K.B. Role of complement and other innate immune mechanisms in the removal of apoptotic cells. Curr. Dir. Autoimmun. 2006, 9, 120–142.
- Anliker-Ort, M.; Dingemanse, J.; van den Anker, J.; Kaufmann, P. Treatment of rare inflammatory kidney diseases: Drugs targeting the terminal complement pathway. Front. Immunol. 2020, 11, 599417.
- Dempsey, P.W.; Allison, M.E.; Akkaraju, S.; Goodnow, C.C.; Fearon, D.T. C3d of complement as a molecular adjuvant: Bridging innate and acquired immunity. Science 1996, 271, 348–350.
- Chen, J.Y.; Cortes, C.; Ferreira, V.P. Properdin: A multifaceted molecule involved in inflammation and diseases. Mol. Immunol. 2018, 102, 58–72.
- Mukherjee, P.; Thomas, S.; Pasinetti, G.M. Complement anaphylatoxin c5a neuroprotects through regulation of glutamate receptor subunit 2 in vitro and in vivo. J. Neuroinflamm. 2008, 5, 5.
- Ling, M.; Murali, M. Analysis of the complement system in the clinical immunology laboratory. Clin. Lab. Med. 2019, 39, 579–590.
- Garred, P.; Genster, N.; Pilely, K.; Bayarri-Olmos, R.; Rosbjerg, A.; Ma, Y.J.; Skjoedt, M.O. A journey through the lectin pathway of complement-mbl and beyond. Immunol. Rev. 2016, 274, 74–97.
- Pouw, R.B.; Ricklin, D. Tipping the balance: Intricate roles of the complement system in disease and therapy. Semin. Immunopathol. 2021, 43, 757–771.
- Bossi, F.; Fischetti, F.; Pellis, V.; Bulla, R.; Ferrero, E.; Mollnes, T.E.; Regoli, D.; Tedesco, F. Platelet-activating factor and kinin-dependent vascular leakage as a novel functional activity of the soluble terminal complement complex. J. Immunol. 2004, 173, 6921–6927.
- Chen, Y.; Yang, C.; Jin, N.; Xie, Z.; Tang, Y.; Fei, L.; Jia, Z.; Wu, Y. Terminal complement complex c5b-9-treated human monocyte-derived dendritic cells undergo maturation and induce th1 polarization. Eur. J. Immunol. 2007, 37, 167–176.
- Jozsi, M.; Barlow, P.N.; Meri, S. Editorial: Function and dysfunction of complement factor h. Front. Immunol. 2021, 12, 831044.
- Johnson, E.; Berge, V.; Hogasen, K. Formation of the terminal complement complex on agarose beads: Further evidence that vitronectin (complement s-protein) inhibits c9 polymerization. Scand. J. Immunol. 1994, 39, 281–285.
- Tschopp, J.; Chonn, A.; Hertig, S.; French, L.E. Clusterin, the human apolipoprotein and complement inhibitor, binds to complement c7, c8 beta, and the b domain of c9. J. Immunol. 1993, 151, 2159–2165.
- Pryzdial, E.L.G.; Leatherdale, A.; Conway, E.M. Coagulation and complement: Key innate defense participants in a seamless web. Front. Immunol. 2022, 13, 918775.
- Lenoir, G.; D’Ambrosio, J.M.; Dieudonne, T.; Copic, A. Transport pathways that contribute to the cellular distribution of phosphatidylserine. Front. Cell Dev. Biol. 2021, 9, 737907.
- Protty, M.B.; Jenkins, P.V.; Collins, P.W.; O’Donnell, V.B. The role of procoagulant phospholipids on the surface of circulating blood cells in thrombosis and haemostasis. Open Biol. 2022, 12, 210318.
- Heijnen, H.; van der Sluijs, P. Platelet secretory behaviour: As diverse as the granules … Or not? J. Thromb. Haemost. 2015, 13, 2141–2151.
- Komiyama, Y.; Pedersen, A.H.; Kisiel, W. Proteolytic activation of human factors ix and x by recombinant human factor viia: Effects of calcium, phospholipids, and tissue factor. Biochemistry 1990, 29, 9418–9425.
- Pryzdial, E.L.G. Maestro tissue factor reaches new height. Blood 2017, 130, 1604–1605.
- Lu, G.; Broze, G.J., Jr.; Krishnaswamy, S. Formation of factors ixa and xa by the extrinsic pathway: Differential regulation by tissue factor pathway inhibitor and antithrombin iii. J. Biol. Chem. 2004, 279, 17241–17249.
- Kamikubo, Y.; Mendolicchio, G.L.; Zampolli, A.; Marchese, P.; Rothmeier, A.S.; Orje, J.N.; Gale, A.J.; Krishnaswamy, S.; Gruber, A.; Ostergaard, H.; et al. Selective factor viii activation by the tissue factor-factor viia-factor xa complex. Blood 2017, 130, 1661–1670.
- Mast, A.E.; Ruf, W. Regulation of coagulation by tissue factor pathway inhibitor: Implications for hemophilia therapy. J. Thromb. Haemost. JTH 2022, 20, 1290–1300.
- Olson, S.T.; Richard, B.; Izaguirre, G.; Schedin-Weiss, S.; Gettins, P.G. Molecular mechanisms of antithrombin-heparin regulation of blood clotting proteinases. A paradigm for understanding proteinase regulation by serpin family protein proteinase inhibitors. Biochimie 2010, 92, 1587–1596.
- Schuijt, T.J.; Bakhtiari, K.; Daffre, S.; Deponte, K.; Wielders, S.J.; Marquart, J.A.; Hovius, J.W.; van der Poll, T.; Fikrig, E.; Bunce, M.W.; et al. Factor xa activation of factor v is of paramount importance in initiating the coagulation system: Lessons from a tick salivary protein. Circulation 2013, 128, 254–266.
- Krishnaswamy, S.; Nesheim, M.E.; Pryzdial, E.L.; Mann, K.G. Assembly of prothrombinase complex. Methods Enzymol. 1993, 222, 260–280.
- Shamanaev, A.; Emsley, J.; Gailani, D. Proteolytic activity of contact factor zymogens. J. Thromb. Haemost. JTH 2021, 19, 330–341.
- Mailer, R.K.; Rangaswamy, C.; Konrath, S.; Emsley, J.; Renne, T. An update on factor xii-driven vascular inflammation. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119166.
- Kannemeier, C.; Shibamiya, A.; Nakazawa, F.; Trusheim, H.; Ruppert, C.; Markart, P.; Song, Y.; Tzima, E.; Kennerknecht, E.; Niepmann, M.; et al. Extracellular rna constitutes a natural procoagulant cofactor in blood coagulation. Proc. Natl. Acad. Sci. USA 2007, 104, 6388–6393.
- von Bruhl, M.L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Kollnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835.
- Muller, F.; Mutch, N.J.; Schenk, W.A.; Smith, S.A.; Esterl, L.; Spronk, H.M.; Schmidbauer, S.; Gahl, W.A.; Morrissey, J.H.; Renne, T. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell 2009, 139, 1143–1156.
- Renne, T. The procoagulant and proinflammatory plasma contact system. Semin. Immunopathol. 2012, 34, 31–41.
- Pryzdial, E.L.G.; Lee, F.M.H.; Lin, B.H.; Carter, R.L.R.; Tegegn, T.Z.; Belletrutti, M.J. Blood coagulation dissected. Transfus. Apher. Sci. 2018, 57, 449–457.
- Maas, C.; Renne, T. Coagulation factor xii in thrombosis and inflammation. Blood 2018, 131, 1903–1909.
- Brown, N.J.; Gainer, J.V.; Stein, C.M.; Vaughan, D.E. Bradykinin stimulates tissue plasminogen activator release in human vasculature. Hypertension 1999, 33, 1431–1435.
- Yin, W.; Ghebrehiwet, B.; Weksler, B.; Peerschke, E.I. Classical pathway complement activation on human endothelial cells. Mol. Immunol. 2007, 44, 2228–2234.
- Nayak, A.; Ferluga, J.; Tsolaki, A.G.; Kishore, U. The non-classical functions of the classical complement pathway recognition subcomponent c1q. Immunol. Lett. 2010, 131, 139–150.
- Hamad, O.A.; Nilsson, P.H.; Wouters, D.; Lambris, J.D.; Ekdahl, K.N.; Nilsson, B. Complement component c3 binds to activated normal platelets without preceding proteolytic activation and promotes binding to complement receptor 1. J. Immunol. 2010, 184, 2686–2692.
- Saggu, G.; Cortes, C.; Emch, H.N.; Ramirez, G.; Worth, R.G.; Ferreira, V.P. Identification of a novel mode of complement activation on stimulated platelets mediated by properdin and c3(h2o). J. Immunol. 2013, 190, 6457–6467.
- Monsinjon, T.; Gasque, P.; Chan, P.; Ischenko, A.; Brady, J.J.; Fontaine, M.C. Regulation by complement c3a and c5a anaphylatoxins of cytokine production in human umbilical vein endothelial cells. FASEB J. 2003, 17, 1003–1014.
- Propson, N.E.; Roy, E.R.; Litvinchuk, A.; Kohl, J.; Zheng, H. Endothelial c3a receptor mediates vascular inflammation and blood-brain barrier permeability during aging. J. Clin. Investig. 2021, 131, e140966.
- Shivshankar, P.; Li, Y.D.; Mueller-Ortiz, S.L.; Wetsel, R.A. In response to complement anaphylatoxin peptides c3a and c5a, human vascular endothelial cells migrate and mediate the activation of b-cells and polarization of t-cells. FASEB J. 2020, 34, 7540–7560.
- Foreman, K.E.; Vaporciyan, A.A.; Bonish, B.K.; Jones, M.L.; Johnson, K.J.; Glovsky, M.M.; Eddy, S.M.; Ward, P.A. C5a-induced expression of p-selectin in endothelial cells. J. Clin. Investig. 1994, 94, 1147–1155.
- Fang, W.; Guo, Z.H.; Zhang, B.Q.; Wu, X.F.; Li, P.; Lv, F.L.; Su, L. . Zhongguo Wei Zhong Bing Ji Jiu Yi Xue Chin. Crit. Care Med. Zhongguo Weizhongbing Jijiuyixue 2009, 21, 168–171.
- Bongoni, A.K.; Lu, B.; McRae, J.L.; Salvaris, E.J.; Toonen, E.J.M.; Vikstrom, I.; Baz Morelli, A.; Pearse, M.J.; Cowan, P.J. Complement-mediated damage to the glycocalyx plays a role in renal ischemia-reperfusion injury in mice. Transplant. Direct 2019, 5, e341.
- Wojta, J.; Huber, K.; Valent, P. New aspects in thrombotic research: Complement induced switch in mast cells from a profibrinolytic to a prothrombotic phenotype. Pathophysiol. Haemost. Thromb. 2003, 33, 438–441.
- Gulla, K.C.; Gupta, K.; Krarup, A.; Gal, P.; Schwaeble, W.J.; Sim, R.B.; O’Connor, C.D.; Hajela, K. Activation of mannan-binding lectin-associated serine proteases leads to generation of a fibrin clot. Immunology 2010, 129, 482–495.
- Krarup, A.; Gulla, K.C.; Gal, P.; Hajela, K.; Sim, R.B. The action of mbl-associated serine protease 1 (masp1) on factor xiii and fibrinogen. Biochim. et Biophys. Acta 2008, 1784, 1294–1300.
- Hamilton, K.K.; Hattori, R.; Esmon, C.T.; Sims, P.J. Complement proteins c5b-9 induce vesiculation of the endothelial plasma membrane and expose catalytic surface for assembly of the prothrombinase enzyme complex. J. Biol. Chem. 1990, 265, 3809–3814.
- Wiedmer, T.; Esmon, C.T.; Sims, P.J. Complement proteins c5b-9 stimulate procoagulant activity through platelet prothrombinase. Blood 1986, 68, 875–880.
- Amara, U.; Flierl, M.A.; Rittirsch, D.; Klos, A.; Chen, H.; Acker, B.; Bruckner, U.B.; Nilsson, B.; Gebhard, F.; Lambris, J.D.; et al. Molecular intercommunication between the complement and coagulation systems. J. Immunol. 2010, 185, 5628–5636.
- Huber-Lang, M.; Sarma, J.V.; Zetoune, F.S.; Rittirsch, D.; Neff, T.A.; McGuire, S.R.; Lambris, J.D.; Warner, R.L.; Flierl, M.A.; Hoesel, L.M.; et al. Generation of c5a in the absence of c3: A new complement activation pathway. Nat. Med. 2006, 12, 682–687.
- Krisinger, M.J.; Goebeler, V.; Lu, Z.; Meixner, S.C.; Myles, T.; Pryzdial, E.L.; Conway, E.M. Thrombin generates previously unidentified c5 products that support the terminal complement activation pathway. Blood 2012, 120, 1717–1725.
- Polley, M.J.; Nachman, R. The human complement system in thrombin-mediated platelet function. J. Exp. Med. 1978, 147, 1713–1726.
- Polley, M.J.; Nachman, R.L. Human complement in thrombin-mediated platelet function: Uptake of the c5b-9 complex. J. Exp. Med. 1979, 150, 633–645.
- Dobo, J.; Szakacs, D.; Oroszlan, G.; Kortvely, E.; Kiss, B.; Boros, E.; Szasz, R.; Zavodszky, P.; Gal, P.; Pal, G. Masp-3 is the exclusive pro-factor d activator in resting blood: The lectin and the alternative complement pathways are fundamentally linked. Sci. Rep. 2016, 6, 31877.
- Oroszlan, G.; Kortvely, E.; Szakacs, D.; Kocsis, A.; Dammeier, S.; Zeck, A.; Ueffing, M.; Zavodszky, P.; Pal, G.; Gal, P.; et al. Masp-1 and masp-2 do not activate pro-factor d in resting human blood, whereas masp-3 is a potential activator: Kinetic analysis involving specific masp-1 and masp-2 inhibitors. J. Immunol. 2016, 196, 857–865.
- Lidington, E.A.; Haskard, D.O.; Mason, J.C. Induction of decay-accelerating factor by thrombin through a protease-activated receptor 1 and protein kinase c-dependent pathway protects vascular endothelial cells from complement-mediated injury. Blood 2000, 96, 2784–2792.
- Foley, J.H.; Walton, B.L.; Aleman, M.M.; O’Byrne, A.M.; Lei, V.; Harrasser, M.; Foley, K.A.; Wolberg, A.S.; Conway, E.M. Complement activation in arterial and venous thrombosis is mediated by plasmin. eBioMedicine 2016, 5, 175–182.
- Ward, P.A. A plasmin-split fragment of c′3 as a new chemotactic factor. J. Exp. Med. 1967, 126, 189–206.
- Mannes, M.; Dopler, A.; Zolk, O.; Lang, S.J.; Halbgebauer, R.; Hochsmann, B.; Skerra, A.; Braun, C.K.; Huber-Lang, M.; Schrezenmeier, H.; et al. Complement inhibition at the level of c3 or c5: Mechanistic reasons for ongoing terminal pathway activity. Blood 2021, 137, 443–455.
- Wetsel, R.A.; Kolb, W.P. Expression of c5a-like biological activities by the fifth component of human complement (c5) upon limited digestion with noncomplement enzymes without release of polypeptide fragments. J. Exp. Med. 1983, 157, 2029–2048.
- DiScipio, R.G. The activation of the alternative pathway c3 convertase by human plasma kallikrein. Immunology 1982, 45, 587–595.
- Saito, A. Plasma kallikrein is activated on dermatan sulfate and cleaves factor h. Biochem. Biophys. Res. Commun. 2008, 370, 646–650.
- Ellis, V.; Scully, M.; MacGregor, I.; Kakkar, V. Inhibition of human factor xa by various plasma protease inhibitors. Biochim. Biophys. Acta 1982, 701, 24–31.
- Osterud, B.; Miller-Andersson, M.; Abildgaard, U.; Prydz, H. The effect of antithrombin iii on the activity of the coagulation factors vii, ix and x. Thromb. Haemost. 1976, 35, 295–304.
- Parej, K.; Dobo, J.; Zavodszky, P.; Gal, P. The control of the complement lectin pathway activation revisited: Both c1-inhibitor and antithrombin are likely physiological inhibitors, while alpha2-macroglobulin is not. Mol. Immunol. 2013, 54, 415–422.
- Ziccardi, R.J. Activation of the early components of the classical complement pathway under physiologic conditions. J. Immunol. 1981, 126, 1769–1773.
- Rossi, V.; Cseh, S.; Bally, I.; Thielens, N.M.; Jensenius, J.C.; Arlaud, G.J. Substrate specificities of recombinant mannan-binding lectin-associated serine proteases-1 and -2. J. Biol. Chem. 2001, 276, 40880–40887.
- Kerr, F.K.; Thomas, A.R.; Wijeyewickrema, L.C.; Whisstock, J.C.; Boyd, S.E.; Kaiserman, D.; Matthews, A.Y.; Bird, P.I.; Thielens, N.M.; Rossi, V.; et al. Elucidation of the substrate specificity of the masp-2 protease of the lectin complement pathway and identification of the enzyme as a major physiological target of the serpin, c1-inhibitor. Mol. Immunol. 2008, 45, 670–677.
- Ratnoff, O.D. Some relationships among hemostasis, fibrinolytic phenomena, immunity, and the inflammatory response. Adv. Immunol. 1969, 10, 145–227.
- Maroney, S.A.; Ellery, P.E.; Mast, A.E. Alternatively spliced isoforms of tissue factor pathway inhibitor. Thromb. Res. 2010, 125 (Suppl. 1), S52–S56.
- Mast, A.E. Tissue factor pathway inhibitor: Multiple anticoagulant activities for a single protein. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 9–14.
- Keizer, M.P.; Pouw, R.B.; Kamp, A.M.; Patiwael, S.; Marsman, G.; Hart, M.H.; Zeerleder, S.; Kuijpers, T.W.; Wouters, D. Tfpi inhibits lectin pathway of complement activation by direct interaction with masp-2. Eur. J. Immunol. 2015, 45, 544–550.
- Puy, C.; Pang, J.; Reitsma, S.E.; Lorentz, C.U.; Tucker, E.I.; Gailani, D.; Gruber, A.; Lupu, F.; McCarty, O.J.T. Cross-talk between the complement pathway and the contact activation system of coagulation: Activated factor xi neutralizes complement factor h. J. Immunol. 2021, 206, 1784–1792.
- Chen, L.J.; Liu, D.T.; Tam, P.O.; Chan, W.M.; Liu, K.; Chong, K.K.; Lam, D.S.; Pang, C.P. Association of complement factor h polymorphisms with exudative age-related macular degeneration. Mol. Vis. 2006, 12, 1536–1542.
- Thangaraj, S.S.; Christiansen, S.H.; Graversen, J.H.; Sidelmann, J.J.; Hansen, S.W.K.; Bygum, A.; Gram, J.B.; Palarasah, Y. Contact activation-induced complex formation between complement factor h and coagulation factor xiia. J. Thromb. Haemost. JTH 2020, 18, 876–884.
- Feng, S.; Liang, X.; Cruz, M.A.; Vu, H.; Zhou, Z.; Pemmaraju, N.; Dong, J.F.; Kroll, M.H.; Afshar-Kharghan, V. The interaction between factor h and von willebrand factor. PLoS ONE 2013, 8, e73715.
- Nolasco, L.; Nolasco, J.; Feng, S.; Afshar-Kharghan, V.; Moake, J. Human complement factor h is a reductase for large soluble von willebrand factor multimers—Brief report. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2524–2528.
- Rayes, J.; Roumenina, L.T.; Dimitrov, J.D.; Repesse, Y.; Ing, M.; Christophe, O.; Jokiranta, T.S.; Halbwachs-Mecarelli, L.; Borel-Derlon, A.; Kaveri, S.V.; et al. The interaction between factor h and vwf increases factor h cofactor activity and regulates vwf prothrombotic status. Blood 2014, 123, 121–125.
- Turner, N.; Nolasco, L.; Nolasco, J.; Sartain, S.; Moake, J. Thrombotic microangiopathies and the linkage between von willebrand factor and the alternative complement pathway. Semin. Thromb. Hemost. 2014, 40, 544–550.
- Feng, S.; Liang, X.; Kroll, M.H.; Chung, D.W.; Afshar-Kharghan, V. Von willebrand factor is a cofactor in complement regulation. Blood 2015, 125, 1034–1037.
- Bajzar, L.; Manuel, R.; Nesheim, M.E. Purification and characterization of tafi, a thrombin-activable fibrinolysis inhibitor. J. Biol. Chem. 1995, 270, 14477–14484.
- Campbell, W.D.; Lazoura, E.; Okada, N.; Okada, H. Inactivation of c3a and c5a octapeptides by carboxypeptidase r and carboxypeptidase n. Microbiol. Immunol. 2002, 46, 131–134.
- Delvaeye, M.; Noris, M.; De Vriese, A.; Esmon, C.T.; Esmon, N.L.; Ferrell, G.; Del-Favero, J.; Plaisance, S.; Claes, B.; Lambrechts, D.; et al. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2009, 361, 345–357.
- Heurich, M.; Preston, R.J.; O’Donnell, V.B.; Morgan, B.P.; Collins, P.W. Thrombomodulin enhances complement regulation through strong affinity interactions with factor h and c3b-factor h complex. Thromb. Res. 2016, 145, 84–92.
- Tateishi, K.; Imaoka, M.; Matsushita, M. Dual modulating functions of thrombomodulin in the alternative complement pathway. Biosci. Trends 2016, 10, 231–234.
- Bu, F.; Maga, T.; Meyer, N.C.; Wang, K.; Thomas, C.P.; Nester, C.M.; Smith, R.J. Comprehensive genetic analysis of complement and coagulation genes in atypical hemolytic uremic syndrome. J. Am. Soc. Nephrol. JASN 2014, 25, 55–64.

