Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | João Martins Gama | -- | 1693 | 2024-01-18 10:46:49 | | | |
| 2 | Rita Xu | Meta information modification | 1693 | 2024-01-19 02:37:07 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Gama, J.M.; Almeida, R.; Oliveira, R.C.; Casanova, J. Inferior Vena Cava Leiomyosarcoma. Encyclopedia. Available online: https://encyclopedia.pub/entry/54030 (accessed on 25 July 2026).
Gama JM, Almeida R, Oliveira RC, Casanova J. Inferior Vena Cava Leiomyosarcoma. Encyclopedia. Available at: https://encyclopedia.pub/entry/54030. Accessed July 25, 2026.
Gama, João Martins, Rui Almeida, Rui Caetano Oliveira, José Casanova. "Inferior Vena Cava Leiomyosarcoma" Encyclopedia, https://encyclopedia.pub/entry/54030 (accessed July 25, 2026).
Gama, J.M., Almeida, R., Oliveira, R.C., & Casanova, J. (2024, January 18). Inferior Vena Cava Leiomyosarcoma. In Encyclopedia. https://encyclopedia.pub/entry/54030
Gama, João Martins, et al. "Inferior Vena Cava Leiomyosarcoma." Encyclopedia. Web. 18 January, 2024.
Copy Citation
Leiomyosarcomas (LMSs) are malignant neoplasms of soft muscle differentiation that can be classified into five distinct groups according to site-related origin: intra-abdominal, subcutaneous or deep soft tissue of the limbs, cutaneous, external genitalia, and vascular.
leiomyosarcoma
inferior vena cava leiomyosarcoma
vascular
1. Introduction
Leiomyosarcoma (LMS) is a malignant neoplasm of soft muscle differentiation [1]. Although uncommon, it is one of the most common sarcomas. LMSs of the soft tissue can be grouped into five distinctive categories according to site-related origin: intra-abdominal, subcutaneous or deep soft tissue of the limbs, cutaneous, external genitalia, and vascular. The distinction is important since there are clinical differences between these subgroups [2]. Intra-abdominal and deep soft tissue are the most common subgroups, and the least common is the vascular LMS group. Although rare, LMS is the most common malignant tumor involving the vascular system [3]. Vascular LMS originates in the walls of medium-sized or large blood vessels, usually from the inferior vena cava. This distinction is fundamental since it reflects distinct biological origins, molecular changes, and prognoses.
Inferior vena cava LMS was first described by Leopold Perl and Rudolph Virchow in 1871 [4]. Since then, many discoveries have been made and a clearer image of this interesting entity has emerged.
2. Epidemiology
It is estimated that LMS of the soft tissue represents up to 15% of all soft tissue sarcomas [5]. LMS of vascular origin is rare, and most of the knowledge gathered so far comes from isolated case reports or small series.
It is estimated that vascular LMS represents 5% of soft tissue LMS, and half arise from the inferior vena cava or veins of the lower limbs. Leiomyosarcomas are five times more common in veins than arteries [6]. Autopsy-based studies estimated an incidence of 1/7000-34,000 autopsies [7]. Currently, less than 400 cases are described in the literature.
Older adults are usually the most affected, with a median age at diagnosis of 56 years (range: 34–75) [8], but interestingly, more than three-quarters of all vena cava leiomyosarcomas occur in women [8][9]. The hormonal influence on growth and proliferation of smooth muscle tissue might explain this. LMS is the most common malignancy in the vascular system in adults, and albeit rare, some cases have been reported in the pediatric age [10].
The distribution of vascular leiomyosarcomas is grossly inversely proportional to the pressure in the vascular bed [7][11]. Vascular leiomyosarcomas are more common in large veins, where the pressure is lower [12]; the most common location is in the inferior vena cava, followed by other large veins. Less commonly, vascular LMSs can arise in the pulmonary artery and in large systemic arteries [6][11]. In 1973, Kevorkian and Cento [11] conducted an extensive review of all of the cases of vascular leiomyosarcomas published in the literature, and out of a total of 86 patients, 33 were found in the inferior vena cava, 35 in other large veins, 10 in the pulmonary artery, and 8 cases in large systemic arteries. LMS arising in an arteriovenous fistula has also been reported [13].
3. Clinical Presentation
The clinical symptoms are diverse, non-specific, dependent on the location of the tumor, the growth rate, and the development of collateral blood flow [6]. When leiomyosarcomas develop in the superior vena cava segment, symptoms manifest as Budd–Chiari syndrome with hepatomegaly, jaundice, ascites, and nausea. In the middle segment, the symptoms are associated with abdominal discomfort and sometimes related to vascular compromise (e.g., edema of the lower limbs) [14]. In the inferior segment, the symptoms appear relatively late (e.g., edema, nausea, and back pain) [15][16]. Rarer presenting symptoms are recurrent pulmonary embolisms and metastases. LMSs may remain asymptomatic and are often incidentally diagnosed [11].
4. Etiology and Pathogenesis
Not much is known about the predisposing factors and etiology of vascular LMS, and the experience gathered from leiomyosarcomas from other sites is not always directly applicable to the soft tissue and vascular counterpart [7].
Leiomyosarcomas have a complex karyotype with complex numeric and structural anomalies, and multiple genes have been implicated in its pathogenesis [17], with significant mutational heterogeneity and frequent copy number variations with no characteristic chromosomal rearrangements [18]. In a study by Chudasama et al., chromothripsis was reported in 35% of LMSs. Losses of chromosome regions encoding for tumor suppressor genes such as TP53, PTEN, CDH1, and MYOCD have been reported [18][19], as well as genes involved in DNA homologous recombination repair (BRCA2 and ATM are a frequent target of deletions). Other recurrently mutated genes are chromatin modifiers (RBL2, DNMT3A, and KAT6B), cytokine receptors (ALK, FGFR2, FLT3, and LIFR), and transcriptional regulators (PAX3, FOXO1, CDX2, and SUFU) [18].
Alternative telomere lengthening, with alterations in telomere maintenance genes such as ATRX, RBL2, and SP100, were identified in 78% of leiomyosarcomas [18]. In the same study, anomalies in the Retinoblastoma–cyclin D1 pathway and TP53 were found in almost every single case of LMS.
5. Imaging Diagnosis
Radiology is essential for the diagnosis, and since the symptoms are rather unspecific, the clinical differential diagnosis is broad, encompassing entities such as liposarcomas and lymphomas [20]. Invariably, the clinician will order an imaging exam such as ultrasonography (US), computerized tomography (CT), magnetic resonance imaging (MRI), or positron emission tomography (PET). US is rather unspecific [21], so CT is usually the method of choice for the first approach, enabling imaging-guided biopsy [22][23].
MRI has no specific signal; it is usually hypointense in T1 and intermediate in T2 but may be helpful in assessing necrosis [24]. MRI has a higher soft tissue resolution, allowing for a better definition of the vascular structures involved, and does not expose the patients to radiation [25]. MRI can also be combined with angiography for better local tumor evaluation. Contrast-enhanced cavography MRI can be used to differentiate an intraluminal mass from a thrombus and will also allow for the determination of the degree of obstruction and the development of collateral circulation [26].
PET has been proposed as a valuable indicator of tumor grade. A study by Punt et al. has demonstrated a correlation between a higher standard uptake value (SUVmax) and higher histological grade and tumor size [27]. In addition, it is a useful tool for assessing distant metastases [25]. Therefore, PET is valuable for a pre-operative decision.
Despite the value of radiology, pathology is fundamental and the gold standard in assessing LMS [28].
6. Gross and Histologic Features
On gross examination, the majority are extraluminal (76%), with minimal or even absent luminal growth. Tumor size can range from 2 to 30 cm [8][9].
Histologically, the cells are elongated in shape, with abundant and eosinophilic cytoplasm, centrally placed nuclei, and blunt-ended edges, sometimes called “cigar-shaped”. Architecturally, the cells are arranged in fascicles [1]. Cytoplasmatic and perinuclear haloes are frequently observed [29]. An example can be seen in Figure 1.
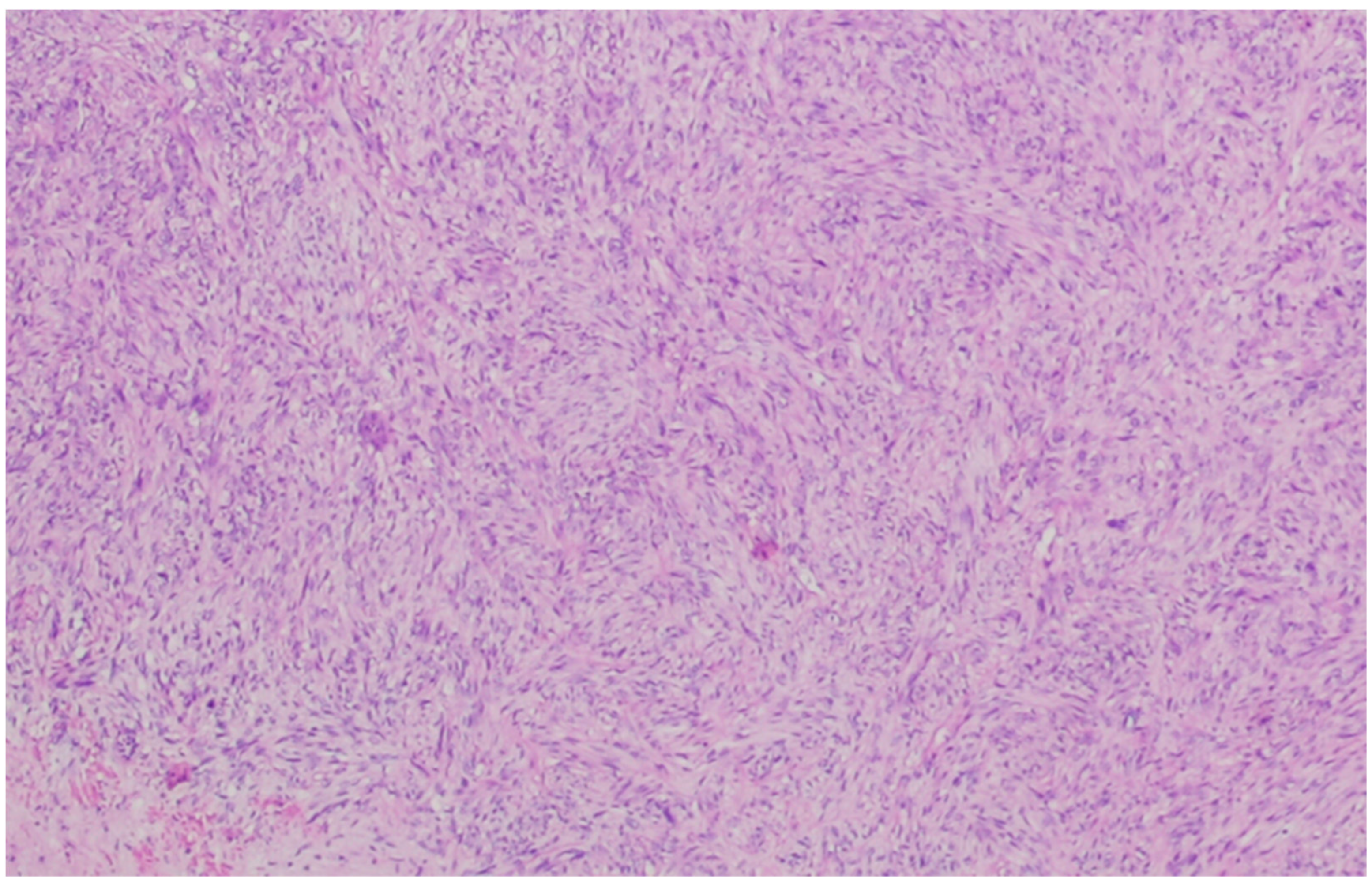
Figure 1. Leiomyosarcoma composed of cells with a fascicular architecture. The tumor cells are elongated with an eosinophilic cytoplasm. The cells, although atypical, resemble normal smooth cells, indicating mild atypia.
At higher magnification, longitudinally oriented myofibrils are a common characteristic. They can clump, giving a clotted appearance of the cytoplasm [7].
As leiomyosarcomas become less differentiated, they lose the resemblance to the normal counterpart. In well-differentiated tumors, the cells are arranged in fascicles, there is mild nuclear polymorphism, and nuclei are centrally located and have a low mitotic rate. In moderately differentiated tumors, the mitotic rate, nuclear polymorphism, and nuclear hyperchromasia are increased. In higher grade tumors, the nuclei often lose the central location, and a fascicular architecture and multinucleated cells are common [8].
This kind of cellular changes can be seen in Figure 2 and Figure 3.

Figure 2. In this leiomyosarcoma, there is a pronounced atypia with pseudoinclusions and pleomorphic, hyperchromatic, and bizarre nuclei. The preserved vascular lumen can be recognized in the central region of the photo (arrow).

Figure 3. In this tumor, the cells are markedly pleomorphic with multinucleation.
Morphologically, there seems to be no histologic difference between vascular LMS and other types of soft tissue LMS [29]. The intimal layer is usually intact, but protrusions into the lumen can also be seen [29].
Immunohistochemistry is helpful in diagnosing leiomyosarcoma and is based on demonstrating myoid differentiation with muscle markers. α-smooth muscle actin (α-SMA), desmin, heavy chain muscle actin, and h-caldesmon, which are myoid markers, are expressed in leiomyosarcomas [30]. α-SMA and muscle-specific actin are considered the most sensitive markers [31]. Desmin is expressed in around 70% of leiomyosarcomas [32][33][34]. It is important to take into consideration not only the expression but also the type of expression of these antibodies: diffuse expression of desmin is indicative of myoid differentiation, but the focal expression of actin or desmin can also be found in myofibroblasts [35][36]. An example of the immunostaining is represented in Figure 4.

Figure 4. Diffuse staining for α-SMA (left side) and desmin (right side). Abbreviations: α-SMA—α-smooth muscle actin.
There are interesting differences in the immunostaining characteristics of leiomyosarcomas arising from vascular smooth muscle compared to leiomyosarcomas from different locations. Vascular leiomyosarcomas are more often desmin negative and h-caldesmon positive than leiomyosarcomas arising from soft tissue [36][37].
The expression of keratins, which is usually regarded as evidence of epithelial differentiation, can be detected in leiomyosarcomas [37]. A cross-reaction with the antibodies used to detect cytokeratins was first suspected to be the cause of immunoreactivity, but it has been demonstrated that cytokeratins are present both in non-neoplastic smooth muscle and in smooth muscle tumors [38][39]. The expression of cytokeratins is limited to low molecular weight keratins [40]. Epithelial membrane antigen (EMA) immunoreactivity can occur in up to 45–60% of cases. When present, staining for cytokeratins is usually focal, but diffuse expression was seen in 11% of cytokeratins and 6% with EMA [37][40]. A dot-like pattern of cytokeratin expression has been described, and it is associated, at least focally, with a diffuse or a fibrillary pattern [37]. There is no correlation between the expression of cytokeratins and the location, sex, age, histological grade, and histological features of LMS, but EMA expression is more common in vascular LMS when compared to LMS of the soft tissue, skin, and uterus [37].
CD34 [41], S100 [42], and HMB45 [43] can also be expressed. Estrogen and progesterone receptors were previously suggested as adjunct (and helpful) markers to distinguish between retroperitoneal leiomyosarcomas and leiomiomas [44], but it was found that they can be expressed in both uterine and extrauterine LMS [45][46], therefore not being useful in this distinction.
Histologically, the differential diagnosis for leiomyosarcoma can be extensive, including other spindle cell lesions such as malignant peripheral nerve sheath tumors, synovial sarcomas, leiomyomas, schwannomas, benign cellular myofibroblastic tumors, gastrointestinal stromal tumor, synovial sarcoma, and inflammatory myofibroblastic tumors, as well as other high-grade malignancies, in poorly differentiated cases.
References
- IARC. WHO Classification of Soft Tissue and Bone Tumours. WHO Classification of Tumours Soft Tissue and Bone Tumours. 2020. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8167394/ (accessed on 6 August 2023).
- Fletcher, C.D.M. Diagnostic Histopathology of Tumors, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2020.
- Roland, C.L.; Boland, G.M.; Demicco, E.G.; Lusby, K.; Ingram, D.; May, C.D.; Kivlin, C.M.; Watson, K.; Sanaa, G.A.A.; Wang, W.L.; et al. Clinical Observations and Molecular Variables of Primary Vascular Leiomyosarcoma. JAMA Surg. 2016, 151, 347–354. Available online: https://pubmed.ncbi.nlm.nih.gov/26629783/ (accessed on 31 July 2023).
- Perl, L.; Virchow, R. Ein Fall von Sarkom der Vena cava inferior. Arch Pathol. Anat. Physiol. Klin. Med. 1871, 53, 378–383. Available online: https://link.springer.com/article/10.1007/BF01957198 (accessed on 29 July 2023).
- Mastrangelo, G.; Coindre, J.M.; Ducimetière, F.; Dei Tos, A.P.; Fadda, E.; Blay, J.Y.; Buja, A.; Fedeli, U.; Cegolon, L.; Frasson, A.; et al. Incidence of soft tissue sarcoma and beyond. Cancer 2012, 118, 5339–5348. Available online: https://onlinelibrary.wiley.com/doi/full/10.1002/cncr.27555 (accessed on 31 July 2023).
- Müller, A.M.; Chromik, A.M.; Bolik, B.; Müller, K.M.; Mittelkötter, U. Leiomyosarkom der vena cava inferior: Übersicht zu einem seltenen krankheitsbild. Pathologe 2005, 26, 153–158. Available online: https://link.springer.com/article/10.1007/s00292-004-0745-y (accessed on 29 July 2023).
- Goldblum, J.R.; Weiss, S.W.; Folpe, A.L. Enzinger and Weiss’s Soft Tissue Tumors, 7th ed.; Elsevier: Amsterdam, The Netherlands, 2019.
- Leiomyosarcoma of the Inferior Vena Cava—Hines—1999—Cancer—Wiley Online Library. Available online: https://acsjournals.onlinelibrary.wiley.com/doi/pdf/10.1002/%28SICI%291097-0142%2819990301%2985%3A5%3C1077%3A%3AAID-CNCR10%3E3.0.CO%3B2-0 (accessed on 29 July 2023).
- International Registry of Inferior Vena Cava Leiomyosarcoma: Analysis of a World Series on 218 Patients—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/8920790/ (accessed on 30 July 2023).
- Yoshizawa, K.; Ohno, Y.; Kurata, T.; Takagi, Y.; Kasai, T.; Takizawa, M.; Soyejima, Y. Primary leiomyosarcoma of the inferior vena cava in a pediatric case: A case report and literature review. Surg. Case Rep. 2023, 9, 52. Available online: https://pmc/articles/PMC10079787/ (accessed on 6 April 2023).
- Kevorkian, J.; Cento, D.P. Leiomyosarcoma of large arteries and veins. Surgery 1973, 73, 390–400. Available online: http://www.surgjournal.com/article/0039606073903073/fulltext (accessed on 27 July 2023).
- Italiano, A.; Toulmonde, M.; Stoeckle, E.; Kind, M.; Kantor, G.; Coindre, J.M.; Bui, B. Clinical outcome of leiomyosarcomas of vascular origin: Comparison with leiomyosarcomas of other origin. Ann. Oncol. 2010, 21, 1915–1921. Available online: http://www.annalsofoncology.org/article/S0923753419400628/fulltext (accessed on 2 August 2023).
- Weinreb, W.; Steinfeld, A.; Rodil, J.; Esparza, A.; Trebbin, W. Leiomyosarcoma Arising in an Arteriovenous Fistula. Available online: https://pubmed.ncbi.nlm.nih.gov/6861080/ (accessed on 30 July 2023).
- Hashimoto, H.; Daimaru, Y.; Tsuneyoshi, M.; Enjoji, M. Leiomyosarcoma of the External Soft Tissues A Clinicopathologic, lmmunohistochemical, and Electron Microscopic Study Materials and Methods. Available online: https://onlinelibrary.wiley.com/terms-and-conditions (accessed on 29 July 2023).
- Mingoli, A.; Feldhaus, R.J.; Cavallaro, A.; Stipa, S. Leiomyosarcoma of the inferior vena cava: Analysis and search of world literature on 141 patients and report of three new cases. J. Vasc. Surg. 1991, 14, 688–699.
- Gomes, J.F.; Vieira, I.; Mendes, J.; Donaire, D.; Almeida, R. Leiomyosarcoma of Inferior Vena Cava in an Immunocompetent Young-adult Female Patient. J. Coll. Physicians Surg. Pak. 2022, 32, 1353–1355. Available online: https://pubmed.ncbi.nlm.nih.gov/36205287/ (accessed on 23 September 2023).
- Nilbert, M.; Mandahl, N.; Heim, S.; Rydholm, A.; Helm, G.; Willén, H.; Baldetorp, B.; Mitelman, F. Complex karyotypic changes, including rearrangements of 12q13 and 14q24, in two leiomyosarcomas. Cancer Genet. Cytogenet. 1990, 48, 217–223. Available online: https://pubmed.ncbi.nlm.nih.gov/2397453/ (accessed on 31 July 2023).
- Chudasama, P.; Mughal, S.S.; Sanders, M.A.; Hübschmann, D.; Chung, I.; Deeg, K.I.; Wong, S.H.; Rabe, S.; Hlenvjak, M.; Zapatka, M.; et al. Integrative genomic and transcriptomic analysis of leiomyosarcoma. Nat. Commun. 2018, 9, 1–15. Available online: https://www.nature.com/articles/s41467-017-02602-0 (accessed on 31 July 2023).
- Gladdy, R.A.; Qin, L.X.; Moraco, N.; Agaram, N.P.; Brennan, M.F.; Singer, S. Predictors of Survival and Recurrence in Primary Leiomyosarcoma. Ann. Surg. Oncol. 2013, 20, 1851. Available online: https://pmc/articles/PMC3657306/ (accessed on 28 August 2023).
- Rusu, C.B.; Gorbatâi, L.; Szatmari, L.; Koren, R.; Bungărdean, C.I.; Feciche, B.O.; Bumbulut, C.; Andras, I.M.; Rahota, R.; Telecan, T.; et al. Leiomyosarcoma of the inferior vena cava. Our experience and a review of the literature. Rom J. Morphol. Embryol. 2020, 61, 227. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7728114/ (accessed on 23 September 2023).
- Wu, X.; Zhou, P.; Li, K. Contrast-enhanced ultrasonography of intraluminal inferior vena cava leiomyosarcoma: A case report. J. Clin. Ultrasound. 2020, 48, 357–361. Available online: https://pubmed.ncbi.nlm.nih.gov/32027023/ (accessed on 23 September 2023).
- Webb, E.M.; Wang, Z.J.; Westphalen, A.C.; Nakakura, E.K.; Coakley, F.V.; Yeh, B.M. Can CT features differentiate between inferior vena cava leiomyosarcomas and primary retroperitoneal masses? Am. J. Roentgenol. 2013, 200, 205–209. Available online: https://www.ajronline.org/doi/10.2214/AJR.11.7476 (accessed on 23 September 2023).
- Mastoraki, A.; Leotsakos, G.; Mastoraki, S.; Papanikolaou, I.S.; Danias, N.; Smyrniotis, V.; Arkadapoulos, N. Challenging diagnostic and therapeutic modalities for leiomyosarcoma of inferior vena cava. Int. J. Surg. 2015, 13, 92–95. Available online: https://pubmed.ncbi.nlm.nih.gov/25489949/ (accessed on 23 September 2023).
- Sessa, B.; Iannicelli, E.; Caterino, S.; D’Angelo, F.; Milione, M.; Ziparo, V.; David, V. Imaging of leiomyosarcoma of the inferior vena cava: Comparison of 2 cases and review of the literature. Cancer Imaging 2010, 10, 80. Available online: https://pmc/articles/PMC2842181/ (accessed on 23 September 2023).
- Ronchi, B.; Peña, G.A.; Sacchi, C. PET/MR: Primary inferior vena cava leiomyosarcoma. Eur. J. Hybrid Imaging 2022, 6, 1–8. Available online: https://ejhi.springeropen.com/articles/10.1186/s41824-022-00144-3 (accessed on 23 September 2023).
- Kim, J.T.; Kwon, T.; Cho, Y.; Shin, S.; Lee, S.; Moon, D. Multidisciplinary treatment and long-term outcomes in six patients with leiomyosarcoma of the inferior vena cava. J. Korean Surg. Soc. 2012, 82, 101–109. Available online: https://pubmed.ncbi.nlm.nih.gov/22347712/ (accessed on 23 September 2023).
- Punt, S.E.W.; Eary, J.F.; O’Sullivan, J.; Conrad, E.U. Fluorodeoxyglucose positron emission tomography in leiomyosarcoma: Imaging characteristics. Nucl. Med. Commun. 2009, 30, 546–549. Available online: https://pubmed.ncbi.nlm.nih.gov/19440162/ (accessed on 23 September 2023).
- Gronchi, A.; Miah, A.B.; Dei Tos, A.P.; Abecassis, N.; Bajpai, J.; Bauer, S.; Biagini, R.; Bielak, S.; Blay, J.Y.; Bolle, S.; et al. Soft tissue and visceral sarcomas: ESMO-EURACAN-GENTURIS Clinical Practice Guidelines for diagnosis, treatment and follow-up☆. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2021, 32, 1348–1365. Available online: https://pubmed.ncbi.nlm.nih.gov/34303806/ (accessed on 23 September 2023).
- Farshid, G.; Pradhan, M.; Goldblum, J.; Weiss, S.W. Leiomyosarcoma of somatic soft tissues: A tumor of vascular origin with multivariate analysis of outcome in 42 cases. Am. J. Surg. Pathol. 2002, 26, 14–24. Available online: https://pubmed.ncbi.nlm.nih.gov/11756765/ (accessed on 29 July 2023).
- Tsukada, T.; McNutt, M.A.; Ross, R.; Gown, A.M. HHF35, a muscle actin-specific monoclonal antibody. II. Reactivity in normal, reactive, and neoplastic human tissues. Am. J. Pathol. 1987, 127, 389. Available online: https://pmc/articles/PMC1899748/?report=abstract (accessed on 29 July 2023).
- Carvalho, J.C.; Thomas, D.G.; Lucas, D.R. Cluster analysis of immunohistochemical markers in leiomyosarcoma delineates specific anatomic and gender subgroups. Cancer 2009, 115, 4186–4195. Available online: https://pubmed.ncbi.nlm.nih.gov/19626649/ (accessed on 2 August 2023).
- Azumi, N.; Ben-Ezra, J.; Battifora, H. Immunophenotypic diagnosis of leiomyosarcomas and rhabdomyosarcomas with monoclonal antibodies to muscle-specific actin and desmin in formalin-fixed tissue. Mod. Pathol. 1988, 1, 469–474.
- Pollock, L.; Rampling, D.; Greenwald, S.E.; Malone, M. Desmin expression in rhabdomyosarcoma: Influence of the desmin clone and immunohistochemical method. J. Clin. Pathol. 1995, 48, 535–538. Available online: https://jcp.bmj.com/content/48/6/535 (accessed on 29 July 2023).
- Truong, L.D.; Rangdaeng, S.; Cagle, P.; Ro, J.Y.; Hawkins, H.; Font, R.L. The diagnostic utility of desmin. A study of 584 cases and review of the literature. Am. J. Clin. Pathol. 1990, 93, 305–314. Available online: https://pubmed.ncbi.nlm.nih.gov/1689936/ (accessed on 29 July 2023).
- Dabbs, D.J. Diagnostic Immunohistochemistry, 6th ed.; Elsevier: Amsterdam, The Netherlands, 2022.
- Matsuyama, A.; Hisaoka, M.; Hashimoto, H. Vascular leiomyosarcoma: Clinicopathology and immunohistochemistry with special reference to a unique smooth muscle phenotype. Pathol. Int. 2010, 60, 212–216. Available online: https://pubmed.ncbi.nlm.nih.gov/20403047/ (accessed on 29 July 2023).
- Iwata, J.; Fletcher, C.D.M. Immunohistochemical detection of cytokeratin and epithelial membrane antigen in leiomyosarcoma: A systematic study of 100 cases. Pathol. Int. 2000, 50, 7–14. Available online: https://onlinelibrary.wiley.com/doi/full/10.1046/j.1440-1827.2000.01001.x (accessed on 29 July 2023).
- Traweek, S.T.; Liu, J.; Battifora, H. Keratin gene expression in non-epithelial tissues. Detection with polymerase chain reaction. Am. J. Pathol. 1993, 142, 1111. Available online: https://pmc/articles/PMC1886881/?report=abstract (accessed on 28 October 2023).
- Gown, A.M.; Boyd, H.C.; Chang, Y.; Ferguson, M.; Reichler, B.; Tippens, D. Smooth muscle cells can express cytokeratins of “simple” epithelium. Immunocytochemical and biochemical studies in vitro and in vivo. Am. J. Pathol. 1988, 132, 223. Available online: https://pmc/articles/PMC1880728/?report=abstract (accessed on 28 October 2023).
- Immunoreactivity for Cytokeratin and Epithelial Membrane Antigen in Leiomyosarcoma—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/2454091/ (accessed on 29 July 2023).
- Fisher, C. Immunohistochemistry in diagnosis of soft tissue tumours. Histopathology 2011, 58, 1001–1012. Available online: https://pubmed.ncbi.nlm.nih.gov/21143519/ (accessed on 30 July 2023).
- Kanamori, T.; Takakura, K.; Mandai, M.; Kariya, M.; Fukuhara, K.; Sakaguchi, M.; Huh, N.H.; Saito, K.; Sakurai, T.; Fujita, J.; et al. Increased expression of calcium-binding protein S100 in human uterine smooth muscle tumours. Mol. Hum. Reprod. 2004, 10, 735–742. Available online: https://pubmed.ncbi.nlm.nih.gov/15322223/ (accessed on 30 July 2023).
- Silva, E.G.; Bodurka, D.C.; Scouros, M.A.; Ayala, A. A uterine leiomyosarcoma that became positive for HMB45 in the metastasis. Ann. Diagn. Pathol. 2005, 9, 43–45.
- Paal, E.; Miettinen, M. Retroperitoneal Leiomyomas: A Clinicopathologic and Immunohi: The American Journal of Surgical Pathology. Am. J. Surg. Pathol. 2001, 25, 1355–1363. Available online: https://journals.lww.com/ajsp/Abstract/2001/11000/Retroperitoneal_Leiomyomas__A_Clinicopathologic.2.aspx (accessed on 30 July 2023).
- Kelley, T.W.; Borden, E.C.; Goldblum, J.R. Estrogen and progesterone receptor expression in uterine and extrauterine leiomyosarcomas: An immunohistochemical study. Appl. Immunohistochem Mol. Morphol. AIMM 2004, 12, 338–341. Available online: https://pubmed.ncbi.nlm.nih.gov/15536333/ (accessed on 30 July 2023).
- Leiomyosarcoma of the Pulmonary Veins: The American Journal of Surgical Pathology. Available online: https://journals.lww.com/ajsp/Abstract/1999/09000/Leiomyosarcoma_of_the_Pulmonary_Veins.11.aspx (accessed on 30 July 2023).
More
Information
Subjects:
Pathology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
540
Revisions:
2 times
(View History)
Update Date:
19 Jan 2024
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No