Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Zhifeng Wang | -- | 4183 | 2024-01-17 09:10:41 | | | |
| 2 | Lindsay Dong | Meta information modification | 4183 | 2024-01-19 02:24:38 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Wang, X.; Xu, X.; Wang, Z. Post-Translational Role of UFMylation in Physiology and Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/53951 (accessed on 02 August 2026).
Wang X, Xu X, Wang Z. Post-Translational Role of UFMylation in Physiology and Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/53951. Accessed August 02, 2026.
Wang, Xingde, Xingzhi Xu, Zhifeng Wang. "Post-Translational Role of UFMylation in Physiology and Disease" Encyclopedia, https://encyclopedia.pub/entry/53951 (accessed August 02, 2026).
Wang, X., Xu, X., & Wang, Z. (2024, January 17). Post-Translational Role of UFMylation in Physiology and Disease. In Encyclopedia. https://encyclopedia.pub/entry/53951
Wang, Xingde, et al. "Post-Translational Role of UFMylation in Physiology and Disease." Encyclopedia. Web. 17 January, 2024.
Copy Citation
Ubiquitin-fold modifier 1 (UFM1) is a newly identified ubiquitin-like protein that has been conserved during the evolution of multicellular organisms. In a similar manner to ubiquitin, UFM1 can become covalently linked to the lysine residue of a substrate via a dedicated enzymatic cascade. Although a limited number of substrates have been identified so far, UFM1 modification (UFMylation) has been demonstrated to play a vital role in a variety of cellular activities, including mammalian development, ribosome biogenesis, the DNA damage response, endoplasmic reticulum stress responses, immune responses, and tumorigenesis.
post-translational modification
UFMylation
ubiquitin-like proteins
1. Introduction
Post-translational modification (PTM) refers to the addition of chemical groups to one or more amino acid residues; such additions can substantially change the biological activity of the target protein [1]. To date, more than 500 different protein PTMs have been identified, including phosphorylation, glycosylation, acetylation, and ubiquitination [2]. Focusing on the latter, ubiquitin (Ub) is a small protein weighing approximately 8.5 kDa and comprising 76 amino acids. Ub is widely distributed in all eukaryotic cells and has been highly conserved during evolution. Indeed, yeast and human ubiquitin differ by only three amino acids. Ubiquitination refers to the covalent binding of Ub to target proteins, and generally requires the synergistic action of three ubiquitinating enzymes: E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzyme, and E3 ubiquitin-ligase [3]. First, ubiquitin is activated by E1 using energy provided by ATP hydrolysis, then transferred to E2, and, finally, covalently linked to the lysine residues of substrates with the aid of E3. Ubiquitination is a tightly regulated and reversible process: deubiquitinating enzymes (DUBs) can reverse ubiquitination by hydrolyzing the peptide or isopeptide bonds between ubiquitin molecules or between ubiquitin and substrate proteins [3]. Ubiquitination helps to regulate numerous biological processes, encompassing immune responses [4][5], the DNA damage response [6][7], cell cycle regulation [8], autophagy [9], epigenetic modulation [10], cellular apoptosis [11], and protein degradation [12], by regulating protein structures, interactions, activities, and even subcellular localizations [3].
The many ubiquitin-like proteins (UBLs) identified to date include small ubiquitin-like modifiers (SUMOs), neural precursor cell-expressed developmentally downregulated 8 (NEDD8), and interferon-stimulated gene 15 (ISG15). Although most UBLs do not necessarily share notable sequence homology with Ub, they all share a similar tertiary structure [13]. Similar to Ub, UBLs can covalently bind to target proteins (UBLylation) through a series of enzymatic reactions, similar to those involved in ubiquitination, to confer different biological functions to the substrate [14].
Ubiquitin-fold modifier 1 (UFM1) is a novel UBL formed from an 85-amino acid precursor (pro-UFM1) that is translated in human cells. UFM1 is evolutionarily conserved in multicellular organisms, but is absent in yeast [15]. Similar to Ub, the C-terminal serine and cysteine of the UFM1 precursor can be removed using specific proteases to expose the C-terminal glycine, leaving mature UFM1 to directly and covalently attach to the lysine residues of target substrates [16]. UFM1 is a unique UBL as it has only one glycine residue at its C-terminus.

2. UFMylation
Similar to ubiquitination, UFM1 covalently binds to its target proteins through a cascade involving three enzymes. As described above, the UFM1 gene is translated into a precursor form and the C-terminal glycine must be exposed by UFM1-specific proteases (UfSPs) before subsequent enzymatic reactions can occur [16]. First, when ATP is present, the E1-like enzyme UBA5 activates the mature UFM1, which forms a high-energy thioester bond between the catalytic cysteine (Cys250) of UBA5 and the exposed C-terminal glycine of UFM1. Next, the E2-like enzyme UFC1 interacts with the UFC1 binding domain of UBA5, which transfers the activated UFM1 to UFC1 by forming a similar thioester bond between UFM1 and the catalytic cysteine (Cys 116) of UFC1. Finally, UFM1 is coupled to the lysine residues of its target proteins in a process mediated by the E3-like enzyme UFL1 [17]. Another similarity with ubiquitination is that UFMylation is also reversible. In addition to maturing UFM1, UfSPs also cleave UFM1 from its target protein, thereby rendering UFM1 and its substrates recyclable (Figure 1) [16]. Numerous verified substrates of UFM1 have been discovered and the impact of UFMylation on their functions elucidated.
Figure 1. The enzyme cascade of UFMylation (A) and five key biological functions of UFMylation (B).
2.1. UfSPs
Pro-UFM1 maturation and the removal of UFMylation from substrates are mediated by the cysteine proteases UfSP1 and UfSP2, respectively. It was, until recently, thought that UfSP2 was the only active protease because UfSP1 apparently lacks a catalytic domain [16]. However, two independent groups recently found that UfSP1 actually utilizes a non-canonical start codon (217CUG) upstream of its canonical counterpart (445AUG) to initiate translation, which produces a catalytically active UfSP1. Cong et al. reported that both UfSP1 and UfSP2 can mediate the maturation of pro-UFM1 and the de-UFMylation of substrate proteins [18]. By contrast, a study by Kulathu et al.’s group indicated that UfSP2, but not UfSP1, de-UFMylates the ribosomal subunit RPL26, while UfSP1 removes the constitutively autoinhibitory UFMylation of UFC1, thereby promoting the activation of UFMylation [19].
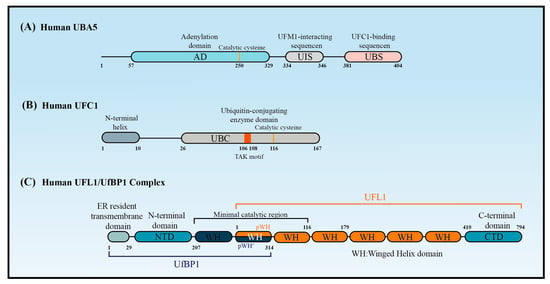
2.2. UBA5
The canonical E1 enzymes that mediate ubiquitination have conserved adenylation domains, catalytic cysteine domains, and ubiquitin-fold domains. By contrast, as a member of the non-canonical E1 enzyme family, UBA5 does not contain a catalytic cysteine domain. Instead, its catalytic cysteine active site (Cys250) is located in its adenylation domain [20] (Figure 2A). This domain comprises an eight-stranded beta sheet that is surrounded by helices [21] and it promotes UBA5 homodimer formation with pseudo-two-fold symmetry [22].
Short and long UBA5 are two distinct isoforms of UBA5 encoded by the human genome, with the latter distinguished by the presence of a 56-amino acid extension at the N-terminal adjacent to the adenylation domain. Structural and biochemical studies indicated that the binding ratio of UBA5 to ATP in the presence of the N-terminus (long UBA5) is 1:1 rather than the 2:1 that occurs in its absence (short UBA5). This finding indicates that the N-terminus greatly increases the affinity of UBA5 for ATP, thereby promoting UFM1 activation at low ATP concentrations [23]. The N-terminal extension also enhances the thermal stability of UBA5 and promotes the faster transfer of UFM1 to UFC1 through a conformational change occurring at the N-terminus of UBA5 when ATP binds to the UBA5-UFM1 complex. Therefore, ATP and the binding of UFM1 to UBA5 stabilize the UBA5 homodimer, enhance UBA5 stability, and promote UFM1 transfer to UFC1.
2.3. UFC1
UFC1 is mainly localized in the nucleus, with a minor proportion located in the cytoplasm [15]. UFC1 lacks some features that are conserved in other E2s, such as the catalytic histidine–proline–asparagine (HPN) motif, which suggests it has a unique mechanism of function and modulation [24]. Some ubiquitin E2 enzymes bind to Ub to promote E2 dimerization, stabilize E2s in their active-closed state, and enhance their catalytic efficiency [3]. It is unknown whether UFC1 activity is regulated by similar mechanisms.
Although UFC1 differs from the other E2 enzymes, it contains a catalytic core structural domain that is conserved in all E2 enzymes. This catalytic core domain presents as a flexible loop formed of approximately 10 amino acid residues and confers strong solvent accessibility [25]. The loop encloses the intermediate active cysteine residue (Cys116), which undergoes a trans-esterification reaction upon the transfer of UFM1 from UBA5 to UFC1 [25] (Figure 2B).
UFC1 may transfer UFM1 to substrates in association with UFL1/UfBP1. Data from in vitro assays confirmed that UFC1 can transfer UFM1 to free cysteines but not free lysines, indicating that UFC1 cannot transfer UFM1 to substrates directly. UFC1 together with UFL1/UfBP1 can, however, transfer UFM1 to free lysines [26].
2.4. UFL1 and UfBP1
UFL1 contains a transmembrane domain enabling its primary localization on the cytoplasmic side of the endoplasmic reticulum (ER) membrane. UFL1 can, however, also be found in the cytoplasm and nucleus due to the presence of a nuclear localization signal [27]. In the UFMylation system, UFL1 is the only E3 ligase identified so far that aids the transfer of UFM1 to target substrates [28].
Hundreds of E3 ligases that participate in ubiquitination are classified into three types according to domain structures. RING E3s contain the very interesting new gene domain, while HECT E3s contain a domain that is homologous to the E6-AP C-terminus domain, and RBR E3s contain a RING-between-RING domain. RING E3s transfer ubiquitin directly from the E2s to the substrates without binding to the Ub, while HECT and RBR E3s require Ub to first form a thioester bond with the conserved cysteine before the Ub is transferred to the substrates [29].
Interestingly, UFL1 cannot function properly alone, but requires UfBP1 to promote its stability and activity. UfBP1, also known as C20orf116 or DDRGK1, is a UFM1-interacting protein composed of 314 amino acids that include a C-terminal proteasome-COP9-initiation factor domain. UfBP1 also contains a transmembrane domain and is localized on the cytoplasmic side of the ER membrane, where it exists in a complex with UFL1 [30].
Structure predictions have shown that UFL1 and UfBP1 form a heterodimer composed of several winged helix (WH) domain repeats [26] (Figure 2C). UFL1 has an N-terminal helix followed by a partial WH (pWH) and five WH domains, which are important for its E3 ligase activity. UfBP1 has an N-terminal transmembrane segment, a long helical region followed by a WH (WH1′) domain, and a partial WH (pWH′) domain [31]. The partial pWH domain at the N-terminus of UFL1 complements the partial pWH′ domain at the C-terminus of UfBP1 to form a composite WH (pWH-pWH′) domain that is essential for complex formation and protein stability [31].
CDK5RAP3 is another protein that is consistently associated with UFL1. Because CDK5RAP3 always functions as a substrate adaptor [32], it is thought that CDK5RAP3, together with UFL1/UfBP1, forms part of an integral E3 ligase complex [33][34][35]. CDK5RAP3 binds to the ligase complex of UFL1/UfBP1 and restricts its E3 ligase activity. CDK5RAP3 functions as a specificity determinant, inhibiting ligase activity in the absence of a substrate and directing ligase activity toward the ribosomal subunit RPL26 [26].
Figure 2. The structure of UBA5, UFC1, and UFL1/UfBP1 complex. Schematic of (A) the key domains of UBA5, (B) the key domain features of UFC1, and (C) the domains of the UFL1-UfBP1 E3 ligase complex.
3. Biological Functions of UFMylation
3.1. UFMylation and the DNA Damage Response
DNA damage caused by endogenous or exogenous factors seriously impairs genomic integrity but can be rescued via DNA damage response pathways [36]. DNA double-strand breaks (DSBs), which are extremely toxic to cells, are repaired almost exclusively by homologous recombination (HR) and non-homologous end-joining. Emerging evidence indicates that UFMylation has numerous important roles in mediating the cellular response to DSBs, thus contributing to the maintenance of genome stability and preventing tumorigenesis [37].
3.1.1. MER11 UFMylation
UFL1 co-localizes with γH2AX, a marker of DSBs, during UV- and IR-induced DNA damage. In addition, UFM1 and UFL1 are immediately recruited to laser-induced DSBs [38][39]. These observations have prompted us to speculate that UFMylation has a role in the DSB response. At the initiation of the DSB response, the MRE11-RAD50-NBS1 (MRN) complex re-localizes to the damage sites in what is regarded as the first and most important step that activates the kinase of ataxia telangiectasia mutated (ATM) [36]. UFL1 depletion inhibited the activation of ATM after DSB formation [37], indicating that UFMylation plays important roles in ATM activation. The interaction between UFL1 and the MRN complex, together with the decreased recruitment and stability of the MRN complex resulting from UFL1 depletion, led to the identification of MRE11 UFMylation on K282 [37][38]. MRE11 is a core factor in the MRN complex and binds directly to RAD50 and NBS1. This complex is integrally recruited to damage sites, which promotes ATM activation and DNA end resection, thereby promoting HR repair [36].
The function of MRE11 UFMylation in the DSB response is also mediated by UfSP2 [40]. Evidence from human cell lines showed that UfSP2 is phosphorylated at serine 374/381 by ATM, which promotes its release from the MRN complex after the formation of DSBs [38][40]. This release enhances MRE11 UFMylation and ATM activation, which, in turn, further promotes UfSP2 phosphorylation. UfSP2 is dephosphorylated by the phosphatase WIP1, which promotes UfSP2 recruitment to DSBs, leading to the de-UFMylation of MRE11 and H4, and suppresses ATM activation [40].
3.1.2. Histone H4 UFMylation
Another mechanism by which UFMylation regulates DSB-induced ATM activation involves UFL1 phosphorylation at serine 462 by ATM. This event promotes UFL1 recruitment to sites of DNA damage. UFL1 catalyzes the mono-UFMylation of histone H4 on K31 [39], which can be recognized by the UFMylation reader serine/threonine kinase 38 (STK38) [41]. STK38 contains a UFM1-binding motif that when mutated at conserved amino acids can no longer interact with UFMylated H4. STK38 recruitment to DNA damage sites also depends on the monoacylation of H4 and is critical for the subsequent recruitment of SUV39H1. SUV39H1 catalyzes the trimethylation of H3K9 (H3K9me3), which binds to and recruits Tip60. In turn, Tip60 acetylates ATM and promotes its activation [39]. The UFMylation of H4 is also regulated by UfSP2 and its ATM-induced phosphorylation (see MRE11 above).
MRE11 UFMylation at DSBs is first catalyzed by UFL1 to promote MRN complex recruitment and ATM activation. Active ATM then phosphorylates UFL1 and UfSP2. The phosphorylation of UFL1 enhances its recruitment to sites of DNA damage, while the phosphorylation of UfSP2 releases it from the MRN complex, and both processes further increase MRE11 UFMylation. UFL1 and UfSP2 phosphorylation also leads to enhanced H4 UFMylation and, later, ATM activation. The phosphatase WIP1 de-phosphorylates UfSP2, which de-UFMylates MRE11 and H4, and finally reverses ATM activation.
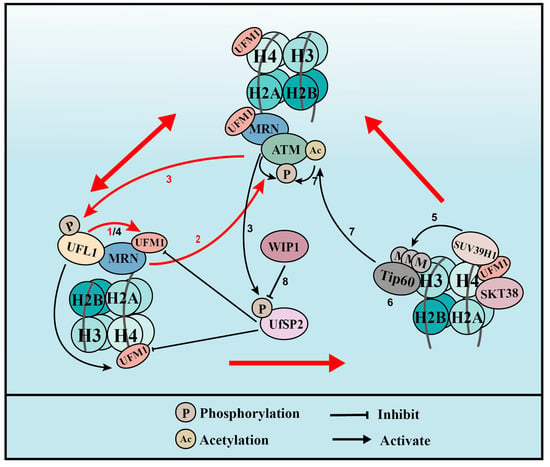
UFMylation therefore promotes ATM activation though two distinct positive feedback loops: one is WIP1-UFL1/UfSP2-MRE11-ATM-UFL1/UfSP2 phosphorylation and the other is WIP1-UFL1/UfSP2-H4-STK38-SUV39H1-H3K9me3-Tip60-ATM acetylation-ATM phosphorylation-UFL1/UfSP2 phosphorylation (Figure 3).
Figure 3. UFMylation of MRE11 and H4 promotes ATM activation (for the purposes of the diagram, MRE11 is abbreviated to MRN complex). (1) Upon DNA damage, UFL1 is recruited to chromatin via MRE11 to UFMylate MRE11, which promotes the formation and recruitment of the MRE11-RAD50-NBS1 (MRN) complex. (2) UFMylated MRE11, together with RAD50 and NBS1, activates ATM. (3) Activated ATM further phosphorylates UFL1 and UfSP2, which promotes UFL1 recruitment, while inhibiting UfSP2 recruitment. (4) As a positive feedback loop, phosphorylated UFL1 further enhances MRE11 UFMylation and UFMylates H4. (5) UFMylated H4 can be recognized by the UFMylation reader serine/threonine kinase 38 (STK38), which is critical for the subsequent recruitment of the methyltransferase SUV39H1. (6) SUV39H1 trimethylates lysine 9 of histone H3 (H3K9me3), which recruits Tip60. (7) Tip60 acetylates ataxia telangiectasia mutated (ATM) and promotes ATM activation. (8) Lastly, the phosphatase WIP1 de-phosphorylates UfSP2 and reverses ATM activation. Red arrows indicate the two positive feedback loops.
3.2. UFMylation in ER Metabolism
3.2.1. ER Stress
ER stress is caused by an overload of un/mis-folded proteins in the ER. In mammalian cells, three ER transmembrane proteins act as sensors of ER stress: activating transcription factor 6, inositol-required enzyme 1α (IRE1α), and PKR-like ER kinase (PERK) [42]. The accumulation of un/mis-folded proteins triggers ER stress and activates un/mis-folded-protein response (UPR) signaling to the ER to enhance its protein processing capacity. Therefore, the amount of UPR signaling is representative of ER stress levels. The un/mis-folded-proteins are then degraded through a type of ubiquitin-proteasome degradation known as ER-associated degradation (ERAD) [42]. Damaged ER can also be removed through an autophagy process called ER-phagy [43]. Thus, ERAD and ER-phagy are the predominant pathways involved in alleviating ER stress.
As mentioned above, UFL1 and UfBP1 are localized on the ER membrane, which suggests that UFMylation is involved in ER stress. The first evidence that UFMylation is related to ER stress was obtained in studies to determine the involvement of ER stress in the development of heart disease [44]. Later, numerous studies showed that the UFMylation substrate UfBP1, an UFL1 partner, could respond to ER stress [28][45][46], and CDK5RAP3 could sense proteotoxic stress in the ER lumen by forming a tripartite receptor complex with the ER-associated UFL1/UfBP1 [47]. In another example, cisplatin induced a stress-related increase in UFL1 expression in granulosa cells and enhanced ER stress, processes which were exacerbated by UFL1 knockdown and alleviated by UFL1 overexpression [48]. Others have reported that UFMylation modulates UPR signaling.
CDK5RAP3, another UFL1 partner, is also involved in ER stress, especially the expression of XBP1 and PERK [49]. During the interphase of the cell cycle, microtubules are predominantly nucleated at the centrosome (microtubule organizing centers; MTOC) by γ-tubulin ring complex (γTuRC) proteins. Most γTuRCs are activated by structural rearrangement, phosphorylation, or binding to modulating proteins accumulated in MTOCs [49]. The interaction of ER membranes with newly formed microtubules could promote ER expansion and help to restore ER homeostasis. UFL1 can interact with CDK5RAP3 and form a complex with γTuRCs [50][51], negatively regulating microtubule nucleation at interphase centrosomes. In mammalian cells, ER network rearrangements largely depend on interactions with dynamic microtubules. Therefore, an UFL1/CDK5RAP3 deletion induces ER stress and the release of γTuRC proteins, which in turn stimulates microtubule nucleation. Thus, the interaction between the ER and newly formed microtubules promotes ER enlargement to restore ER homeostasis.
3.2.2. ER-Phagy
UFMylation has an important role in ER-phagy. Data from several recent studies have confirmed that UFMylation regulates ER degradation through lysosomes, thus furthering our understanding of the mechanisms by which UFMylation regulates ER stress. A human genome-wide screen indicated that UfBP1 is an ER-phagy regulator [52]. Many factors associated with the ribosome and translational quality control, such as RPL26 and RPN1, were identified as UFMylation substrates that mediate ER-associated autophagy [52][53][54]. NADH-cytochrome b5 reductase 3 (CYB5R3) is also UFMylated, and this mediates its degradation by lysosomes [55].
CDK5RAP3 has been proposed to function as both a substrate adaptor that directs UFMylation toward target substrates, such as ribosomal protein RPL26 [26], and an adaptor protein for UFMylation-dependent ER-phagy [47][55]. The results from one study showed that CDK5RAP3 depletion increased the amounts of GFP-UFM1-conjugated CYB5R3 and UfBP1, while treatment with bafilomycin A1 to suppress autophagy had no effect [55]. These results support the idea that CDK5RAP3 promotes UFMylation-mediated ER-phagy.
The UFMylation of ribosomal proteins is believed to control the ribosomal stress response and ribosome-mediated protein translation quality. Indeed, UFL1 interacts with ribosomes, and the UFMylation substrate screening of ribosomal proteins showed that many ribosomal subunits are UFMylated [56]. Similarly, UfBP1-dependent UFMylation substrate screening with nutrient starvation led to the identification of several ribosomal subunits, ribosome-associated factors, and ER-resident translocon proteins as UFMylation substrates, including RPL7A, RPLP0, RPL10A, RPL30, RPL19, and RPN1 [52]. These findings suggest that ribosomal proteins are likely modified by UFMylation, although most candidates remain to be confirmed experimentally.
RPL26 is UFMylated at K132 and K134 [53][54] when it is located at the ER surface, as the UFL1/CDK5RAP3/UfBP1 E3 complex is restricted to the ER membrane [53]. This modification can be upregulated after treatment with the protein translation inhibitor anisomycin, indicating that ribosome arrest during cotranslational translocation in the ER is a specific trigger for RPL26 UFMylation [54].
CYB5R3 is another master regulator of ER-phagy and is UFMylated at K214 [55] while it is anchored to the ER membrane. The researchers who used genome-wide CRISPR screening to identify UFL1 and UfBP1 as activators of ER-phagy [52] explored whether CYB5R3 UFMylation is involved in this process. They showed that CYB5R3 contains FAD- and NADH-binding domains and catalyzes the transfer of reducing equivalents from NADH to cytochrome b5, which then acts as an electron donor [57].
3.2.3. Autophagy
Although UFMylation regulates ER-phagy, the involvement of this PTM in general autophagy remains uncertain. Results from one study indicated that UFL1 deficiency impairs autophagy activity. LC3B associates with autophagosome development and maturation [58] and p62/SQSTM1 serves as a bridge between LC3 and polyubiquitinated proteins, which are selectively packaged into autophagosomes. Therefore, LC3B and p62/SQSTM1 reflect the levels of autophagy [59]. Indeed, UFL1 depletion in bone marrow (BM) cells resulted in increased ER stress and an increase in the abundance of LC3B and p62/SQSTM1, indicating that UFMylation regulates ER stress and general autophagy [60]. However, the knockout of UfBP1 in BM cells did not influence the levels of LC3B or p62/SQSTM1 [30]. As UfBP1 normally functions synergistically with UFL1 to promote UFMylation, the opposite effects caused by defects in these two proteins seem contradictory. Interestingly, a genome-wide CRISPR screen of neuroglioma H4 cells identified several novel modulators of p62/SQSTM1, including the UFMylation cascade, which regulates p62/SQSTM1 expression by eliciting a cell-type-specific ER stress response, although few LC3B expression changes were evident when UFM1 was depleted [61].
3.3. UFMylation and Development
UFMylation has an important impact on embryonic development. The complete depletion of UBA5 is embryonically lethal, with most UBA5−/− mice embryos succumbing between 12.5 and 13.5 embryonic days (E12.5-E13.5) after gestation. By contrast, UBA5-heterozygous (UBA5+/−) mice are born healthy and fertile without the emergence of any noticeable pathology for at least 2 years [62]. Similarly, the complete depletion of UFL1 is embryonically lethal, with most UFL1−/− mice embryos succumbing before E11.5, and as early as E10.5; UFL1+/− mice are born healthy [60]. UfBP1 depletion also causes death during embryonic development. While UfBP1+/− mice are born healthy, most UfBP1−/− mice embryos succumb by E12.5 [30]. Finally, CDK5RAP3 depletion also results in embryonic lethality by E8.5 [63].
3.3.1. Erythroid Development
An UFMylation deficiency causes the failed development of erythroid lineages [30][60][62]. An analysis of UBA5−/− mouse embryos at different developmental stages revealed a marked fetal anemia phenotype compared with UBA5+/+ mouse embryos that was rescued by transgenic expression of UBA5 in the erythroid lineage [62]. The loss of UFL1 blocked autophagic degradation and increased mitochondrial mass and ROS production in bone marrow cells, leading to the DNA damage response, p53 activation, and ER stress. This ER stress and the resulting generation of UPR enhanced hematopoietic stem cell death and impaired hematopoietic development, resulting in severe anemia, cytopenia, and ultimately animal death [48].
3.3.2. Skeletal Development
Several studies have indicated the importance of UfBP1 in cartilage growth and development. Alongside this, reports that UfBP1 mutations are involved in spondylo-epi-metaphyseal dysplasia Shohat type (SEMDSH) disease indicate that UfBP1 has important roles in skeletal development. The whole-exome sequencing of four SEMDSH-prone families revealed a splice variant of UfBP1 (c.408 + 1G > A) resulting in a premature stop codon that causes a loss of function [64]. Two unrelated SEMDSH patients were found to carry a different mutation of UfBP1 (G135K), which was associated with a similar phenotype to the UfBP1 (c.408 + 1G > A) mutation. These findings indicate that UfBP1 is associated with SEMDSH and skeletal development [65]. In support of this association, UfBP1 depletion in zebrafish embryos resulted in craniofacial defects, and the deletion of UfBP1 in mouse embryos significantly increased limb bud apoptosis and cell death. Mechanistically, UfBP1 binds directly to SOX9, a major transcription factor for chondroblasts, to inhibit SOX9 ubiquitination and proteasomal degradation. COL2A1, the downstream target of SOX9, is linked to skeletal disorders. Therefore, UfBP1 defects lead to skeletal dysplasia by disturbing the SOX9-COL2A1 axis [64].
3.3.3. Brain Development
Many studies have implicated UFMylation in brain development. Genetic studies have revealed that variants of the human UBA5, UFC1, and UFM1 genes are associated with a number of neurodevelopmental diseases, including infantile-onset encephalopathy [66], autosomal recessive cerebellar ataxia [67], and microcephaly [24]. Using exome sequencing, two groups found two biallelic mutations in UBA5 (A371T and a loss-of-function nonsense mutation) that led to postnatal microcephaly, epilepsy, and spasticity in severe epileptic syndrome patients [66][67][68]. CNS-specific knockout of UFM1 in mice caused neonatal death accompanied by microcephaly and the apoptosis of specific neurons [66].
3.3.4. Development of Other Organs and Tissues
UFMylation is also involved in the development of other organs and tissues. Nephron-tubule-specific UFL1-KO mice presented kidney atrophy and interstitial fibrosis, demonstrating the crucial role of UFL1 in regulating kidney function [69]. Hepatocyte-specific UFL1-KO induced hepatocyte apoptosis and mild steatosis in mice at 2 months of age and hepatocellular ballooning, extensive fibrosis, and steatohepatitis at 6–8 months of age [70]. The deletion of UFL1 in cardiomyocytes and intestinal epithelial cells caused heart failure and an increased susceptibility to experimentally-induced colitis, respectively, suggesting that UFL1 has an essential role in the maintenance of homeostasis in these organs [71][72].
3.4. UFMylation and Immune Response
Recent work has shed light on the important role of UFL1 in antiviral innate immunity after DNA virus infection [73]. UFL1 protein levels were significantly downregulated when peritoneal macrophages were infected with DNA viruses, such as the herpes simplex virus (HSV-1) or vaccinia virus (VACV), which also significantly decreased the mRNA expression of interferon β1, interleukin-6, and tumor necrosis factor. These results suggest that UFL1 promotes antiviral innate immunity. Further studies showed that UFL1 regulates the cGAS-STING pathway through its effects on STING stability. The E3 ligase TRIM29 ubiquitinates STING at K338/347/370, promoting its proteasome-dependent degradation [74][75]. UFL1 competitively binds to STING to inhibit K48-linked ubiquitination, thereby maintaining STING protein stability and ultimately promoting antiviral innate immunity [73].
3.5. UFMylation and Cancers
A comprehensive analysis of genomic alterations in the eight UFMylation family genes (UFM1, UBA5, UFC1, UFL1, UfBP1, CDK5RAP3, UfSP1, and UfSP2) across the TCGA database of 33 cancer types identified 55 recurrent and focal somatic copy number alteration events in UFMylation family genes [76]. Among the UFMylation genes, UfSP2 was frequently deleted in 14 cancer types. Calculations of the frequencies of copy number gain or loss for UFMylation genes in each cancer type revealed that UfSP2 (31%), UFM1 (31%), and UFL1 (28%) had the highest average frequency of copy number loss, whereas UFC1 (34%), UfSP1 (34%), and UfBP1 (30%) had the highest average frequency of copy number gain [76]. In total, 11.08% of the TCGA samples had high-level copy number alterations in at least one of the eight genes [76].
References
- Venne, A.S.; Kollipara, L.; Zahedi, R.P. The next level of complexity: Crosstalk of posttranslational modifications. Proteomics 2014, 14, 513–524.
- Doll, S.; Burlingame, A.L. Mass spectrometry-based detection and assignment of protein posttranslational modifications. ACS Chem. Biol. 2015, 10, 63–71.
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422.
- Soh, S.M.; Kim, Y.-J.; Kim, H.-H.; Lee, H.-R. Modulation of Ubiquitin Signaling in Innate Immune Response by Herpesviruses. Int. J. Mol. Sci. 2022, 23, 492.
- Çetin, G.; Klafack, S.; Studencka-Turski, M.; Krüger, E.; Ebstein, F. The Ubiquitin-Proteasome System in Immune Cells. Biomolecules 2021, 11, 60.
- Hofmann, K. Ubiquitin-binding domains and their role in the DNA damage response. DNA Repair 2009, 8, 544–556.
- Al-Hakim, A.; Escribano-Diaz, C.; Landry, M.C.; O’Donnell, L.; Panier, S.; Szilard, R.K.; Durocher, D. The ubiquitous role of ubiquitin in the DNA damage response. DNA Repair 2010, 9, 1229–1240.
- Dang, F.; Nie, L.; Wei, W. Ubiquitin signaling in cell cycle control and tumorigenesis. Cell Death Differ. 2021, 28, 427–438.
- Chen, R.H.; Chen, Y.H.; Huang, T.Y. Ubiquitin-mediated regulation of autophagy. J. Biomed. Sci. 2019, 26, 80.
- Vaughan, R.M.; Kupai, A.; Rothbart, S.B. Chromatin Regulation through Ubiquitin and Ubiquitin-like Histone Modifications. Trends Biochem. Sci. 2021, 46, 258–269.
- Abbas, R.; Larisch, S. Killing by Degradation: Regulation of Apoptosis by the Ubiquitin-Proteasome-System. Cells 2021, 10, 3465.
- Varshavsky, A. The Ubiquitin System, Autophagy, and Regulated Protein Degradation. Annu. Rev. Biochem. 2017, 86, 123–128.
- Van der Veen, A.G.; Ploegh, H.L. Ubiquitin-like proteins. Annu. Rev. Biochem. 2012, 81, 323–357.
- Cappadocia, L.; Lima, C.D. Ubiquitin-like Protein Conjugation: Structures, Chemistry, and Mechanism. Chem. Rev. 2018, 118, 889–918.
- Komatsu, M.; Chiba, T.; Tatsumi, K.; Iemura, S.; Tanida, I.; Okazaki, N.; Ueno, T.; Kominami, E.; Natsume, T.; Tanaka, K. A novel protein-conjugating system for Ufm1, a ubiquitin-fold modifier. Embo J. 2004, 23, 1977–1986.
- Kang, S.H.; Kim, G.T.; Seong, M.; Baek, S.H.; Seol, J.H.; Bang, O.S.; Ovaa, H.; Tatsumi, K.; Komatsu, M.; Tanaka, K.; et al. Two novel ubiquitin-fold modifier 1 (Ufm1)-specific proteases, UfSP1 and UfSP2. J. Biol. Chem. 2007, 282, 5256–5262.
- Wei, Y.; Xu, X. UFMylation: A Unique & Fashionable Modification for Life. Genom. Proteomics Bioinformatics 2016, 14, 140–146.
- Liang, Q.; Jin, Y.; Xu, S.; Zhou, J.; Mao, J.; Ma, X.; Wang, M.; Cong, Y.-S. Human UFSP1 translated from an upstream near-cognate initiation codon functions as an active UFM1-specific protease. J. Biol. Chem. 2022, 298, 102016.
- Millrine, D.; Cummings, T.; Matthews, S.P.; Peter, J.J.; Magnussen, H.M.; Lange, S.M.; Macartney, T.; Lamoliatte, F.; Knebel, A. Human UFSP1 is an active protease that regulates UFM1 maturation and UFMylation. Cell Rep. 2022, 40, 111168.
- Schulman, B.A.; Harper, J.W. Ubiquitin-like protein activation by E1 enzymes: The apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 2009, 10, 319–331.
- Oweis, W.; Padala, P.; Hassouna, F.; Cohen-Kfir, E.; Gibbs, D.R.; Todd, E.A.; Berndsen, C.E.; Wiener, R. Trans-Binding Mechanism of Ubiquitin-like Protein Activation Revealed by a UBA5-UFM1 Complex. Cell Rep. 2016, 16, 3113–3120.
- Taherbhoy, A.M.; Kaiser, S.E.; Schulman, B.A. Trans mechanism for ubiquitin-like protein transfer in autophagy. Cell Cycle 2012, 11, 635–636.
- Soudah, N.; Padala, P.; Hassouna, F.; Kumar, M.; Mashahreh, B.; Lebedev, A.A.; Isupov, M.N.; Cohen-Kfir, E.; Wiener, R. An N-Terminal Extension to UBA5 Adenylation Domain Boosts UFM1 Activation: Isoform-Specific Differences in Ubiquitin-like Protein Activation. J. Mol. Biol. 2019, 431, 463–478.
- Nahorski, M.S.; Maddirevula, S.; Ishimura, R.; Alsahli, S.; Brady, A.F.; Begemann, A.; Mizushima, T.; Guzmán-Vega, F.J.; Obata, M.; Ichimura, Y.; et al. Biallelic UFM1 and UFC1 mutations expand the essential role of ufmylation in brain development. Brain 2018, 141, 1934–1945.
- Liu, G.; Forouhar, F.; Eletsky, A.; Atreya, H.S.; Aramini, J.M.; Xiao, R.; Huang, Y.J.; Abashidze, M.; Seetharaman, J.; Liu, J.; et al. NMR and X-RAY structures of human E2-like ubiquitin-fold modifier conjugating enzyme 1 (UFC1) reveal structural and functional conservation in the metazoan UFM1-UBA5-UFC1 ubiquination pathway. J. Struct. Funct. Genom. 2009, 10, 127–136.
- Peter, J.J.; Magnussen, H.M.; DaRosa, P.A.; Millrine, D.; Matthews, S.P.; Lamoliatte, F.; Sundaramoorthy, R.; Kopito, R.R.; Kulathu, Y. A non-canonical scaffold-type E3 ligase complex mediates protein UFMylation. EMBO J. 2022, 41, e111015.
- Bacik, J.P.; Walker, J.R.; Ali, M.; Schimmer, A.D.; Dhe-Paganon, S. Crystal structure of the human ubiquitin-activating enzyme 5 (UBA5) bound to ATP: Mechanistic insights into a minimalistic E1 enzyme. J. Biol. Chem. 2010, 285, 20273–20280.
- Tatsumi, K.; Sou, Y.-S.; Tada, N.; Nakamura, E.; Iemura, S.-I.; Natsume, T.; Kang, S.H.; Chung, C.H.; Kasahara, M.; Kominami, E.; et al. A novel type of E3 ligase for the Ufm1 conjugation system. J. Biol. Chem. 2010, 285, 5417–5427.
- Morreale, F.E.; Walden, H. Types of Ubiquitin Ligases. Cell 2016, 165, 248.e1.
- Cai, Y.; Pi, W.; Sivaprakasam, S.; Zhu, X.; Zhang, M.; Chen, J.; Makala, L.; Lu, C.; Wu, J.; Teng, Y.; et al. UFBP1, a Key Component of the Ufm1 Conjugation System, Is Essential for Ufmylation-Mediated Regulation of Erythroid Development. PLoS Genet. 2015, 11, e1005643.
- Zhu, J.; Ma, X.; Jing, Y.; Zhang, G.; Zhang, D.; Mao, Z.; Ma, X.; Liu, H.; Chen, F. P4HB UFMylation regulates mitochondrial function and oxidative stress. Free Radic. Biol. Med. 2022, 18, 277–286.
- Yang, R.; Wang, H.; Kang, B.; Chen, B.; Shi, Y.; Yang, S.; Sun, L.; Liu, Y.; Xiao, W.; Zhang, T.; et al. CDK5RAP3, a UFL1 substrate adaptor, is crucial for liver development. Development 2019, 146, dev169235.
- Wu, J.; Lei, G.; Mei, M.; Tang, Y.; Li, H. A novel C53/LZAP-interacting protein regulates stability of C53/LZAP and DDRGK domain-containing Protein 1 (DDRGK1) and modulates NF-kappaB signaling. J. Biol. Chem. 2010, 285, 15126–15136.
- Banerjee, S.; Kumar, M.; Wiener, R. Decrypting UFMylation: How Proteins Are Modified with UFM1. Biomolecules 2020, 10, 1442.
- Witting, K.F.; Mulder, M.P.C. Highly Specialized Ubiquitin-Like Modifications: Shedding Light into the UFM1 Enigma. Biomolecules 2021, 11, 255.
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078.
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204.
- Wang, Z.; Gong, Y.; Peng, B.; Shi, R.; Fan, D.; Zhao, H.; Zhu, M.; Zhang, H.; Lou, Z.; Zhou, J.; et al. MRE11 UFMylation promotes ATM activation. Nucleic Acids Res. 2019, 47, 4124–4135.
- Qin, B.; Yu, J.; Nowsheen, S.; Zhao, F.; Wang, L.; Lou, Z. UFL1 promotes histone H4 ufmylation and ATM activation. Nat. Commun. 2019, 10, 1242.
- Qin, B.; Yu, J.; Zhao, F.; Huang, J.; Zhou, Q.; Lou, Z. Dynamic recruitment of UFM1-specific peptidase 2 to the DNA double-strand breaks regulated by WIP1. Genome Instab. Dis. 2022, 3, 217–226.
- Qin, B.; Yu, J.; Nowsheen, S.; Zhao, F.; Wang, L.; Lou, Z. STK38 promotes ATM activation by acting as a reader of histone H4 ufmylation. Sci. Adv. 2020, 6, eaax8214.
- Di Conza, G.; Ho, P.C. ER Stress Responses: An Emerging Modulator for Innate Immunity. Cells 2020, 9, 695.
- Gubas, A.; Dikic, I. ER remodeling via ER-phagy. Mol. Cell 2022, 82, 1492–1500.
- Azfer, A.; Niu, J.; Rogers, L.M.; Adamski, F.M.; Kolattukudy, P.E.; Miller, C.; Cai, Y.; Patton, T.; Graves, S.H.; Li, H.; et al. Activation of endoplasmic reticulum stress response during the development of ischemic heart disease. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1411–H1420.
- Yoo, H.M.; Kang, S.H.; Kim, Y.J.; Lee, J.E.; Seong, M.W.; Lee, S.W.; Ka, S.H.; Sou, Y.-S.; Komatsu, M.; Tanaka, K.; et al. Modification of ASC1 by UFM1 is crucial for ERα transactivation and breast cancer development. Mol. Cell 2014, 56, 261–274.
- Lemaire, K.; Moura, R.F.; Granvik, M.; Igoillo-Esteve, M.; Hohmeier, H.E.; Hendrickx, N.; Newgard, C.B.; Waelkens, E.; Cnop, M.; Schuit, F. Ubiquitin fold modifier 1 (UFM1) and its target UFBP1 protect pancreatic beta cells from ER stress-induced apoptosis. PLoS ONE 2011, 6, e18517.
- Stephani, M.; Picchianti, L.; Gajic, A.; Beveridge, R.; Skarwan, E.; de Medina Hernandez, V.; Mohseni, A.; Clavel, M.; Zheng, Y.; Naumann, C.; et al. A cross-kingdom conserved ER-phagy receptor maintains endoplasmic reticulum homeostasis during stress. eLife 2020, 9, e58396.
- Tang, X.; Dong, H.; Fang, Z.; Li, J.; Yang, Q.; Yao, T.; Pan, Z. Ubiquitin-like modifier 1 ligating enzyme 1 relieves cisplatin-induced premature ovarian failure by reducing endoplasmic reticulum stress in granulosa cells. Reprod. Biol. Endocrinol. 2022, 20, 84.
- Zhang, Y.; Zhang, M.; Wu, J.; Lei, W.; Li, H. Transcriptional regulation of the Ufm1 conjugation system in response to disturbance of the endoplasmic reticulum homeostasis and inhibition of vesicle trafficking. PLoS ONE 2012, 7, e48587.
- Klebanovych, A.; Vinopal, S.; Dráberová, E.; Sládková, V.; Sulimenko, T.; Sulimenko, V.; Vosecká, V.; Macůrek, L.; Legido, A.; Dráber, P. C53 Interacting with UFM1-Protein Ligase 1 Regulates Microtubule Nucleation in Response to ER Stress. Cells 2022, 11, 555.
- Hořejší, B.; Vinopal, S.; Sládková, V.; Dráberová, E.; Sulimenko, V.; Sulimenko, T.; Vosecká, V.; Philimonenko, A.; Hozák, P.; Katsetos, C.D.; et al. Nuclear γ-tubulin associates with nucleoli and interacts with tumor suppressor protein C53. J. Cell. Physiol. 2012, 227, 367–382.
- Liang, J.R.; Lingeman, E.; Luong, T.; Ahmed, S.; Muhar, M.; Nguyen, T.; Olzmann, J.A.; Corn, J.E. A Genome-wide ER-phagy Screen Highlights Key Roles of Mitochondrial Metabolism and ER-Resident UFMylation. Cell 2020, 180, 1160–1177.e20.
- Walczak, C.P.; Leto, D.E.; Zhang, L.; Riepe, C.; Muller, R.T.; DaRosa, P.A.; Ingolia, N.T.; Elias, J.E.; Kopito, R.R. Ribosomal protein RPL26 is the principal target of UFMylation. Proc. Natl. Acad. Sci. USA 2019, 116, 1299–1308.
- Wang, L.; Xu, Y.; Rogers, H.; Saidi, L.; Tom Noguchi, C.; Li, H.; Yewdell, J.W.; Guydosh, N.R.; Ye, Y. UFMylation of RPL26 links translocation-associated quality control to endoplasmic reticulum protein homeostasis. Cell Res. 2020, 30, 5–20.
- Ishimura, R.; El-Gowily, A.H.; Noshiro, D.; Komatsu-Hirota, S.; Ono, Y.; Shindo, M.; Hatta, T.; Abe, M.; Uemura, T.; Lee-Okada, H.-C.; et al. The UFM1 system regulates ER-phagy through the ufmylation of CYB5R3. Nat. Commun. 2022, 13, 7857.
- Simsek, D.; Tiu, C.C.; Flynn, R.A.; Byeon, G.W.; Leppek, K.; Xu, A.F.; Chang, H.Y.; Barna, M. The Mammalian Ribo-interactome Reveals Ribosome Functional Diversity and Heterogeneity. Cell 2017, 169, 1051–1065.e18.
- Percy, M.J.; Lappin, T.R. Recessive congenital methaemoglobinaemia: Cytochrome b(5) reductase deficiency. Br. J. Haematol. 2008, 141, 298–308.
- Schaaf, M.B.E.; Keulers, T.G.; Vooijs, M.A.; Rouschop, K.M.A. LC3/GABARAP family proteins: Autophagy-(un)related functions. FASEB J. 2016, 30, 3961–3978.
- Lamark, T.; Svenning, S.; Johansen, T. Regulation of selective autophagy: The p62/SQSTM1 paradigm. Essays Biochem. 2017, 61, 609–624.
- Zhang, M.; Zhu, X.; Zhang, Y.; Cai, Y.; Chen, J.; Sivaprakasam, S.; Gurav, A.; Pi, W.; Makala, L.; Wu, J.; et al. RCAD/Ufl1, a Ufm1 E3 ligase, is essential for hematopoietic stem cell function and murine hematopoiesis. Cell Death Differ. 2015, 22, 1922–1934.
- DeJesus, R.; Moretti, F.; McAllister, G.; Wang, Z.; Bergman, P.; Liu, S.; Frias, E.; Alford, J.; Reece-Hoyes, J.S.; Lindeman, A.; et al. Functional CRISPR screening identifies the ufmylation pathway as a regulator of SQSTM1/p62. eLife 2016, 5, e17290.
- Tatsumi, K.; Yamamoto-Mukai, H.; Shimizu, R.; Waguri, S.; Sou, Y.-S.; Sakamoto, A.; Taya, C.; Shitara, H.; Hara, T.; Chung, C.H.; et al. The Ufm1-activating enzyme Uba5 is indispensable for erythroid differentiation in mice. Nat. Commun. 2011, 2, 181.
- Quintero, M.; Liu, S.; Xia, Y.; Huang, Y.; Zou, Y.; Li, G.; Hu, L.; Singh, N.; Blumberg, R.; Cai, Y.; et al. Cdk5rap3 is essential for intestinal Paneth cell development and maintenance. Cell Death Dis. 2021, 12, 131.
- Egunsola, A.T.; Bae, Y.; Jiang, M.-M.; Liu, D.S.; Chen-Evenson, Y.; Bertin, T.; Chen, S.; Lu, J.T.; Nevarez, L.; Magal, N.; et al. Loss of DDRGK1 modulates SOX9 ubiquitination in spondyloepimetaphyseal dysplasia. J. Clin. Investig. 2017, 127, 1475–1484.
- Franceschi, R.; Iascone, M.; Maitz, S.; Marchetti, D.; Mariani, M.; Selicorni, A.; Soffiati, M.; Maines, E. A missense mutation in DDRGK1 gene associated to Shohat-type spondyloepimetaphyseal dysplasia: Two case reports and a review of literature. Am. J. Med. Genet. A 2022, 188, 2434–2437.
- Muona, M.; Ishimura, R.; Laari, A.; Ichimura, Y.; Linnankivi, T.; Keski-Filppula, R.; Herva, R.; Rantala, H.; Paetau, A.; Pöyhönen, M.; et al. Biallelic Variants in UBA5 Link Dysfunctional UFM1 Ubiquitin-like Modifier Pathway to Severe Infantile-Onset Encephalopathy. Am. J. Hum. Genet. 2016, 99, 683–694.
- Duan, R.; Shi, Y.; Yu, L.; Zhang, G.; Li, J.; Lin, Y.; Guo, J.; Wang, J.; Shen, L.; Jiang, H.; et al. UBA5 Mutations Cause a New Form of Autosomal Recessive Cerebellar Ataxia. PLoS ONE 2016, 11, e0149039.
- Colin, E.; Daniel, J.; Ziegler, A.; Wakim, J.; Scrivo, A.; Haack, T.B.; Khiati, S.; Denommé, A.-S.; Amati-Bonneau, P.; Charif, M.; et al. Biallelic Variants in UBA5 Reveal that Disruption of the UFM1 Cascade Can Result in Early-Onset Encephalopathy. Am. J. Hum. Genet. 2016, 99, 695–703.
- Zhou, Y.; Ye, X.; Zhang, C.; Wang, J.; Guan, Z.; Yan, J.; Xu, L.; Wang, K.; Guan, D.; Liang, Q.; et al. Ufl1 deficiency causes kidney atrophy associated with disruption of endoplasmic reticulum homeostasis. J. Genet. Genom. 2021, 48, 403–410.
- Chen, F.; Sheng, L.; Zhou, T.; Yan, L.; Loveless, R.; Li, H.; Teng, Y.; Cai, Y. Loss of Ufl1/Ufbp1 in hepatocytes promotes liver pathological damage and carcinogenesis through activating mTOR signaling. J. Exp. Clin. Cancer Res. 2023, 42, 110.
- Li, J.; Yue, G.; Ma, W.; Zhang, A.; Zou, J.; Cai, Y.; Tang, X.; Wang, J.; Liu, J.; Li, H.; et al. Ufm1-Specific Ligase Ufl1 Regulates Endoplasmic Reticulum Homeostasis and Protects Against Heart Failure. Circ. Heart. Fail. 2018, 11, e004917.
- Cai, Y.; Zhu, G.; Liu, S.; Pan, Z.; Quintero, M.; Poole, C.J.; Lu, C.; Zhu, H.; Islam, B.; van Riggelen, J.; et al. Indispensable role of the Ubiquitin-fold modifier 1-specific E3 ligase in maintaining intestinal homeostasis and controlling gut inflammation. Cell Discov. 2019, 5, 7.
- Tao, Y.; Yin, S.; Liu, Y.; Li, C.; Chen, Y.; Han, D.; Huang, J.; Xu, S.; Zou, Z.; Yu, Y. UFL1 promotes antiviral immune response by maintaining STING stability independent of UFMylation. Cell Death Differ. 2023, 30, 16–26.
- Li, Q.; Lin, L.; Tong, Y.; Liu, Y.; Mou, J.; Wang, X.; Wang, X.; Gong, Y.; Zhao, Y.; Liu, Y.; et al. TRIM29 negatively controls antiviral immune response through targeting STING for degradation. Cell Discov. 2018, 4, 13.
- Xing, J.; Zhang, A.; Zhang, H.; Wang, J.; Li, X.C.; Zeng, M.-S.; Zhang, Z. TRIM29 promotes DNA virus infections by inhibiting innate immune response. Nat. Commun. 2017, 8, 945.
- Zhou, J.; Ma, X.; Xu, L.; Liang, Q.; Mao, J.; Liu, J.; Wang, M.; Yuan, J.; Cong, Y. Genomic profiling of the UFMylation family genes identifies UFSP2 as a potential tumour suppressor in colon cancer. Clin. Transl. Med. 2021, 11, e642.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
791
Revisions:
2 times
(View History)
Update Date:
19 Jan 2024
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No